
Invadopodia, a Kingdom of Non-Receptor Tyrosine Kinases



Abstract
:
1. Introduction
2. The Secret Life of Invadopodia
3. The Dual Face of Src Kinase
4. Grow or Go
5. The Emperor of All Kinases
6. Migrate or Invade: A Game of Thrones
7. The Other Side of The Coin
8. The Hitchhiker
9. Concluding Remarks and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Weaver, A.M. Invadopodia: Specialized Cell Structures for Cancer Invasion. Clin. Exp. Metastasis 2006, 23, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Weaver, A.M. Invadopodia. Curr. Biol. 2008, 18, 362–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monsky, W.L.; Lin, C.Y.; Aoyama, A.; Kelly, T.; Akiyama, S.K.; Mueller, S.C.; Chen, W.T. A Potential Marker Protease of Invasiveness, Seprase, Is Localized on Invadopodia of Human Malignant Melanoma Cells. Cancer Res. 1994, 54, 5702–5710. [Google Scholar] [PubMed]
- Stylli, S.S.; Kaye, A.H.; Lock, P. Invadopodia: At the Cutting Edge of Tumour Invasion. J. Clin. Neurosci. 2008, 15, 725–737. [Google Scholar] [CrossRef] [PubMed]
- Sutoh, M.; Hashimoto, Y.; Yoneyama, T.; Yamamoto, H.; Hatakeyama, S.; Koie, T.; Okamoto, A.; Yamaya, K.; Saitoh, H.; Funyu, T.; et al. Invadopodia Formation by Bladder Tumor Cells. Oncol. Res. 2010, 19, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Sutoh, M.; Hatakeyama, S.; Hashimoto, Y.; Yoneyama, T.; Koie, T.; Saitoh, H.; Yamaya, K.; Funyu, T.; Nakamura, T.; et al. Requirement for FBP17 in Invadopodia Formation by Invasive Bladder Tumor Cells. J. Urol. 2011, 185, 1930–1938. [Google Scholar] [CrossRef] [PubMed]
- Gil-Henn, H.; Patsialou, A.; Wang, Y.; Warren, M.S.; Condeelis, J.S.; Koleske, A.J. Arg/Abl2 Promotes Invasion and Attenuates Proliferation of Breast Cancer In Vivo. Oncogene 2013, 32, 2622–2630. [Google Scholar] [CrossRef] [Green Version]
- Gligorijevic, B.; Wyckoff, J.; Yamaguchi, H.; Wang, Y.; Roussos, E.T.; Condeelis, J. N-WASP-Mediated Invadopodium Formation Is Involved in Intravasation and Lung Metastasis of Mammary Tumors. J. Cell Sci. 2012, 125, 724–734. [Google Scholar] [CrossRef] [Green Version]
- Eckert, M.A.; Lwin, T.M.; Chang, A.T.; Kim, J.; Danis, E.; Ohno-Machado, L.; Yang, J. Twist1-Induced Invadopodia Formation Promotes Tumor Metastasis. Cancer Cell 2011, 19, 372–386. [Google Scholar] [CrossRef] [Green Version]
- Gligorijevic, B.; Bergman, A.; Condeelis, J. Multiparametric Classification Links Tumor Microenvironments with Tumor Cell Phenotype. PLoS Biol. 2014, 12, e1001995. [Google Scholar] [CrossRef]
- Chen, W.T. Proteolytic Activity of Specialized Surface Protrusions Formed at Rosette Contact Sites of Transformed Cells. J. Exp. Zool. 1989, 251, 167–185. [Google Scholar] [CrossRef]
- Eddy, R.J.; Weidmann, M.D.; Sharma, V.P.; Condeelis, J.S. Tumor Cell Invadopodia: Invasive Protrusions that Orchestrate Metastasis. Trends Cell Biol. 2017, 27, 595–607. [Google Scholar] [CrossRef]
- Sharma, V.P.; Eddy, R.; Entenberg, D.; Kai, M.; Gertler, F.B.; Condeelis, J. Tks5 and SHIP2 Regulate Invadopodium Maturation, but Not Initiation, in Breast Carcinoma Cells. Curr. Biol. 2013, 23, 2079–2089. [Google Scholar] [CrossRef] [Green Version]
- Carmona, G.; Perera, U.; Gillett, C.; Naba, A.; Law, A.L.; Sharma, V.P.; Wang, J.; Wyckoff, J.; Balsamo, M.; Mosis, F.; et al. Lamellipodin Promotes Invasive 3D Cancer Cell Migration via Regulated Interactions with Ena/VASP and SCAR/WAVE. Oncogene 2016, 35, 5155–5169. [Google Scholar] [CrossRef]
- Beaty, B.T.; Sharma, V.P.; Bravo-Cordero, J.J.; Simpson, M.A.; Eddy, R.J.; Koleske, A.J.; Condeelis, J. beta1 Integrin Regulates Arg to Promote Invadopodial Maturation and Matrix Degradation. Mol. Biol. Cell 2013, 24, 1661–1675. [Google Scholar] [CrossRef]
- Oser, M.; Mader, C.C.; Gil-Henn, H.; Magalhaes, M.; Bravo-Cordero, J.J.; Koleske, A.J.; Condeelis, J. Specific Tyrosine Phosphorylation Sites on Cortactin Regulate Nck1-Dependent Actin Polymerization in Invadopodia. J. Cell Sci. 2010, 123, 3662–3673. [Google Scholar] [CrossRef] [Green Version]
- Beaty, B.T.; Wang, Y.; Bravo-Cordero, J.J.; Sharma, V.P.; Miskolci, V.; Hodgson, L.; Condeelis, J. Talin Regulates Moesin-NHE-1 Recruitment to Invadopodia and Promotes Mammary Tumor Metastasis. J. Cell Biol. 2014, 205, 737–751. [Google Scholar] [CrossRef] [Green Version]
- Magalhaes, M.A.; Larson, D.R.; Mader, C.C.; Bravo-Cordero, J.J.; Gil-Henn, H.; Oser, M.; Chen, X.; Koleske, A.J.; Condeelis, J. Cortactin Phosphorylation Regulates Cell Invasion Through a pH-Dependent Pathway. J. Cell Biol. 2011, 195, 903–920. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, B.J.; Gil-Henn, H.; Mader, C.C.; Halo, T.; Yin, T.; Condeelis, J.; Machida, K.; Wu, Y.I.; Koleske, A.J. Phosphorylated Cortactin Recruits Vav2 Guanine Nucleotide Exchange Factor to Activate Rac3 and Promote Invadopodial Function in Invasive Breast Cancer Cells. Mol. Biol. Cell 2017, 28, 1347–1360. [Google Scholar] [CrossRef] [Green Version]
- Donnelly, S.K.; Cabrera, R.; Mao, S.P.H.; Christin, J.R.; Wu, B.; Guo, W.; Bravo-Cordero, J.J.; Condeelis, J.S.; Segall, J.E.; Hodgson, L. Rac3 Regulates Breast Cancer Invasion and Metastasis by Controlling Adhesion and Matrix Degradation. J. Cell Biol. 2017, 216, 4331–4349. [Google Scholar] [CrossRef]
- Schoumacher, M.; Goldman, R.D.; Louvard, D.; Vignjevic, D.M. Actin, Microtubules, and Vimentin Intermediate Filaments Cooperate for Elongation of Invadopodia. J. Cell Biol. 2010, 189, 541–556. [Google Scholar] [CrossRef] [Green Version]
- Monteiro, P.; Rosse, C.; Castro-Castro, A.; Irondelle, M.; Lagoutte, E.; Paul-Gilloteaux, P.; Desnos, C.; Formstecher, E.; Darchen, F.; Perrais, D.; et al. Endosomal WASH and Exocyst Complexes Control Exocytosis of MT1-MMP at Invadopodia. J. Cell Biol. 2013, 203, 1063–1079. [Google Scholar] [CrossRef] [Green Version]
- Sinha, S.; Hoshino, D.; Hong, N.H.; Kirkbride, K.C.; Grega-Larson, N.E.; Seiki, M.; Tyska, M.J.; Weaver, A.M. Cortactin Promotes Exosome Secretion by Controlling Branched Actin Dynamics. J. Cell Biol. 2016, 214, 197–213. [Google Scholar] [CrossRef]
- Yeatman, T.J. A Renaissance for SRC. Nat. Rev. Cancer 2004, 4, 470–480. [Google Scholar] [CrossRef]
- Artym, V.V.; Zhang, Y.; Seillier-Moiseiwitsch, F.; Yamada, K.M.; Mueller, S.C. Dynamic Interactions of Cortactin and Membrane Type 1 Matrix Metalloproteinase at Invadopodia: Defining the Stages of Invadopodia Formation and Function. Cancer Res. 2006, 66, 3034–3043. [Google Scholar] [CrossRef] [Green Version]
- Chan, K.T.; Cortesio, C.L.; Huttenlocher, A. FAK Alters Invadopodia and Focal Adhesion Composition and Dynamics To Regulate Breast Cancer Invasion. J. Cell Biol. 2009, 185, 357–370. [Google Scholar] [CrossRef] [Green Version]
- Oser, M.; Yamaguchi, H.; Mader, C.C.; Bravo-Cordero, J.J.; Arias, M.; Chen, X.; Desmarais, V.; van Rheenen, J.; Koleske, A.J.; Condeelis, J. Cortactin Regulates Cofilin and N-WASp Activities to Control the Stages of Invadopodium Assembly and Maturation. J. Cell Biol. 2009, 186, 571–587. [Google Scholar] [CrossRef] [Green Version]
- Mader, C.C.; Oser, M.; Magalhaes, M.A.; Bravo-Cordero, J.J.; Condeelis, J.; Koleske, A.J.; Gil-Henn, H. An EGFR-Src-Arg-Cortactin Pathway Mediates Functional Maturation of Invadopodia and Breast Cancer Cell Invasion. Cancer Res. 2011, 71, 1730–1741. [Google Scholar] [CrossRef] [Green Version]
- Saini, P.; Courtneidge, S.A. Tks Adaptor Proteins at a Glance. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [Green Version]
- Stylli, S.S.; Stacey, T.T.; Verhagen, A.M.; Xu, S.S.; Pass, I.; Courtneidge, S.A.; Lock, P. Nck Adaptor Proteins Link Tks5 to Invadopodia Actin Regulation and ECM Degradation. J. Cell Sci. 2009, 122, 2727–2740. [Google Scholar] [CrossRef] [Green Version]
- Weaver, A.M. Regulation of Cancer Invasion by Reactive Oxygen Species and Tks Family Scaffold Proteins. Sci. Signal. 2009, 2, pe56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abram, C.L.; Seals, D.F.; Pass, I.; Salinsky, D.; Maurer, L.; Roth, T.M.; Courtneidge, S.A. The Adaptor Protein Fish Associates with Members of the ADAMs Family and Localizes to Podosomes of Src-Transformed Cells. J. Biol. Chem. 2003, 278, 16844–16851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacob, A.; Linklater, E.; Bayless, B.A.; Lyons, T.; Prekeris, R. The Role and Regulation of Rab40b-Tks5 Complex During Invadopodia Formation and Cancer Cell Invasion. J. Cell Sci. 2016, 129, 4341–4353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradley, W.D.; Koleske, A.J. Regulation of Cell Migration and Morphogenesis by Abl-Family Kinases: Emerging Mechanisms and Physiological Contexts. J. Cell Sci. 2009, 122, 3441–3454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patsialou, A.; Wang, Y.; Lin, J.; Whitney, K.; Goswami, S.; Kenny, P.A.; Condeelis, J.S. Selective Gene-Expression Profiling of Migratory Tumor Cells In Vivo Predicts Clinical Outcome in Breast Cancer Patients. Breast Cancer Res. 2012, 14, R139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patsialou, A.; Condeelis, J.S. Metastatic Cells: Moving Onco-Targets. Oncotarget 2014, 5, 3424–3425. [Google Scholar] [CrossRef] [PubMed]
- Meirson, T.; Genna, A.; Lukic, N.; Makhnii, T.; Alter, J.; Sharma, V.P.; Wang, Y.; Samson, A.O.; Condeelis, J.S.; Gil-Henn, H. Targeting Invadopodia-Mediated Breast Cancer Metastasis by Using ABL Kinase Inhibitors. Oncotarget 2018, 9, 22158–22183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipinski, C.A.; Loftus, J.C. Targeting Pyk2 for Therapeutic Intervention. Expert Opin. Ther. Targets 2010, 14, 95–108. [Google Scholar] [CrossRef]
- Selitrennik, M.; Lev, S. PYK2 Integrates Growth Factor and Cytokine Receptors Signaling and Potentiates Breast Cancer Invasion via a Positive Feedback Loop. Oncotarget 2015, 6, 22214–22226. [Google Scholar] [CrossRef] [Green Version]
- Genna, A.; Lapetina, S.; Lukic, N.; Twafra, S.; Meirson, T.; Sharma, V.P.; Condeelis, J.S.; Gil-Henn, H. Pyk2 and FAK Differentially Regulate Invadopodia Formation and Function in Breast Cancer Cells. J. Cell Biol. 2018, 217, 375–395. [Google Scholar] [CrossRef] [Green Version]
- Shoval, O.; Alon, U. SnapShot: Network Motifs. Cell 2010, 143, 326. [Google Scholar] [CrossRef] [Green Version]
- Alon, U. Network motifs: Theory and Experimental Approaches. Nat. Rev. Genet. 2007, 8, 450–461. [Google Scholar] [CrossRef]
- Azeloglu, E.U.; Iyengar, R. Signaling Networks: Information Flow, Computation, and Decision Making. Cold Spring Harb. Perspect. Biol. 2015, 7, a005934. [Google Scholar] [CrossRef] [Green Version]
- Meirson, T.; Gil-Henn, H. Targeting Invadopodia for Blocking Breast Cancer Metastasis. Drug Resist. Update 2018, 39, 1–17. [Google Scholar] [CrossRef]
- Riggs, D.; Yang, Z.; Kloss, J.; Loftus, J.C. The Pyk2 FERM Regulates Pyk2 Complex Formation and Phosphorylation. Cell Signal. 2011, 23, 288–296. [Google Scholar] [CrossRef] [Green Version]
- Kohno, T.; Matsuda, E.; Sasaki, H.; Sasaki, T. Protein-Tyrosine Kinase CAKbeta/PYK2 Is Activated by Binding Ca2+/Calmodulin to FERM F2 alpha2 Helix and Thus Forming Its Dimer. Biochem. J. 2008, 410, 513–523. [Google Scholar] [CrossRef] [Green Version]
- Lu, F.; Sun, J.; Zheng, Q.; Li, J.; Hu, Y.; Yu, P.; He, H.; Zhao, Y.; Wang, X.; Yang, S.; et al. Imaging Elemental Events of Store-Operated Ca(2+) Entry in Invading Cancer Cells With Plasmalemmal Targeted Sensors. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef] [Green Version]
- Burger, K.L.; Learman, B.S.; Boucherle, A.K.; Sirintrapun, S.J.; Isom, S.; Diaz, B.; Courtneidge, S.A.; Seals, D.F. Src-Dependent Tks5 Phosphorylation Regulates Invadopodia-Associated Invasion in Prostate Cancer Cells. Prostate 2014, 74, 134–148. [Google Scholar] [CrossRef] [Green Version]
- Oikawa, T.; Oyama, M.; Kozuka-Hata, H.; Uehara, S.; Udagawa, N.; Saya, H.; Matsuo, K. Tks5-Dependent Formation of Circumferential Podosomes/Invadopodia Mediates Cell-Cell Fusion. J. Cell Biol. 2012, 197, 553–568. [Google Scholar] [CrossRef] [Green Version]
- Lahlou, H.; Sanguin-Gendreau, V.; Zuo, D.; Cardiff, R.D.; McLean, G.W.; Frame, M.C.; Muller, W.J. Mammary Epithelial-Specific Disruption of the Focal Adhesion Kinase Blocks Mammary Tumor Progression. Proc. Natl. Acad. Sci. USA 2007, 104, 20302–20307. [Google Scholar] [CrossRef] [Green Version]
- Pylayeva, Y.; Gillen, K.M.; Gerald, W.; Beggs, H.E.; Reichardt, L.F.; Giancotti, F.G. Ras- and PI3K-Dependent Breast Tumorigenesis in Mice and Humans Requires Focal Adhesion Kinase Signaling. J. Clin. Investig. 2009, 119, 252–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Provenzano, P.P.; Inman, D.R.; Eliceiri, K.W.; Beggs, H.E.; Keely, P.J. Mammary Epithelial-Specific Disruption of Focal Adhesion Kinase Retards Tumor Formation and Metastasis in a Transgenic Mouse Model of Human Breast Cancer. Am. J. Pathol. 2008, 173, 1551–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genna, A.; Gil-Henn, H. FAK Family Kinases: The Yin and Yang of Cancer Cell Invasiveness. Mol. Cell Oncol. 2018, 5, e1449584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.K.; Man, K.; Ng, K.T.; Ho, J.W.; Lim, Z.X.; Cheng, Q.; Lo, C.M.; Poon, R.T.; Fan, S.T. Proline-Rich Tyrosine Kinase 2 (Pyk2) Promotes Proliferation and Invasiveness of Hepatocellular Carcinoma Cells Through C-Src/ERK Activation. Carcinogenesis 2008, 29, 2096–2105. [Google Scholar] [CrossRef] [Green Version]
- Vultur, A.; Buettner, R.; Kowolik, C.; Liang, W.; Smith, D.; Boschelli, F.; Jove, R. SKI-606 (Bosutinib), a Novel Src Kinase Inhibitor, Suppresses Migration and Invasion of Human Breast Cancer Cells. Mol. Cancer Ther. 2008, 7, 1185–1194. [Google Scholar] [CrossRef] [Green Version]
- Hunter, T. Protein Kinases and Phosphatases: The Yin and Yang of Protein Phosphorylation and Signaling. Cell 1995, 80, 225–236. [Google Scholar] [CrossRef] [Green Version]
- Elson, A. Stepping Out of the Shadows: Oncogenic and Tumor-Promoting Protein Tyrosine Phosphatases. Int. J. Biochem. Cell Biol. 2018, 96, 135–147. [Google Scholar] [CrossRef]
- Cortesio, C.L.; Chan, K.T.; Perrin, B.J.; Burton, N.O.; Zhang, S.; Zhang, Z.Y.; Huttenlocher, A. Calpain 2 and PTP1B Function in a Novel Pathway With Src To Regulate Invadopodia Dynamics and Breast Cancer Cell Invasion. J. Cell Biol. 2008, 180, 957–971. [Google Scholar] [CrossRef] [Green Version]
- Weidmann, M.D.; Surve, C.R.; Eddy, R.J.; Chen, X.; Gertler, F.B.; Sharma, V.P.; Condeelis, J.S. Mena(INV) Dysregulates Cortactin Phosphorylation To Promote Invadopodium Maturation. Sci. Rep. 2016, 6, 36142. [Google Scholar] [CrossRef] [Green Version]
- Nordenstedt, H.; White, D.L.; El-Serag, H.B. The Changing Pattern of Epidemiology in Hepatocellular Carcinoma. Dig. Liver Dis. 2010, 42, 206–214. [Google Scholar] [CrossRef] [Green Version]
- Yuki, K.; Hirohashi, S.; Sakamoto, M.; Kanai, T.; Shimosato, Y. Growth and Spread of Hepatocellular Carcinoma. A Review of 240 Consecutive Autopsy Cases. Cancer 1990, 66, 2174–2179. [Google Scholar] [CrossRef]
- Ninio, L.; Nissani, A.; Meirson, T.; Domovitz, T.; Genna, A.; Twafra, S.; Srikanth, K.D.; Dabour, R.; Avraham, E.; Davidovich, A.; et al. Hepatitis C Virus Enhances the Invasiveness of Hepatocellular Carcinoma via EGFR-Mediated Invadopodia Formation and Activation. Cells 2019, 8. [Google Scholar] [CrossRef] [Green Version]
- Harper, K.L.; Sosa, M.S.; Entenberg, D.; Hosseini, H.; Cheung, J.F.; Nobre, R.; Avivar-Valderas, A.; Nagi, C.; Girnius, N.; Davis, R.J.; et al. Mechanism of Early Dissemination and Metastasis in Her2(+) Mammary Cancer. Nature 2016, 540, 588–592. [Google Scholar] [CrossRef]
- Patsialou, A.; Wang, Y.; Pignatelli, J.; Chen, X.; Entenberg, D.; Oktay, M.; Condeelis, J.S. Autocrine CSF1R Signaling Mediates Switching Between Invasion and Proliferation Downstream of TGFbeta in Claudin-Low Breast Tumor Cells. Oncogene 2015, 34, 2721–2731. [Google Scholar] [CrossRef] [Green Version]
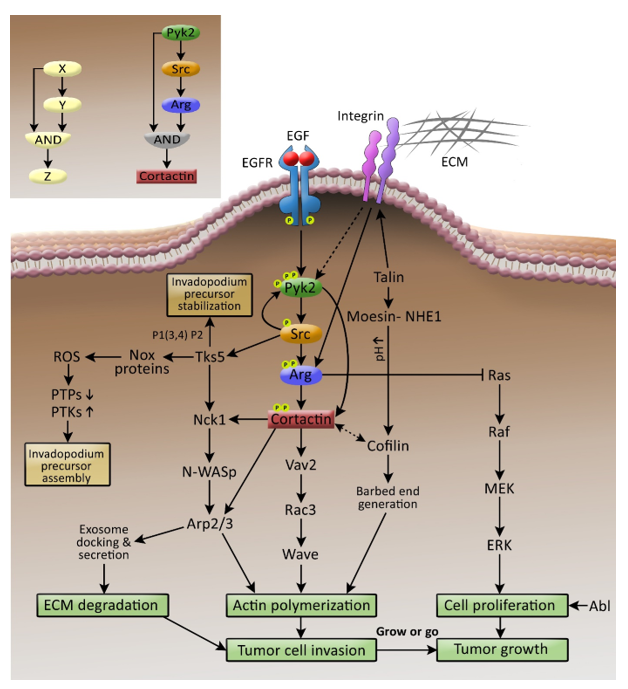

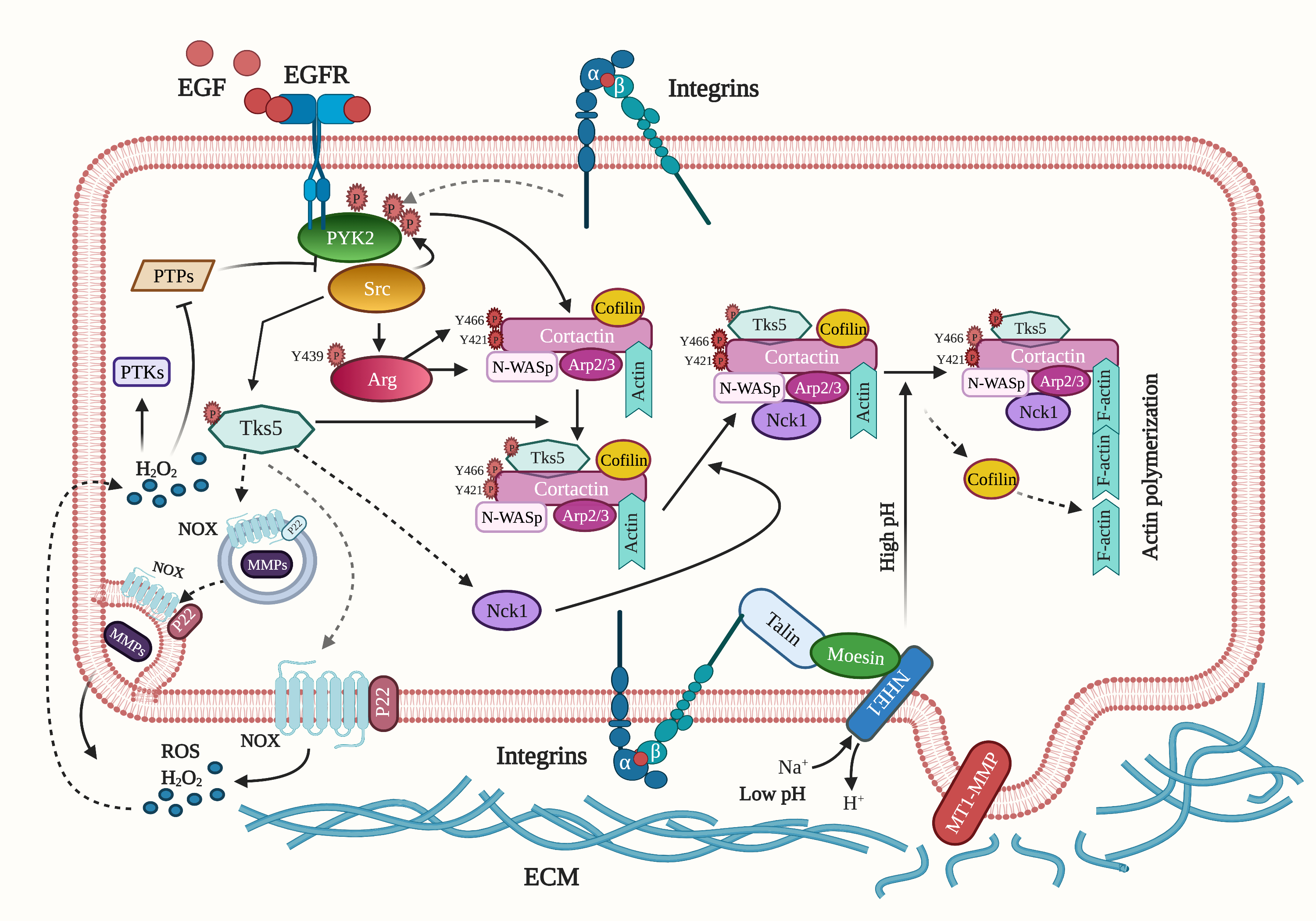{kind=link}
{kind=link}
| NRTK | Target | Role in Invadopodia | Reference |
|---|---|---|---|
| Pyk2 | Src | Pyk2 recruits and activates Src in invadopodia. | [40] |
| Cortactin | Pyk2 tyrosine phosphorylates cortactin both directly and indirectly by recruiting and activating Src, which activates Arg to phosphorylate cortactin. | [40] | |
| FAK | Src | FAK indirectly regulates invadopodium precursor formation and activation by sequestering Src to focal adhesions and away from invadopodia. | [26] |
| Src | Tks5 | Src tyrosine phosphorylates Tks5 to regulate invadopodium precursor formation and stabilization. | [30,48,49] |
| Arg | Src phosphorylates and activates Arg, which phosphorylates cortactin during invadopodium maturation and activation | [28] | |
| Arg | Cortactin | Arg tyrosine phosphorylates cortactin in invadopodia, leading to invadopodium maturation and ECM degradation. | [28] |
| Ras | Arg inhibits the Ras-MAPK pathway to block tumor cell proliferation. | [7] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saha, T.; Gil-Henn, H. Invadopodia, a Kingdom of Non-Receptor Tyrosine Kinases. Cells 2021, 10, 2037. https://doi.org/10.3390/cells10082037
Saha T, Gil-Henn H. Invadopodia, a Kingdom of Non-Receptor Tyrosine Kinases. Cells. 2021; 10(8):2037. https://doi.org/10.3390/cells10082037
Chicago/Turabian StyleSaha, Trishna, and Hava Gil-Henn. 2021. "Invadopodia, a Kingdom of Non-Receptor Tyrosine Kinases" Cells 10, no. 8: 2037. https://doi.org/10.3390/cells10082037
APA StyleSaha, T., & Gil-Henn, H. (2021). Invadopodia, a Kingdom of Non-Receptor Tyrosine Kinases. Cells, 10(8), 2037. https://doi.org/10.3390/cells10082037