
Integral Membrane Protein 2A Is a Negative Regulator of Canonical and Non-Canonical Hedgehog Signalling



Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Lines
2.3. Cell Culture
2.4. Quantitative PCR
2.5. Co-Immunoprecipitation
2.6. Gli-Luciferase Assay
2.7. Mass Spectrometry
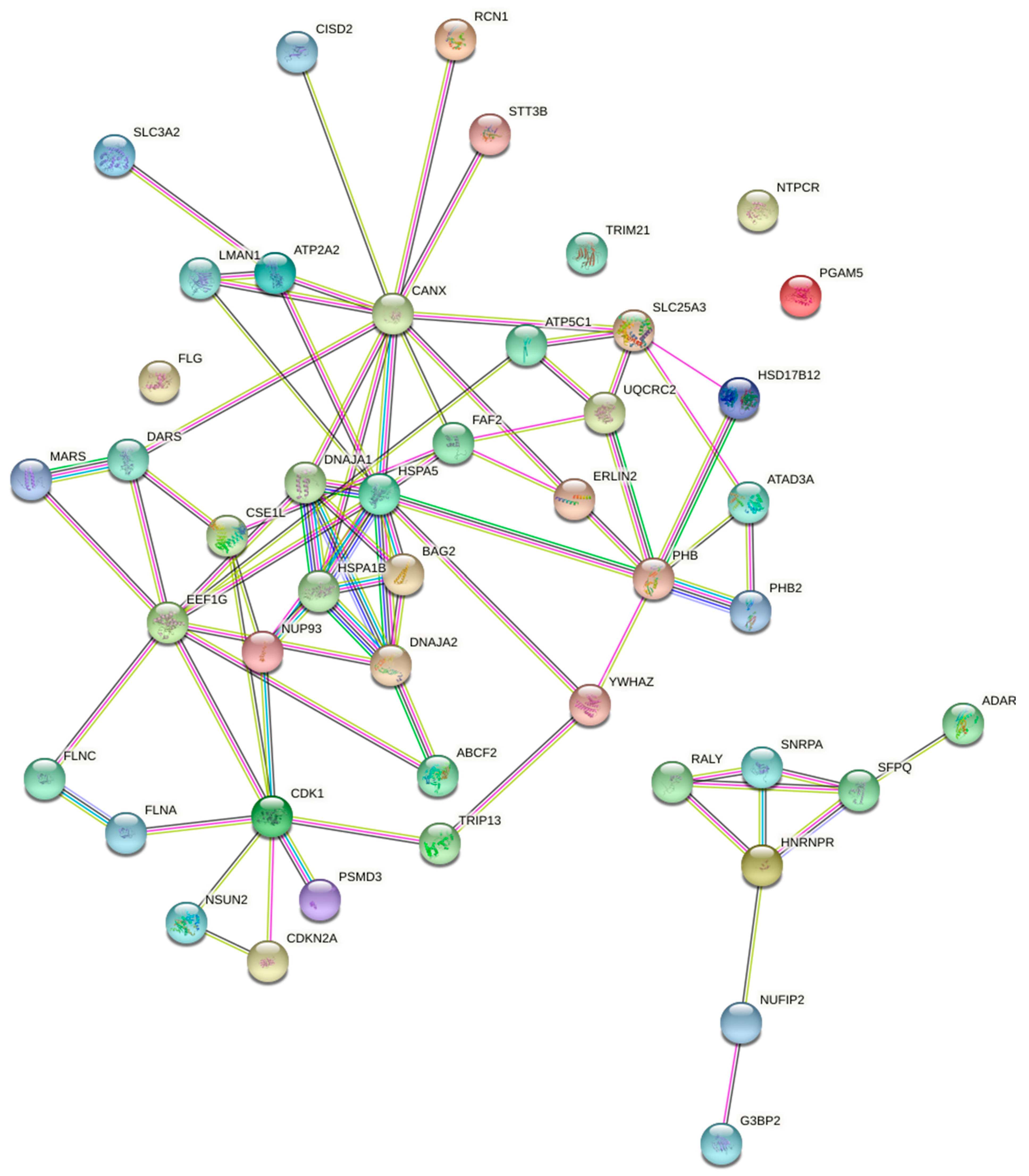
2.8. STRING Protein Association Network
2.9. Statistical Analysis
3. Results
3.1. ITM2A Physically Interacts with the Hedgehog Receptor PTCH1
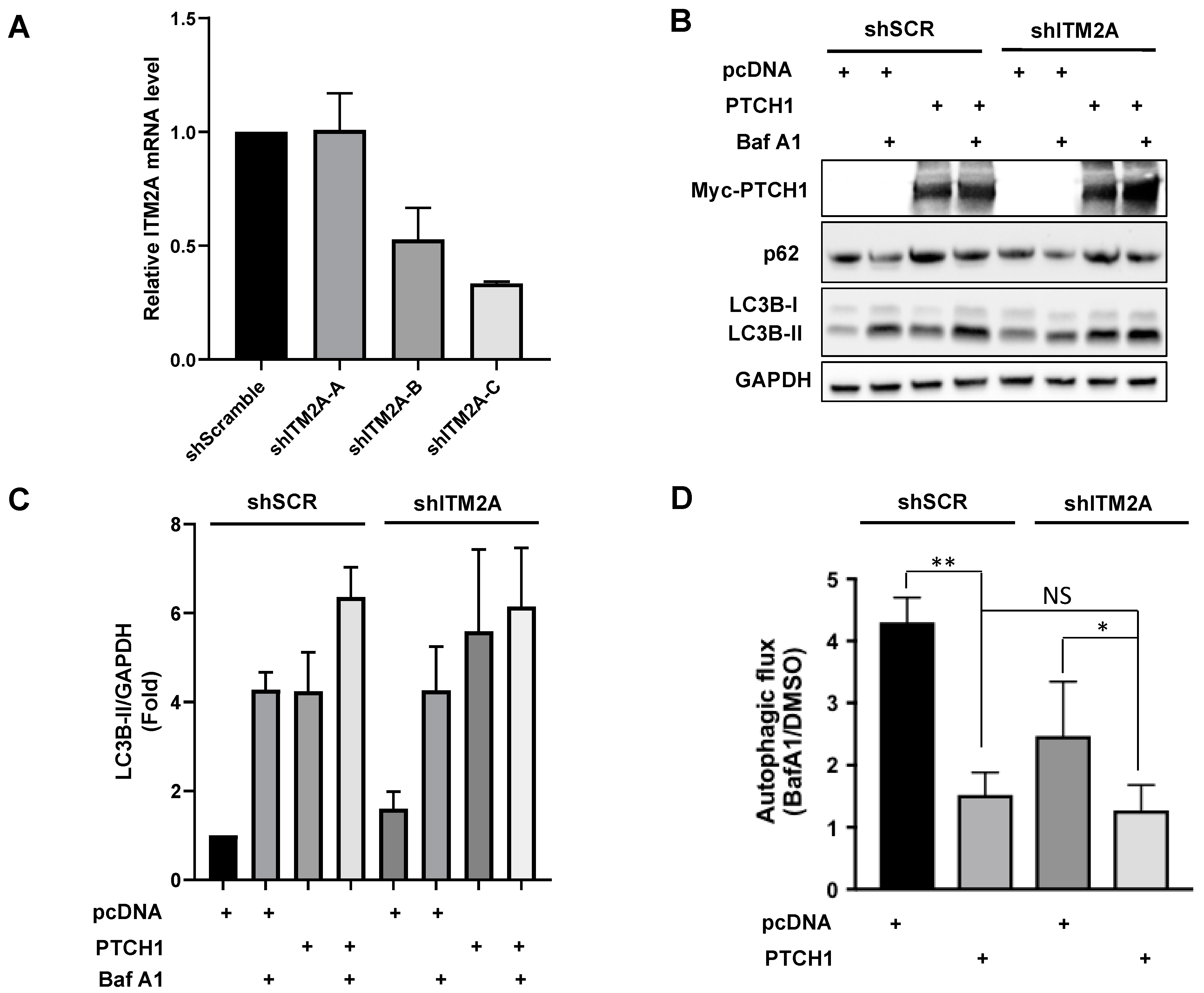
3.2. ITM2A Does Not Mediate PTCH1 Autophagic Flux Inhibition
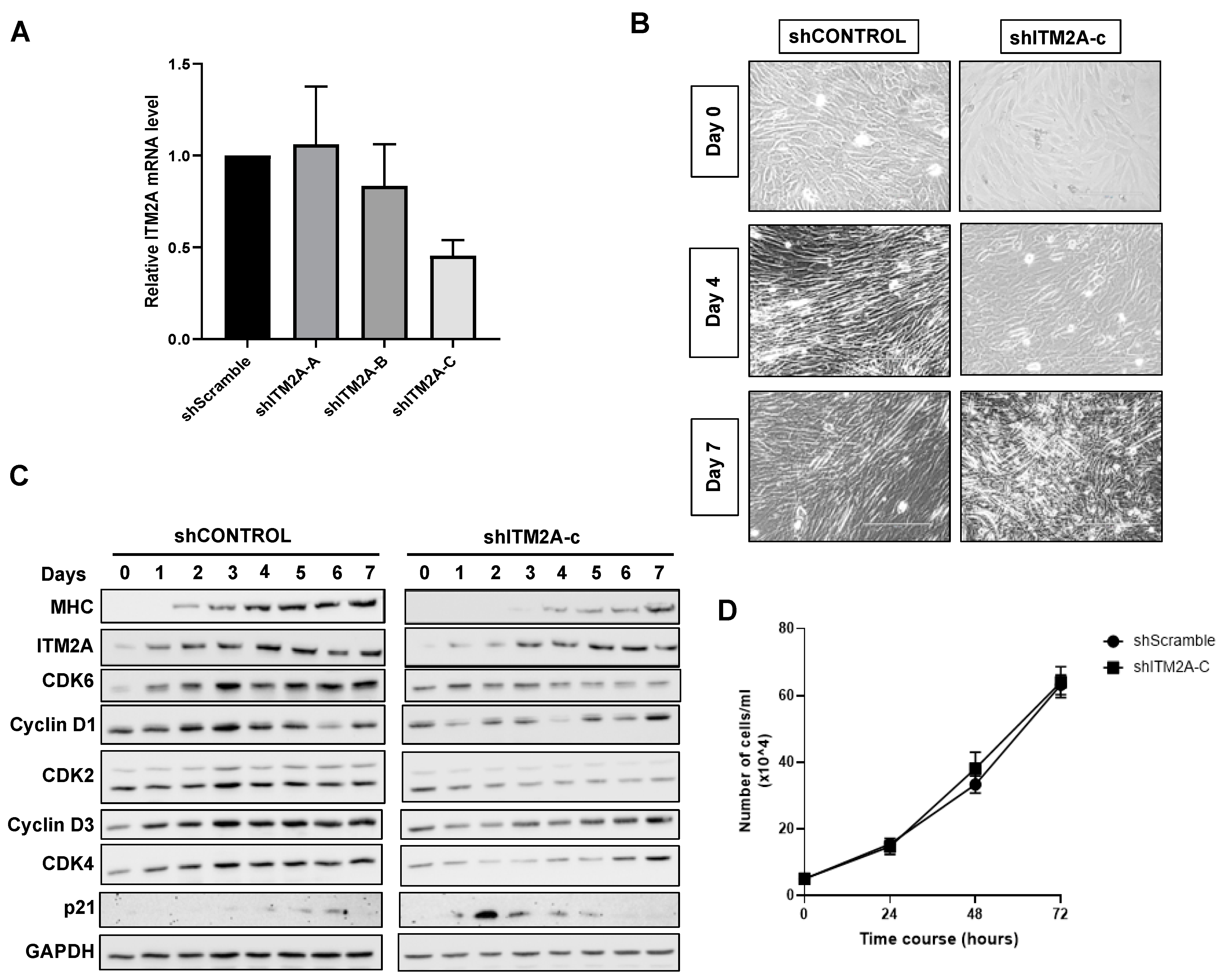
3.3. Increase in ITM2A Levels Accompanies Reduction of Autophagic Flux during Skeletal Muscle Differentiation
3.4. ITM2A Is as a Negative Regulator of Canonical Hh Signalling
3.5. Identification of ITM2A-Interacting Proteins by Mass Spectrometry
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Barakat, M.T.; Humke, E.H.; Scott, M.P. Learning from Jekyll to control Hyde: Hedgehog signaling in development and cancer. Trends Mol. Med. 2010, 16, 337–348. [Google Scholar] [CrossRef]
- Robbins, D.J.; Fei, D.L.; Riobo, N.A. The Hedgehog signal transduction network. Sci. Signal. 2012, 5, re6. [Google Scholar] [CrossRef]
- Teperino, R.; Aberger, F.; Esterbauer, H.; Riobo, N.A.; Pospisilik, J.A. Canonical and non-canonical Hedgehog signaling and the control of metabolism. Sem. Cell Dev. Biol. 2014, 33, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Fleet, A.; Lee, J.P.; Tamachi, A.; Javeed, I.; Hamel, P.A. Activities of the cytoplasmic domains of Patched-1 modulate but are not essential for the regulation of canonical Hedgehog signaling. J. Biol. Chem. 2016, 291, 17557–17568. [Google Scholar] [CrossRef]
- Timmis, A.J. Structure-Function Relationship of the Patched Family of Proteins. Ph.D. Thesis, University of Leeds, Leeds, UK, 16 March 2021. [Google Scholar]
- Kim, J.; Hsia, E.Y.C.; Brigui, A.; Plessis, A.; Beachy, P.A.; Zheng, X. The role of ciliary trafficking in Hedgehog receptor signaling. Sci. Signal. 2015, 8, ra55. [Google Scholar] [CrossRef] [PubMed]
- Thibert, C.; Teillet, M.A.; Lapointe, F.; Mazelin, L.; Le Douarin, N.M.; Mehlen, P. Inhibition of neuroepithelial patched-induced apoptosis by sonic hedgehog. Science 2003, 301, 843–846. [Google Scholar] [CrossRef]
- Mille, F.; Thibert, C.; Fombonne, J.; Rama, N.; Guix, C.; Hayashi, H.; Corset, V.; Reed, J.C.; Mehlen, P. The Patched dependence receptor triggers apoptosis through a DRAL-caspase 9 complex. Nat. Cell Biol. 2009, 11, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Fombonne, J.; Bissey, P.A.; Guix, C.; Sadoul, R.; Thibert, C.; Mehlen, P. Patched dependence receptor triggers apoptosis through ubiquitination of caspase-9. Proc. Natl. Acad. Sci. USA 2012, 109, 10510–10515. [Google Scholar] [CrossRef]
- Chen, X.L.; Chinchilla, P.; Fombonne, J.; Ho, L.; Guix, C.; Keen, J.H.; Mehlen, P.; Riobo, N.A. Patched-1 proapoptotic activity is downregulated by modification of K1413 by the E3 ubiquitin-protein ligase Itchy homolog. Mol. Cell. Biol. 2014, 34, 3855–3866. [Google Scholar] [CrossRef]
- Chen, X.; Morales-Alcala, C.C.; Riobo-Del Galdo, N.A. Autophagic flux is regulated by interaction between the C-terminal domain of PATCHED1 and ATG101. Mol. Cancer Res. 2018, 16, 909–919. [Google Scholar] [CrossRef]
- Namkoong, S.; Lee, K.I.; Lee, J.I.; Park, R.; Lee, E.-J.; Jang, I.-S.; Park, J. The integral membrane protein ITM2A, a transcriptional target of PKA-CREB, regulates autophagic flux via interaction with the vacuolar ATPase. Autophagy 2015, 11, 756–768. [Google Scholar] [CrossRef]
- Zhou, C.; Wang, M.; Yang, J.; Xiong, H.; Wang, Y.; Tang, J. Integral membrane protein 2A inhibits cell growth in human breast cancer via enhancing autophagy induction. Cell Commun. Signal. 2019, 17, 105. [Google Scholar] [CrossRef]
- Mitsui, S.; Osako, Y.; Yuri, K. Mental retardation-related protease, motopsin (prss12), binds to the BRICHOS domain of the integral membrane protein 2a. Cell Biol. Int. 2014, 38, 117–123. [Google Scholar] [CrossRef]
- van den Plas, D.; Merregaert, J. Constitutive overexpression of the integral membrane protein Itm2A enhances myogenic differentiation of C2C12 cells. Cell Biol. Int. 2004, 28, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Kirchner, J.; Bevan, M.J. ITM2A is induced during thymocyte selection and T cell activation and causes downregulation of CD8 when overexpressed in CD4(+) CD8(+) double positive thymocytes. J. Exp. Med. 1999, 190, 217–228. [Google Scholar] [CrossRef]
- Lee, E.J.; Jan, A.T.; Baig, M.H.; Ahmad, K.; Malik, A.; Rabbani, G.; Kim, I.-K.; Lee, Y.H.; Park, S.-Y.; Choi, I. Fibromodulin and regulation of the intricate balance between myoblast differentiation to myocytes or adipocyte-like cells. FASEB J. 2018, 32, 768–781. [Google Scholar] [CrossRef] [PubMed]
- Lagha, M.; Mayeuf-Louchart, A.; Chang, T.; Montarras, D.; Rocancourt, D.; Zalc, A.; Kormish, J.; Zaret, K.S.; Buckingham, M.E.; Relaix, F. Itm2a is a Pax3 target gene, expressed at sites of skeletal muscle formation in vivo. PLoS ONE 2013, 8, e63143. [Google Scholar] [CrossRef] [PubMed]
- Koleva, M.; Kappler, R.; Vogler, M.; Herwig, A.; Fulda, S.; Hahn, H. Pleiotropic effects of sonic hedgehog on muscle satellite cells. Cell Mol. Life Sci. 2005, 62, 1863–1870. [Google Scholar] [CrossRef]
- Gerber, A.N.; Wilson, C.W.; Li, Y.-J.; Chuang, P.-T. The hedgehog regulated oncogenes Gli1 and Gli2 block myoblast differentiation by inhibiting MyoD-mediated transcriptional activation. Oncogene 2007, 26, 1122–1136. [Google Scholar] [CrossRef]
- Mellis, D.; Staines, K.A.; Peluso, S.; Georgiou, I.C.; Dora, N.; Kubiak, M.; Van’t Hof, R.; Grillo, M.; Farquharson, C.; Kinsella, E.; et al. Ubiquitin-protein ligase Ubr5 cooperates with hedgehog signalling to promote skeletal tissue homeostasis. PLoS Genet. 2021, 17, e1009275. [Google Scholar] [CrossRef]
- Zhang, Y.; Bulkley, D.P.; Xin, Y.; Roberts, K.J.; Asarnow, D.E.; Sharma, A.; Myers, B.R.; Cho, W.; Cheng, Y.; Beachy, P.A. Structural basis for the cholesterol transport-like activity of the Hedgehog receptor Patched. Cell 2018, 175, 1352–1364. [Google Scholar] [CrossRef] [PubMed]
- Kee, H.L.; Dishinger, J.F.; Blasius, T.L.; Liu, C.-J.; Morgolis, B.; Verhey, K.J. A size-exclusion permeability barrier and nucleoporins characterize a ciliary pore complex that regulates transport into cilia. Nat. Cell Biol. 2012, 14, 431–437. [Google Scholar] [CrossRef]
- Kutay, U.; Bischoff, F.R.; Kostka, S.; Kraft, R.; Görlich, D. Export of importin alpha from the nucleus is mediated by a specific nuclear transport factor. Cell 1997, 90, 1061–1071. [Google Scholar] [CrossRef]
- Torrado, B.; Graña, M.; Badano, J.L.; Irigoín, F. Ciliary entry of the Hedgehog transcriptional activator Gli2 is mediated by the nuclear import machinery but differs from nuclear transport in being Imp-α/β1-independent. PLoS ONE 2016, 11, e0162033. [Google Scholar] [CrossRef]
- Franklin, D.S.; Xiong, Y. Induction of p18INK4c and its predominant association with CDK4 and CDK6 during myogenic differentiation. Mol. Biol. Cell 1996, 7, 1587–1599. [Google Scholar] [CrossRef][Green Version]
- Williamson, D.L.; Butler, D.C.; Always, S.E. AMPK inhibits myoblast differentiation through a PGC-1alpha-dependent mechanism. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E304–E314. [Google Scholar] [CrossRef] [PubMed]
- Cenciarelli, C.; De Santa, F.; Puri, P.L.; Mattei, E.; Ricci, L.; Bucci, F.; Felsani, A.; Caruso, M. Critical role played by cyclin D3 in the MyoD-mediated arrest of cell cycle during myoblast differentiation. Mol. Cell. Biol. 1999, 19, 5203–5217. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Lukas, J.; Strauss, M.; Bertek, J. Cyclin D3: Requirement for G1/S transition and high abundance in quiescent tissues suggest a dual role in proliferation and differentiation. Oncogene 1998, 17, 1027–1037. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence (5′–3′) |
|---|---|
| ITM2A-A (human) | GATCCCTGCAGCAATTATTCATGATTCAAGGATCATGAATAATTGCTGCAGTTTTT |
| ITM2A-B (human) | GATCCGGAAATTCGTGATGTTATTTCAAGGAACTAACATCACGAATTTCCTTTTT |
| ITM2A-C (human) | GATCCCAAGATCTGTCAAGAGTAATTCAAGAGATTACTCTTGACAGATCTTGTTTTT |
| Itm2a-A (mouse) | GATCCCTAGGCCTCTCATTCATCTTTCAAGAGAAGATGAATGAGAGGCCTAGTTTTT |
| Itm2a-B (mouse) | GATCCGGATCCTGTCAATTCCATTTTCAAGAGAAATGGAATTGACAGGATCCTTTTT |
| Itm2a-C (mouse) | GATCCCAAGCGTGCCATTGACAAATTCAAGAGATTTGTCAATGGCACGCTTGTTTTT |
| Scrambled | Santa Cruz Biotechnology Scrambled shRNA (sc-108060) |
| Name | Species | Sequence (5′–3′) |
|---|---|---|
| ITM2A | Human | Forward: ACTGCTATCTGATGCCCCTCAAT Reverse: GGTCTTCTCGAACCACATAAGTTTG |
| Itm2a | Mouse | Forward: CGCACTGTCCGAGCTCAAAT Reverse: CATCTCCCAGATGAGCCATC |
| Ptc1 | Mouse | Forward: ATGGTCCTGGCTCTGATGAC Reverse: TAGCCCTGTGGTTCTTGTCC |
| GADPH | Human | Forward: TCCCATCACCATCTTCCA Reverse: CATCACGCCACAGTTTCC |
| Gapdh | Mouse | Forward: AGTATGATGACATCAAGAAGG Reverse: ATGGTATTCAAGAGAGTAGGG |
| Antibody | Company | Catalogue |
|---|---|---|
| GFP-tag (mouse monoclonal) | Proteintech | 66002-1-g |
| HA-tag (mouse monoclonal) | Proteintech | 66006-2-Ig |
| MYC-tag (rabbit polyclonal) | Proteintech | 16286-1-AP |
| Protein Name | Function |
|---|---|
| ABCF2 | ATP-binding cassette transporter |
| ACTG1 | Actin cytoskeleton |
| ADAR | Adenosine deaminase, RNA editing |
| ALB | Albumin |
| ATAD3A | Mitochondrial network organization, steroid hormone |
| ATP2A2 | ER-isolation membrane contacts, autophagosome formation |
| ATP5C1 | Mitochondria -ATP production |
| ATXN2L | Stress granule and P-body formation |
| BAG2 | Co-chaperone for HSP70/HSC70 |
| C1orf57 | Nucleotide phosphatase |
| CANX | ER protein folding quality control |
| CDK1 | Cell cycle progression |
| CDKN2A | Negative regulator of proliferation |
| CISD2 | Regulator of autophagy |
| CSE1L | Nuclear export receptor |
| DARS | Aspartate tRNA-ligase |
| DNAJA1 | Co-chaperone of HSC70, mitochondria protein transport |
| DNAJA2 | Co-chaperone of HSC70 |
| EEF1G | Elongation factor, protein synthesis |
| EIF4A1 | Eukaryotic initiation factor |
| ERLIN2 | ERAD, cholesterol homeostasis |
| FAF2 | ERAD |
| FLG | Keratin intermediate filament aggregation |
| FLNA | Branching of actin filaments |
| FLNC | Muscle-specific filamin |
| G3BP2 | Stress granule formation |
| GANAB | Neutral alpha-glucosidase |
| GP96 | ER-chaperone, ERAD |
| HNRNPR | Processing of mRNA precursor in the nucleus |
| HSD17B12 | ER-localised fatty acid elongation |
| HSPA1B | HSP70 chaperone |
| HSPA5 | ER chaperone |
| LMAN1 | ER to Golgi transport |
| MARS | Methionine-tRNA ligase |
| NPM1 | Ribosome biogenesis, ribosome nuclear export |
| NSUN2 | RNA-C5 methyl transferase, tRNA stability, mRNA decay |
| NUFIP2 | Stress granules, transcription control |
| NUP93 | Nuclear pore complex |
| PAI-1 | Serine protease inhibitor |
| PGAM5 | Mitochondrial dynamics |
| PHB | Mitochondrial chaperone, mitophagy receptor |
| PHB-2 | Mitochondrial chaperone, mitophagy receptor |
| PSMD3 | Component of the 26S proteasome |
| RALY | Transcriptional cofactor for cholesterogenesis |
| RCN | ER-post-ER calcium regulatory protein |
| SFPQ | Pre-mRNA splicing factor, myoblast marker |
| SLC25A3 | Cytosol-mitochondria phosphate transport |
| SLC3A2 | Amino acid transport in plasma membrane and lysosomes |
| SNRPA | Pre-mRNA splicing factor |
| STT3B | N-glycosylation of misfolded protein for ERAD |
| TRIM21 | E3 ligase, promotes autophagy, autophagy receptor |
| TRIP13 | Meiosis, chromosomal recombination |
| UQCRC2 | Mitochondrial electron transport chain |
| YWHAZ | Autophagy regulator |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morales-Alcala, C.C.; Georgiou, I.C.; Timmis, A.J.; Riobo-Del Galdo, N.A. Integral Membrane Protein 2A Is a Negative Regulator of Canonical and Non-Canonical Hedgehog Signalling. Cells 2021, 10, 2003. https://doi.org/10.3390/cells10082003
Morales-Alcala CC, Georgiou IC, Timmis AJ, Riobo-Del Galdo NA. Integral Membrane Protein 2A Is a Negative Regulator of Canonical and Non-Canonical Hedgehog Signalling. Cells. 2021; 10(8):2003. https://doi.org/10.3390/cells10082003
Chicago/Turabian StyleMorales-Alcala, Cintli C., Ioanna Ch. Georgiou, Alex J. Timmis, and Natalia A. Riobo-Del Galdo. 2021. "Integral Membrane Protein 2A Is a Negative Regulator of Canonical and Non-Canonical Hedgehog Signalling" Cells 10, no. 8: 2003. https://doi.org/10.3390/cells10082003
APA StyleMorales-Alcala, C. C., Georgiou, I. C., Timmis, A. J., & Riobo-Del Galdo, N. A. (2021). Integral Membrane Protein 2A Is a Negative Regulator of Canonical and Non-Canonical Hedgehog Signalling. Cells, 10(8), 2003. https://doi.org/10.3390/cells10082003