
Interleukin-17A Triggers the Release of Platelet-Derived Factors Driving Vascular Endothelial Cells toward a Pro-Angiogenic State



 and
and
Abstract
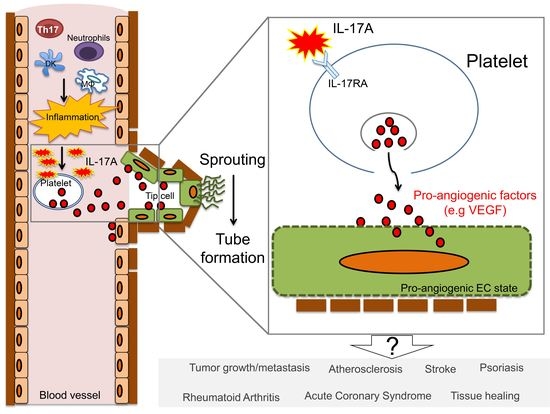
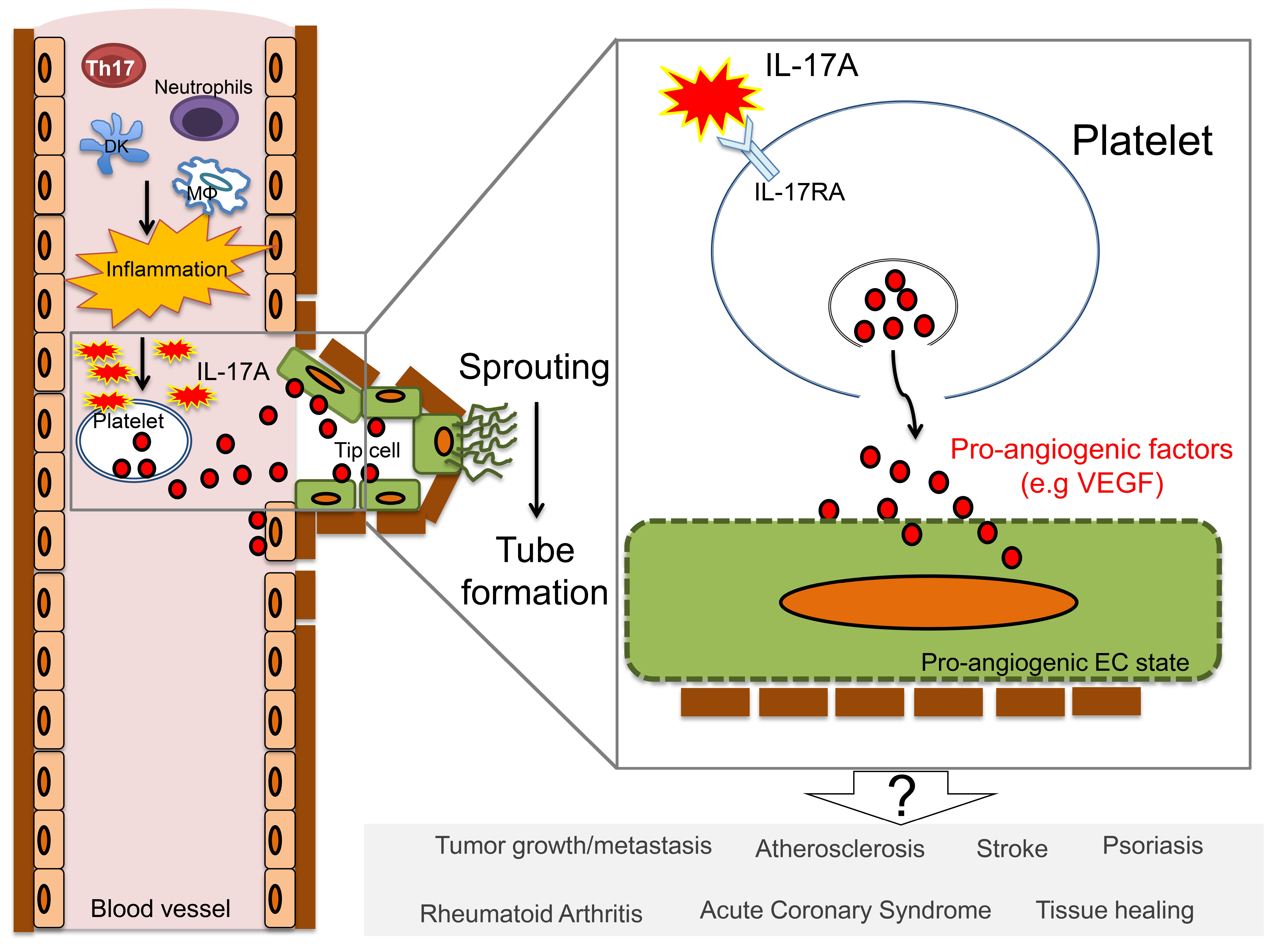{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Preparation and Activation of Human and Murine Platelets
2.2. RT-PCR Analysis of Highly Purified Platelet
2.3. Western Blot
2.4. Light Transmission Aggregometry
2.5. Protein Quantification in Platelet Releasates
2.6. Capillary Tube Formation Assay
2.7. 3-D Sprouting Assay
2.8. Ex Vivo Murine Aortic Ring Assay
2.9. Statistical Analysis
3. Results
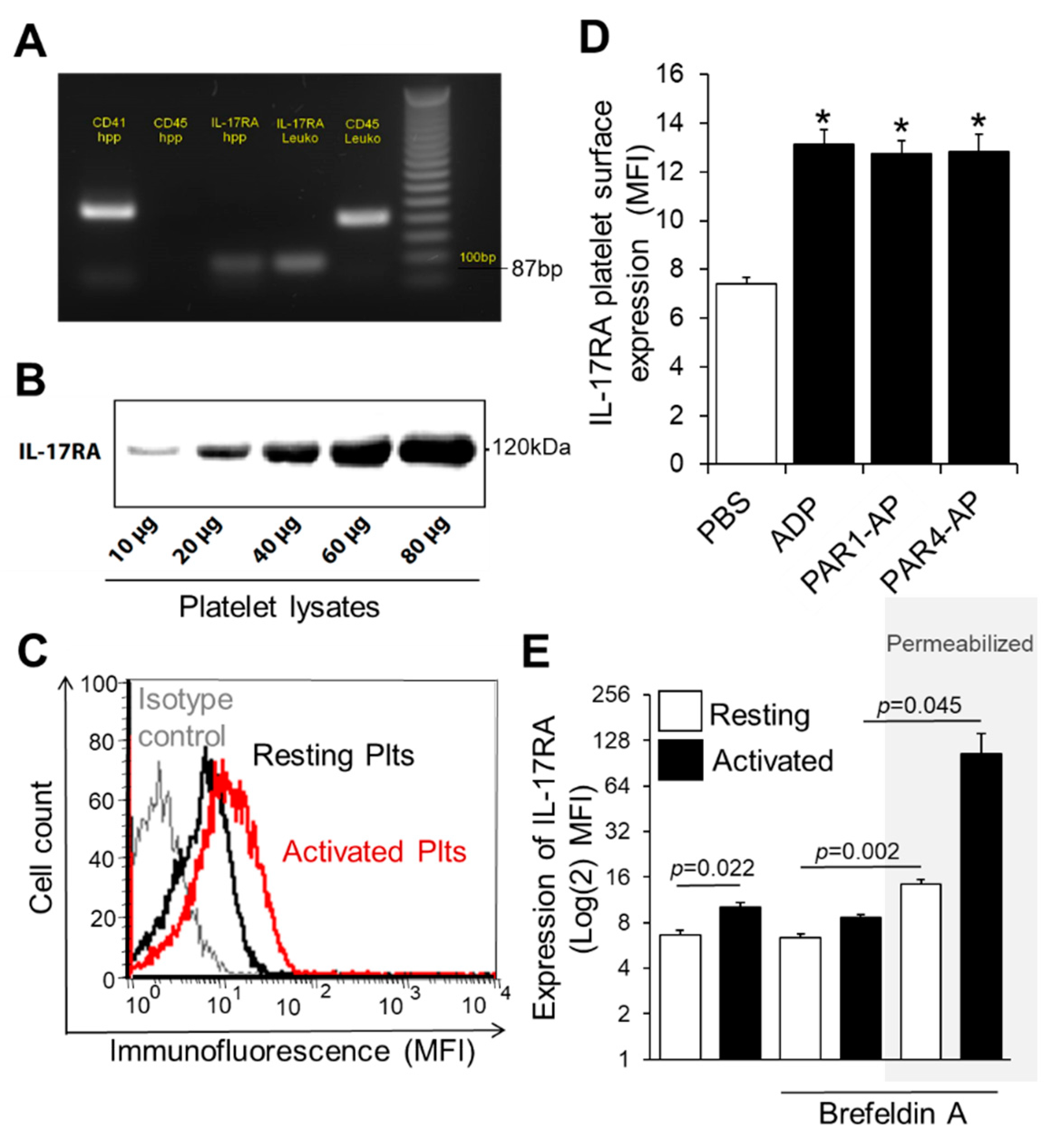
3.1. IL-17RA Is Expressed on Platelet Surface and Induced upon Platelet Activation
3.2. IL-17A Has No Effect over Platelet Aggregation
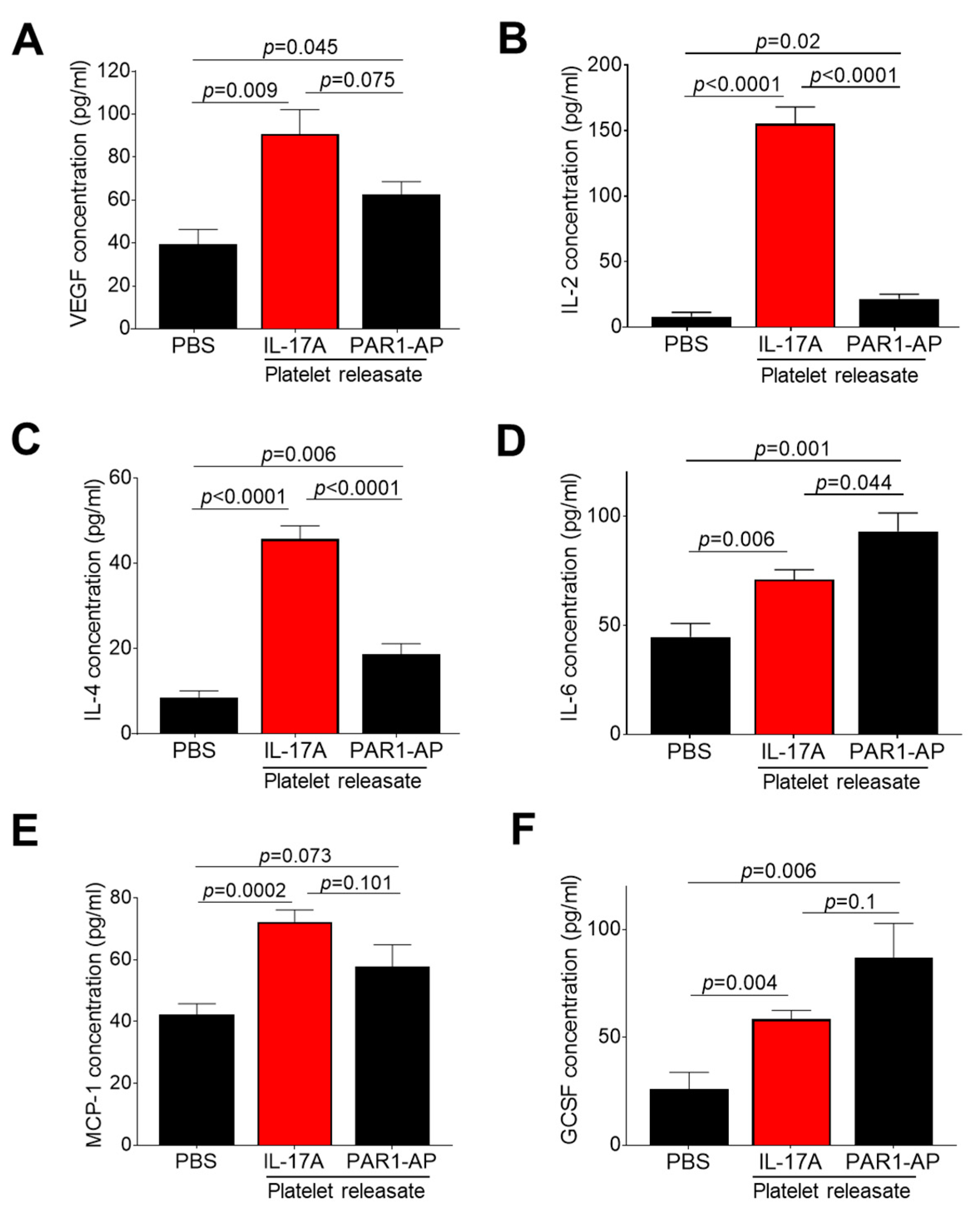
3.3. IL-17A Triggers Release of Several Platelet-Derived Factors with Pro-angiogenic Potential
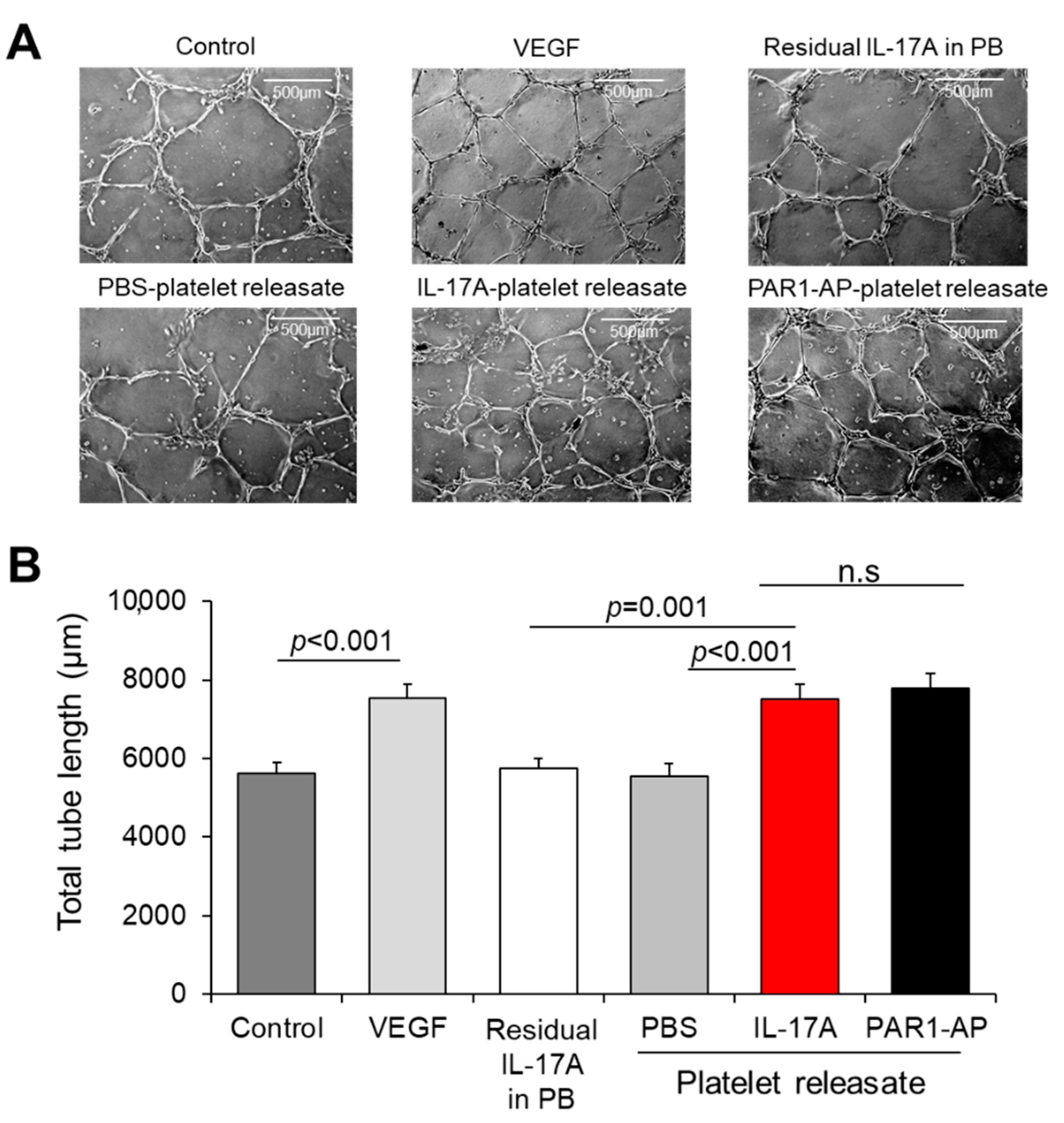
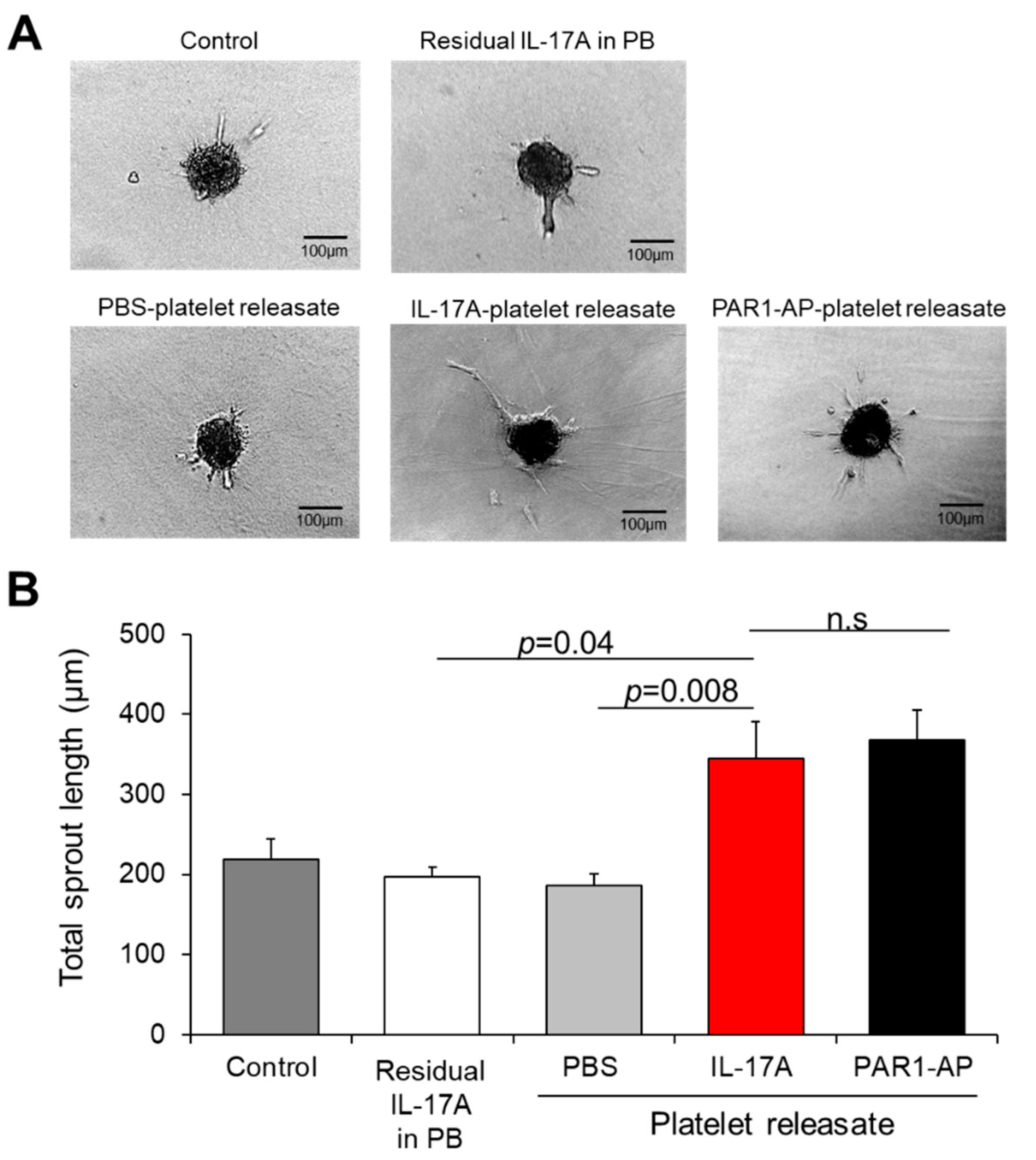
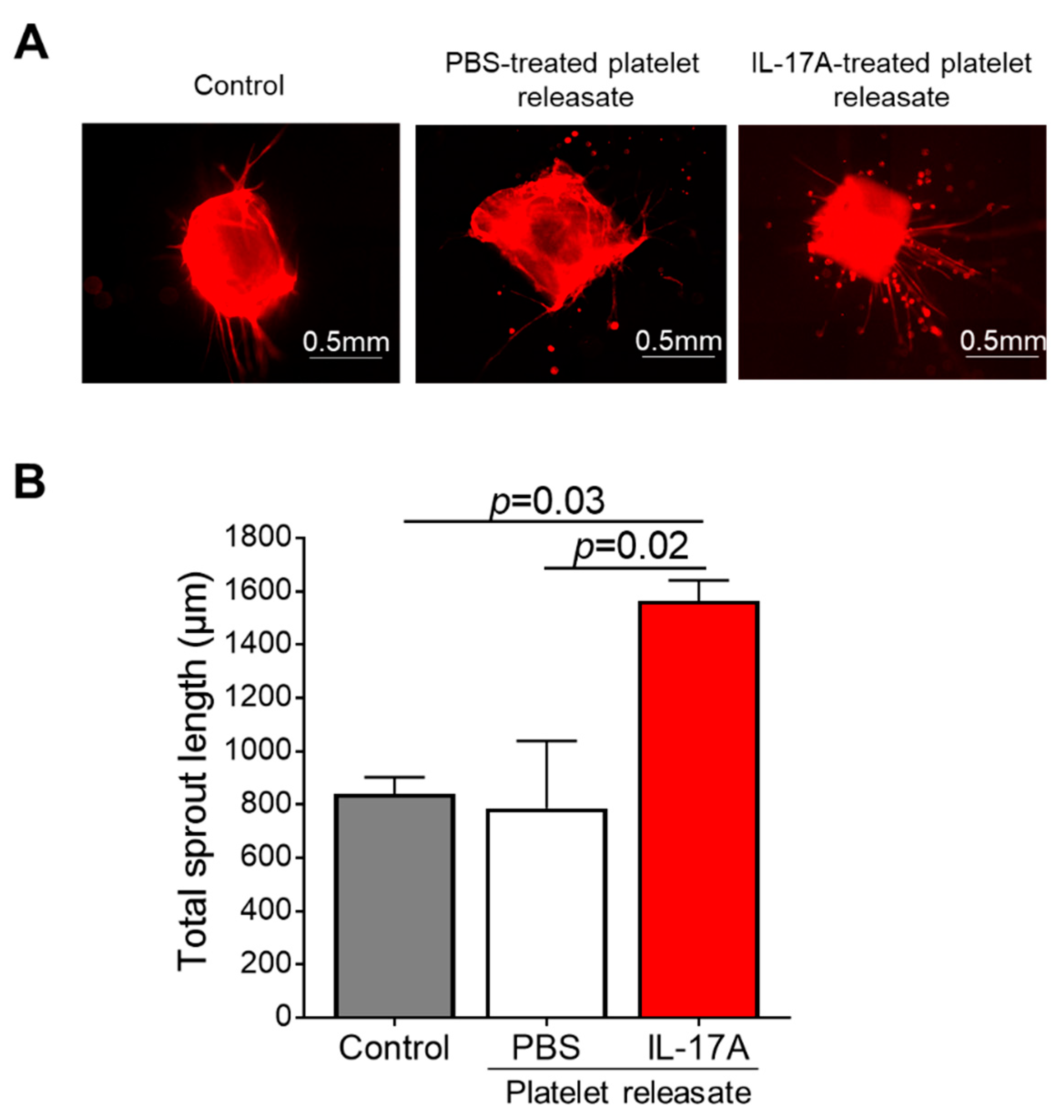
3.4. Releasate of IL-17A-Induced Platelets Has an Overall Pro-angiogenic Potential
4. Discussion
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stakos, D.A.; Stamatelopoulos, K.; Bampatsias, D.; Sachse, M.; Zormpas, E.; Vlachogiannis, N.I.; Tual-Chalot, S.; Stellos, K. The Alzheimer’s Disease Amyloid-Beta Hypothesis in Cardiovascular Aging and Disease: JACC Focus Seminar. J. Am. Coll. Cardiol. 2020, 75, 952–967. [Google Scholar] [CrossRef] [PubMed]
- Stellos, K.; Sauter, R.; Fahrleitner, M.; Grimm, J.; Stakos, D.; Emschermann, F.; Panagiota, V.; Gnerlich, S.; Perk, A.; Schonberger, T.; et al. Binding of oxidized low-density lipoprotein on circulating platelets is increased in patients with acute coronary syndromes and induces platelet adhesion to vascular wall in vivo–brief report. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2017–2020. [Google Scholar] [CrossRef] [PubMed]
- Bultmann, A.; Li, Z.; Wagner, S.; Peluso, M.; Schonberger, T.; Weis, C.; Konrad, I.; Stellos, K.; Massberg, S.; Nieswandt, B.; et al. Impact of glycoprotein VI and platelet adhesion on atherosclerosis—A possible role of fibronectin. J. Mol. Cell. Cardiol. 2010, 49, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Schulz, C.; Penz, S.; Hoffmann, C.; Langer, H.; Gillitzer, A.; Schneider, S.; Brandl, R.; Seidl, S.; Massberg, S.; Pichler, B.; et al. Platelet GPVI binds to collagenous structures in the core region of human atheromatous plaque and is critical for atheroprogression in vivo. Basic Res. Cardiol. 2008, 103, 356–367. [Google Scholar] [CrossRef]
- Rath, D.; Chatterjee, M.; Borst, O.; Muller, K.; Stellos, K.; Mack, A.F.; Bongartz, A.; Bigalke, B.; Langer, H.; Schwab, M.; et al. Expression of stromal cell-derived factor-1 receptors CXCR4 and CXCR7 on circulating platelets of patients with acute coronary syndrome and association with left ventricular functional recovery. Eur. Heart J. 2014, 35, 386–394. [Google Scholar] [CrossRef]
- Stellos, K.; Panagiota, V.; Kogel, A.; Leyhe, T.; Gawaz, M.; Laske, C. Predictive value of platelet activation for the rate of cognitive decline in Alzheimer’s disease patients. J. Cereb. Blood Flow Metab. 2010, 30, 1817–1820. [Google Scholar] [CrossRef]
- Geisler, T.; Mueller, K.; Aichele, S.; Bigalke, B.; Stellos, K.; Htun, P.; Ninci, E.; Fateh-Moghadam, S.; May, A.E.; Gawaz, M. Impact of inflammatory state and metabolic control on responsiveness to dual antiplatelet therapy in type 2 diabetics after PCI: Prognostic relevance of residual platelet aggregability in diabetics undergoing coronary interventions. Clin. Res. Cardiol. 2010, 99, 743–752. [Google Scholar] [CrossRef]
- Stellos, K.; Bigalke, B.; Stakos, D.; Henkelmann, N.; Gawaz, M. Platelet-bound P-selectin expression in patients with coronary artery disease: Impact on clinical presentation and myocardial necrosis, and effect of diabetes mellitus and anti-platelet medication. J. Thromb. Haemost. 2010, 8, 205–207. [Google Scholar] [CrossRef] [PubMed]
- Geisler, T.; Zurn, C.; Simonenko, R.; Rapin, M.; Kraibooj, H.; Kilias, A.; Bigalke, B.; Stellos, K.; Schwab, M.; May, A.E.; et al. Early but not late stent thrombosis is influenced by residual platelet aggregation in patients undergoing coronary interventions. Eur. Heart J. 2010, 31, 59–66. [Google Scholar] [CrossRef]
- Bigalke, B.; Stellos, K.; Geisler, T.; Kremmer, E.; Seizer, P.; May, A.E.; Lindemann, S.; Melms, A.; Luft, A.; Gawaz, M. Expression of platelet glycoprotein VI is associated with transient ischemic attack and stroke. Eur. J. Neurol. 2010, 17, 111–117. [Google Scholar] [CrossRef]
- Geisler, T.; Grass, D.; Bigalke, B.; Stellos, K.; Drosch, T.; Dietz, K.; Herdeg, C.; Gawaz, M. The Residual Platelet Aggregation after Deployment of Intracoronary Stent (PREDICT) score. J. Thromb. Haemost. 2008, 6, 54–61. [Google Scholar] [CrossRef]
- Geisler, T.; Anders, N.; Paterok, M.; Langer, H.; Stellos, K.; Lindemann, S.; Herdeg, C.; May, A.E.; Gawaz, M. Platelet response to clopidogrel is attenuated in diabetic patients undergoing coronary stent implantation. Diabetes Care 2007, 30, 372–374. [Google Scholar] [CrossRef][Green Version]
- Geisler, T.; Langer, H.; Wydymus, M.; Gohring, K.; Zurn, C.; Bigalke, B.; Stellos, K.; May, A.E.; Gawaz, M. Low response to clopidogrel is associated with cardiovascular outcome after coronary stent implantation. Eur. Heart J. 2006, 27, 2420–2425. [Google Scholar] [CrossRef]
- Htun, P.; Fateh-Moghadam, S.; Tomandl, B.; Handschu, R.; Klinger, K.; Stellos, K.; Garlichs, C.; Daniel, W.; Gawaz, M. Course of platelet activation and platelet-leukocyte interaction in cerebrovascular ischemia. Stroke 2006, 37, 2283–2287. [Google Scholar] [CrossRef]
- Massberg, S.; Brand, K.; Gruner, S.; Page, S.; Muller, E.; Muller, I.; Bergmeier, W.; Richter, T.; Lorenz, M.; Konrad, I.; et al. A critical role of platelet adhesion in the initiation of atherosclerotic lesion formation. J. Exp. Med. 2002, 196, 887–896. [Google Scholar] [CrossRef]
- Huo, Y.; Schober, A.; Forlow, S.B.; Smith, D.F.; Hyman, M.C.; Jung, S.; Littman, D.R.; Weber, C.; Ley, K. Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein E. Nat. Med. 2003, 9, 61–67. [Google Scholar] [CrossRef]
- Gawaz, M.; Langer, H.; May, A.E. Platelets in inflammation and atherogenesis. J. Clin. Investig. 2005, 115, 3378–3384. [Google Scholar] [CrossRef]
- Kisucka, J.; Butterfield, C.E.; Duda, D.G.; Eichenberger, S.C.; Saffaripour, S.; Ware, J.; Ruggeri, Z.M.; Jain, R.K.; Folkman, J.; Wagner, D.D. Platelets and platelet adhesion support angiogenesis while preventing excessive hemorrhage. Proc. Natl. Acad. Sci. USA 2006, 103, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Italiano, J.E., Jr.; Richardson, J.L.; Patel-Hett, S.; Battinelli, E.; Zaslavsky, A.; Short, S.; Ryeom, S.; Folkman, J.; Klement, G.L. Angiogenesis is regulated by a novel mechanism: Pro- and antiangiogenic proteins are organized into separate platelet alpha granules and differentially released. Blood 2008, 111, 1227–1233. [Google Scholar] [CrossRef] [PubMed]
- Heimark, R.L.; Twardzik, D.R.; Schwartz, S.M. Inhibition of endothelial regeneration by type-beta transforming growth factor from platelets. Science 1986, 233, 1078–1080. [Google Scholar] [CrossRef] [PubMed]
- Fujimi, S.; MacConmara, M.P.; Maung, A.A.; Zang, Y.; Mannick, J.A.; Lederer, J.A.; Lapchak, P.H. Platelet depletion in mice increases mortality after thermal injury. Blood 2006, 107, 4399–4406. [Google Scholar] [CrossRef]
- Ma, L.; Elliott, S.N.; Cirino, G.; Buret, A.; Ignarro, L.J.; Wallace, J.L. Platelets modulate gastric ulcer healing: Role of endostatin and vascular endothelial growth factor release. Proc. Natl. Acad. Sci. USA 2001, 98, 6470–6475. [Google Scholar] [CrossRef]
- Lesurtel, M.; Graf, R.; Aleil, B.; Walther, D.J.; Tian, Y.; Jochum, W.; Gachet, C.; Bader, M.; Clavien, P.A. Platelet-derived serotonin mediates liver regeneration. Science 2006, 312, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Stellos, K.; Bigalke, B.; Borst, O.; Pfaff, F.; Elskamp, A.; Sachsenmaier, S.; Zachmann, R.; Stamatelopoulos, K.; Schonberger, T.; Geisler, T.; et al. Circulating platelet-progenitor cell coaggregate formation is increased in patients with acute coronary syndromes and augments recruitment of CD34+ cells in the ischaemic microcirculation. Eur. Heart J. 2013, 34, 2548–2556. [Google Scholar] [CrossRef]
- Stellos, K.; Panagiota, V.; Gnerlich, S.; Borst, O.; Bigalke, B.; Gawaz, M. Expression of junctional adhesion molecule-C on the surface of platelets supports adhesion, but not differentiation, of human CD34 cells in vitro. Cell. Physiol. Biochem. 2012, 29, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Stellos, K.; Langer, H.; Gnerlich, S.; Panagiota, V.; Paul, A.; Schonberger, T.; Ninci, E.; Menzel, D.; Mueller, I.; Bigalke, B.; et al. Junctional adhesion molecule A expressed on human CD34+ cells promotes adhesion on vascular wall and differentiation into endothelial progenitor cells. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1127–1136. [Google Scholar] [CrossRef]
- Langer, H.F.; Stellos, K.; Steingen, C.; Froihofer, A.; Schonberger, T.; Kramer, B.; Bigalke, B.; May, A.E.; Seizer, P.; Muller, I.; et al. Platelet derived bFGF mediates vascular integrative mechanisms of mesenchymal stem cells in vitro. J. Mol. Cell. Cardiol. 2009, 47, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Langer, H.F.; Daub, K.; Braun, G.; Schonberger, T.; May, A.E.; Schaller, M.; Stein, G.M.; Stellos, K.; Bueltmann, A.; Siegel-Axel, D.; et al. Platelets recruit human dendritic cells via Mac-1/JAM-C interaction and modulate dendritic cell function in vitro. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1463–1470. [Google Scholar] [CrossRef] [PubMed]
- Daub, K.; Langer, H.; Seizer, P.; Stellos, K.; May, A.E.; Goyal, P.; Bigalke, B.; Schonberger, T.; Geisler, T.; Siegel-Axel, D.; et al. Platelets induce differentiation of human CD34+ progenitor cells into foam cells and endothelial cells. FASEB J. 2006, 20, 2559–2561. [Google Scholar] [CrossRef]
- Sidiropoulou, S.; Papadaki, S.; Tsouka, A.N.; Koutsaliaris, I.K.; Chantzichristos, V.G.; Pantazi, D.; Paschopoulos, M.E.; Hansson, K.M.; Tselepis, A.D. The Effect of Platelet-Rich Plasma on Endothelial Progenitor Cell Functionality. Angiology 2021, 3319721998895. [Google Scholar] [CrossRef]
- Tatsidou, P.T.; Chantzichristos, V.G.; Tsoumani, M.E.; Sidiropoulou, S.; Ntalas, I.V.; Goudevenos, J.A.; Stellos, K.; Tselepis, A.D. Circulating progenitor cells and their interaction with platelets in patients with an acute coronary syndrome. Platelets 2019, 30, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Chantzichristos, V.G.; Gkrozou, F.; Stellos, K.; Paschopoulos, M.E.; Tselepis, A.D. Comparative Anti-Platelet Profiling Reveals a Potent Anti-Aggregatory Effect of CD34+ Progenitor Cell-Derived Late-Outgrowth Endothelial Cells in vitro. J. Vasc. Res. 2018, 55, 13–25. [Google Scholar] [CrossRef]
- Stellos, K.; Kopf, S.; Paul, A.; Marquardt, J.U.; Gawaz, M.; Huard, J.; Langer, H.F. Platelets in regeneration. Semin. Thromb. Hemost. 2010, 36, 175–184. [Google Scholar] [CrossRef]
- Bakogiannis, C.; Sachse, M.; Stamatelopoulos, K.; Stellos, K. Platelet-derived chemokines in inflammation and atherosclerosis. Cytokine 2019, 122, 154157. [Google Scholar] [CrossRef] [PubMed]
- Jorbenadze, R.; Schleicher, E.; Bigalke, B.; Stellos, K.; Gawaz, M. Expression of platelet-bound stromal-cell derived factor-1 (SDF-1) and number of CD34(+) progenitor cells in patients with congestive heart failure. Platelets 2014, 25, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Stellos, K.; Rahmann, A.; Kilias, A.; Ruf, M.; Sopova, K.; Stamatelopoulos, K.; Jorbenadze, R.; Weretka, S.; Geisler, T.; Gawaz, M.; et al. Expression of platelet-bound stromal cell-derived factor-1 in patients with non-valvular atrial fibrillation and ischemic heart disease. J. Thromb. Haemost. 2012, 10, 49–55. [Google Scholar] [CrossRef]
- Stellos, K.; Bigalke, B.; Langer, H.; Geisler, T.; Schad, A.; Kogel, A.; Pfaff, F.; Stakos, D.; Seizer, P.; Muller, I.; et al. Expression of stromal-cell-derived factor-1 on circulating platelets is increased in patients with acute coronary syndrome and correlates with the number of CD34+ progenitor cells. Eur. Heart J. 2009, 30, 584–593. [Google Scholar] [CrossRef]
- Stellos, K.; Langer, H.; Daub, K.; Schoenberger, T.; Gauss, A.; Geisler, T.; Bigalke, B.; Mueller, I.; Schumm, M.; Schaefer, I.; et al. Platelet-derived stromal cell-derived factor-1 regulates adhesion and promotes differentiation of human CD34+ cells to endothelial progenitor cells. Circulation 2008, 117, 206–215. [Google Scholar] [CrossRef]
- Chatterjee, M.; Huang, Z.; Zhang, W.; Jiang, L.; Hultenby, K.; Zhu, L.; Hu, H.; Nilsson, G.P.; Li, N. Distinct platelet packaging, release, and surface expression of proangiogenic and antiangiogenic factors on different platelet stimuli. Blood 2011, 117, 3907–3911. [Google Scholar] [CrossRef]
- Klement, G.L.; Yip, T.T.; Cassiola, F.; Kikuchi, L.; Cervi, D.; Podust, V.; Italiano, J.E.; Wheatley, E.; Abou-Slaybi, A.; Bender, E.; et al. Platelets actively sequester angiogenesis regulators. Blood 2009, 113, 2835–2842. [Google Scholar] [CrossRef]
- Taleb, S.; Romain, M.; Ramkhelawon, B.; Uyttenhove, C.; Pasterkamp, G.; Herbin, O.; Esposito, B.; Perez, N.; Yasukawa, H.; Van Snick, J.; et al. Loss of SOCS3 expression in T cells reveals a regulatory role for interleukin-17 in atherosclerosis. J. Exp. Med. 2009, 206, 2067–2077. [Google Scholar] [CrossRef]
- Smith, E.; Prasad, K.M.; Butcher, M.; Dobrian, A.; Kolls, J.K.; Ley, K.; Galkina, E. Blockade of interleukin-17A results in reduced atherosclerosis in apolipoprotein E-deficient mice. Circulation 2010, 121, 1746–1755. [Google Scholar] [CrossRef]
- Erbel, C.; Chen, L.; Bea, F.; Wangler, S.; Celik, S.; Lasitschka, F.; Wang, Y.; Bockler, D.; Katus, H.A.; Dengler, T.J. Inhibition of IL-17A attenuates atherosclerotic lesion development in apoE-deficient mice. J. Immunol. 2009, 183, 8167–8175. [Google Scholar] [CrossRef]
- Lin, W.W.; Karin, M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J. Clin. Investig. 2007, 117, 1175–1183. [Google Scholar] [CrossRef]
- Numasaki, M.; Lotze, M.T.; Sasaki, H. Interleukin-17 augments tumor necrosis factor-alpha-induced elaboration of proangiogenic factors from fibroblasts. Immunol. Lett. 2004, 93, 39–43. [Google Scholar] [CrossRef]
- Numasaki, M.; Watanabe, M.; Suzuki, T.; Takahashi, H.; Nakamura, A.; McAllister, F.; Hishinuma, T.; Goto, J.; Lotze, M.T.; Kolls, J.K.; et al. IL-17 enhances the net angiogenic activity and in vivo growth of human non-small cell lung cancer in SCID mice through promoting CXCR-2-dependent angiogenesis. J. Immunol. 2005, 175, 6177–6189. [Google Scholar] [CrossRef] [PubMed]
- Eid, R.E.; Rao, D.A.; Zhou, J.; Lo, S.F.; Ranjbaran, H.; Gallo, A.; Sokol, S.I.; Pfau, S.; Pober, J.S.; Tellides, G. Interleukin-17 and interferon-gamma are produced concomitantly by human coronary artery-infiltrating T cells and act synergistically on vascular smooth muscle cells. Circulation 2009, 119, 1424–1432. [Google Scholar] [CrossRef]
- Gaffen, S.L. Structure and signalling in the IL-17 receptor family. Nat. Rev. Immunol. 2009, 9, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Shen, F.; Crellin, N.K.; Ouyang, W. The IL-17 pathway as a major therapeutic target in autoimmune diseases. Ann. N. Y. Acad. Sci. 2011, 1217, 60–76. [Google Scholar] [CrossRef] [PubMed]
- Kolls, J.K.; Linden, A. Interleukin-17 family members and inflammation. Immunity 2004, 21, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Maione, F.; Cicala, C.; Liverani, E.; Mascolo, N.; Perretti, M.; D’Acquisto, F. IL-17A increases ADP-induced platelet aggregation. Biochem. Biophys. Res. Commun. 2011, 408, 658–662. [Google Scholar] [CrossRef]
- von Hundelshausen, P.; Weber, C. Platelets as immune cells: Bridging inflammation and cardiovascular disease. Circ. Res. 2007, 100, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Randriamboavonjy, V.; Isaak, J.; Elgheznawy, A.; Pistrosch, F.; Fromel, T.; Yin, X.; Badenhoop, K.; Heide, H.; Mayr, M.; Fleming, I. Calpain inhibition stabilizes the platelet proteome and reactivity in diabetes. Blood 2012, 120, 415–423. [Google Scholar] [CrossRef]
- Ahrens, I.; Chen, Y.C.; Topcic, D.; Bode, M.; Haenel, D.; Hagemeyer, C.E.; Seeba, H.; Duerschmied, D.; Bassler, N.; Jandeleit-Dahm, K.A.; et al. HMGB1 binds to activated platelets via the receptor for advanced glycation end products and is present in platelet rich human coronary artery thrombi. Thromb. Haemost. 2015, 114, 994–1003. [Google Scholar] [CrossRef] [PubMed]
- Stellos, K.; Gatsiou, A.; Stamatelopoulos, K.; Perisic Matic, L.; John, D.; Lunella, F.F.; Jae, N.; Rossbach, O.; Amrhein, C.; Sigala, F.; et al. Adenosine-to-inosine RNA editing controls cathepsin S expression in atherosclerosis by enabling HuR-mediated post-transcriptional regulation. Nat. Med. 2016, 22, 1140–1150. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.; Robinson, S.D.; Lechertier, T.; Barber, P.R.; Tavora, B.; D’Amico, G.; Jones, D.T.; Vojnovic, B.; Hodivala-Dilke, K. Use of the mouse aortic ring assay to study angiogenesis. Nat. Protoc. 2011, 7, 89–104. [Google Scholar] [CrossRef] [PubMed]
- Starnes, T.; Broxmeyer, H.E.; Robertson, M.J.; Hromas, R. Cutting edge: IL-17D, a novel member of the IL-17 family, stimulates cytokine production and inhibits hemopoiesis. J. Immunol. 2002, 169, 642–646. [Google Scholar] [CrossRef] [PubMed]
- Welti, J.; Loges, S.; Dimmeler, S.; Carmeliet, P. Recent molecular discoveries in angiogenesis and antiangiogenic therapies in cancer. J. Clin. Investig. 2013, 123, 3190–3200. [Google Scholar] [CrossRef]
- Lindemann, S.; Tolley, N.D.; Dixon, D.A.; McIntyre, T.M.; Prescott, S.M.; Zimmerman, G.A.; Weyrich, A.S. Activated platelets mediate inflammatory signaling by regulated interleukin 1beta synthesis. J. Cell Biol. 2001, 154, 485–490. [Google Scholar] [CrossRef]
- Weyrich, A.S.; Dixon, D.A.; Pabla, R.; Elstad, M.R.; McIntyre, T.M.; Prescott, S.M.; Zimmerman, G.A. Signal-dependent translation of a regulatory protein, Bcl-3, in activated human platelets. Proc. Natl. Acad. Sci. USA 1998, 95, 5556–5561. [Google Scholar] [CrossRef]
- Gidlof, O.; van der Brug, M.; Ohman, J.; Gilje, P.; Olde, B.; Wahlestedt, C.; Erlinge, D. Platelets activated during myocardial infarction release functional miRNA, which can be taken up by endothelial cells and regulate ICAM1 expression. Blood 2013, 121, 3908–3917. [Google Scholar] [CrossRef]
- Etulain, J.; Fondevila, C.; Negrotto, S.; Schattner, M. Platelet-mediated angiogenesis is independent of VEGF and fully inhibited by aspirin. Br. J. Pharmacol. 2013, 170, 255–265. [Google Scholar] [CrossRef]
- Takahashi, H.; Numasaki, M.; Lotze, M.T.; Sasaki, H. Interleukin-17 enhances bFGF-, HGF- and VEGF-induced growth of vascular endothelial cells. Immunol. Lett. 2005, 98, 189–193. [Google Scholar] [CrossRef]
- Coenen, D.M.; Mastenbroek, T.G.; Cosemans, J. Platelet interaction with activated endothelium: Mechanistic insights from microfluidics. Blood 2017, 130, 2819–2828. [Google Scholar] [CrossRef] [PubMed]
- Mirshahi, F.; Pourtau, J.; Li, H.; Muraine, M.; Trochon, V.; Legrand, E.; Vannier, J.; Soria, J.; Vasse, M.; Soria, C. SDF-1 activity on microvascular endothelial cells: Consequences on angiogenesis in in vitro and in vivo models. Thromb. Res. 2000, 99, 587–594. [Google Scholar] [CrossRef]
- Imhof, B.A.; Aurrand-Lions, M. Angiogenesis and inflammation face off. Nat. Med. 2006, 12, 171–172. [Google Scholar] [CrossRef] [PubMed]
- Silvestre, J.S.; Mallat, Z.; Duriez, M.; Tamarat, R.; Bureau, M.F.; Scherman, D.; Duverger, N.; Branellec, D.; Tedgui, A.; Levy, B.I. Antiangiogenic effect of interleukin-10 in ischemia-induced angiogenesis in mice hindlimb. Circ. Res. 2000, 87, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Bugert, P.; Dugrillon, A.; Gunaydin, A.; Eichler, H.; Kluter, H. Messenger RNA profiling of human platelets by microarray hybridization. Thromb. Haemost. 2003, 90, 738–748. [Google Scholar]
- Dittrich, M.; Birschmann, I.; Mietner, S.; Sickmann, A.; Walter, U.; Dandekar, T. Platelet protein interactions: Map, signaling components, and phosphorylation groundstate. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1326–1331. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gatsiou, A.; Sopova, K.; Tselepis, A.; Stellos, K. Interleukin-17A Triggers the Release of Platelet-Derived Factors Driving Vascular Endothelial Cells toward a Pro-Angiogenic State. Cells 2021, 10, 1855. https://doi.org/10.3390/cells10081855
Gatsiou A, Sopova K, Tselepis A, Stellos K. Interleukin-17A Triggers the Release of Platelet-Derived Factors Driving Vascular Endothelial Cells toward a Pro-Angiogenic State. Cells. 2021; 10(8):1855. https://doi.org/10.3390/cells10081855
Chicago/Turabian StyleGatsiou, Aikaterini, Kateryna Sopova, Alexandros Tselepis, and Konstantinos Stellos. 2021. "Interleukin-17A Triggers the Release of Platelet-Derived Factors Driving Vascular Endothelial Cells toward a Pro-Angiogenic State" Cells 10, no. 8: 1855. https://doi.org/10.3390/cells10081855
APA StyleGatsiou, A., Sopova, K., Tselepis, A., & Stellos, K. (2021). Interleukin-17A Triggers the Release of Platelet-Derived Factors Driving Vascular Endothelial Cells toward a Pro-Angiogenic State. Cells, 10(8), 1855. https://doi.org/10.3390/cells10081855