
Effects of Reactive Oxygen and Nitrogen Species on TrkA Expression and Signalling: Implications for proNGF in Aging and Alzheimer’s Disease


{kind=link}
{kind=link}
Abstract
1. Nerve Growth Factor Receptors and Signalling
2. Basal Forebrain Cholinergic Neurons: Dependence on NGF and Implications in Alzheimer’s Disease
3. Oxidative and Nitrative Stress in Neurodegenerative Disease
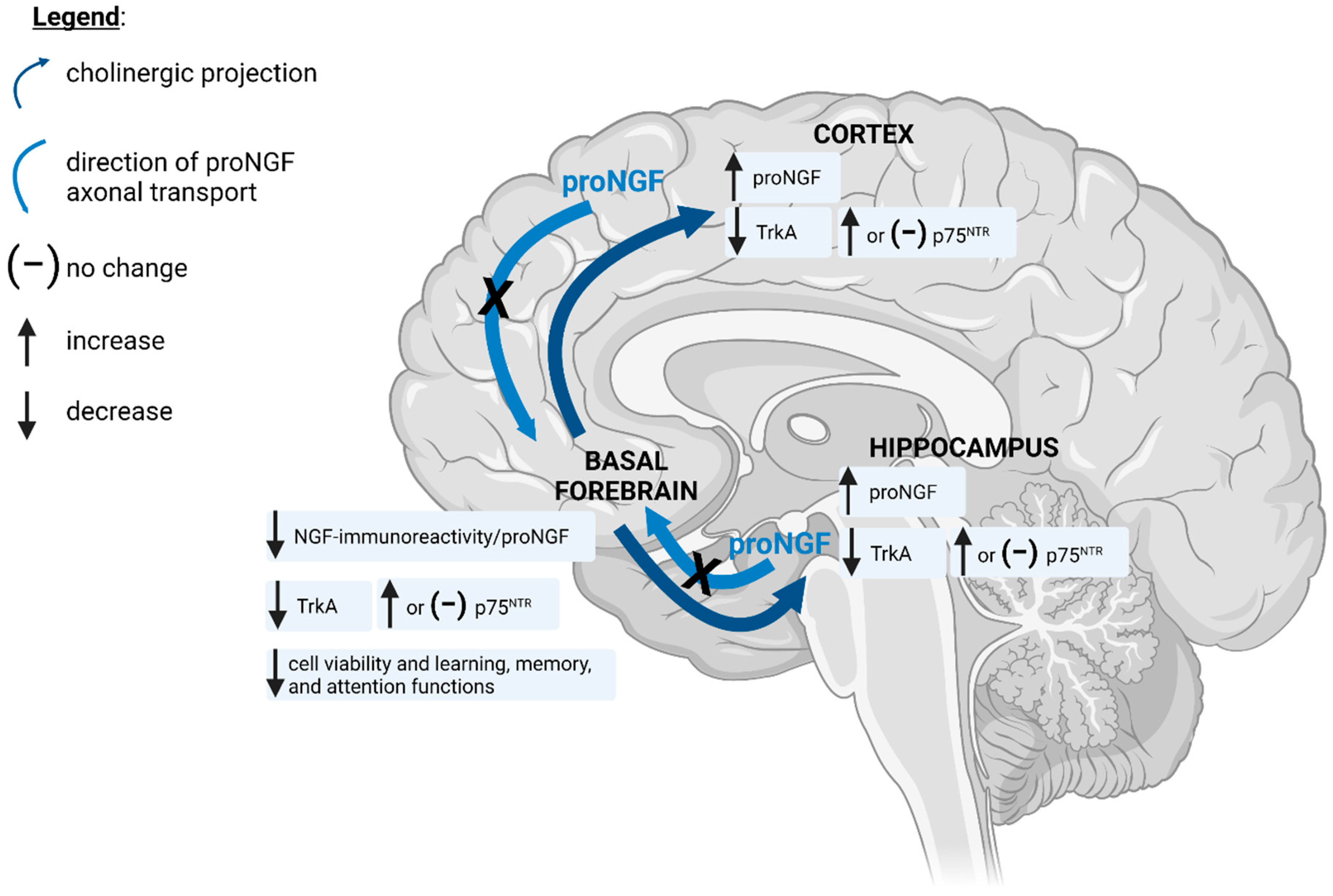
4. ROS/RNS-Induced Deficits in BFCN Viability and Cognitive Function Are Associated with Nitration of proNGF and Reduced TrkA Expression and Signalling
5. Oxidative and Nitrative Stress Alter the Expression and Signalling of proNGF Receptors
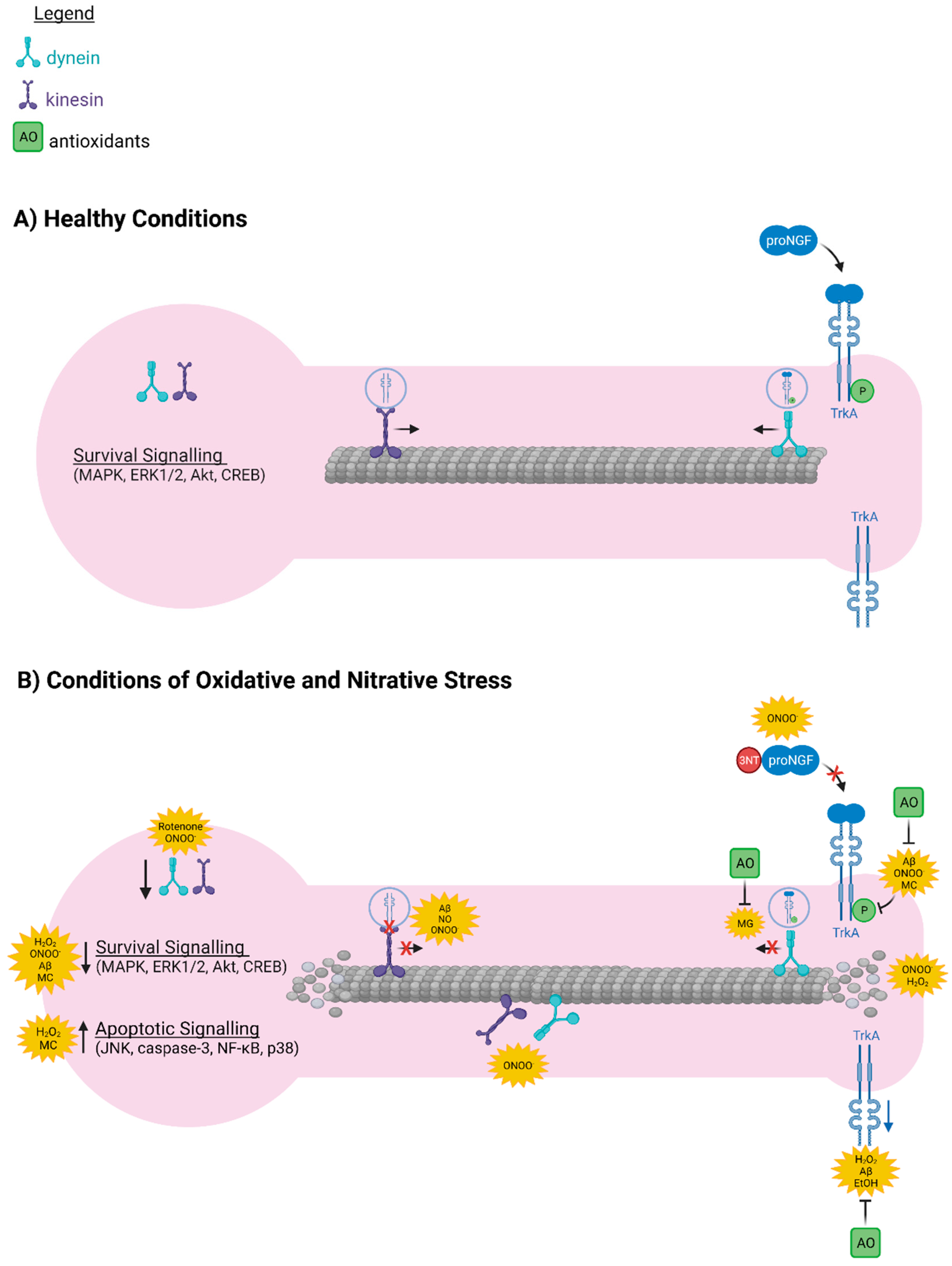
6. Oxidative and Nitrative Stress Interfere with the Axonal Transport Machinery Required for Retrograde Transport of proNGF
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Frade, J.M.; Barde, Y.-A. Nerve Growth Factor: Two Receptors, Multiple Functions. BioEssays 1998, 20, 137–145. [Google Scholar] [CrossRef]
- Thoenen, H.; Barde, Y.A. Physiology of Nerve Growth Factor. Physiol. Rev. 1980, 60, 1284–1335. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, D.R.; Martin-Zanca, D.; Parada, L.F. Tyrosine Phosphorylation and Tyrosine Kinase Activity of the Trk Proto-Oncogene Product Induced by NGF. Nature 1991, 350, 158–160. [Google Scholar] [CrossRef] [PubMed]
- Klesse, L.J.; Meyers, K.A.; Marshall, C.J.; Parada, L.F. Nerve Growth Factor Induces Survival and Differentiation through Two Distinct Signaling Cascades in PC12 Cells. Oncogene 1999, 18, 2055–2068. [Google Scholar] [CrossRef][Green Version]
- Kim, U.H.; Fink, D.; Kim, H.S.; Park, D.J.; Contreras, M.L.; Guroff, G.; Rhee, S.G. Nerve Growth Factor Stimulates Phosphorylation of Phospholipase C-Gamma in PC12 Cells. J. Biol. Chem. 1991, 266, 1359–1362. [Google Scholar] [CrossRef]
- Vetter, M.L.; Martin-Zanca, D.; Parada, L.F.; Bishop, J.M.; Kaplan, D.R. Nerve Growth Factor Rapidly Stimulates Tyrosine Phosphorylation of Phospholipase C-Gamma 1 by a Kinase Activity Associated with the Product of the Trk Protooncogene. Proc. Nat. Acad. Sci. USA 1991, 88, 5650–5654. [Google Scholar] [CrossRef]
- Kaplan, D.R.; Miller, F.D. Neurotrophin Signal Transduction in the Nervous System. Curr. Opin. Neurobiol. 2000, 10, 381–391. [Google Scholar] [CrossRef]
- Stephens, R.M.; Loeb, D.M.; Copeland, T.D.; Pawson, T.; Greene, L.A.; Kaplan, D.R. Trk Receptors Use Redundant Signal Transduction Pathways Involving SHC and PLC-Γ1 to Mediate NGF Responses. Neuron 1994, 12, 691–705. [Google Scholar] [CrossRef]
- Obermeier, A.; Bradshaw, R.A.; Seedorf, K.; Choidas, A.; Schlessinger, J.; Ullrich, A. Neuronal Differentiation Signals Are Controlled by Nerve Growth Factor Receptor/Trk Binding Sites for SHC and PLC Gamma. EMBO J. 1994, 13, 1585–1590. [Google Scholar] [CrossRef]
- Xing, J.; Kornhauser, J.M.; Xia, Z.; Thiele, E.A.; Greenberg, M.E. Nerve Growth Factor Activates Extracellular Signal-Regulated Kinase and P38 Mitogen-Activated Protein Kinase Pathways to Stimulate CREB Serine 133 Phosphorylation. Mol. Cell. Biol. 1998, 18, 1946–1955. [Google Scholar] [CrossRef]
- Saura, C.A.; Valero, J. The Role of CREB Signaling in Alzheimer’s Disease and Other Cognitive Disorders. Rev. Neurosci. 2011, 22, 153–169. [Google Scholar] [CrossRef] [PubMed]
- Roux, P.P.; Barker, P.A. Neurotrophin Signaling through the P75 Neurotrophin Receptor. Prog. Neurobiol. 2002, 67, 203–233. [Google Scholar] [CrossRef]
- Mahadeo, D.; Kaplan, L.; Chao, M.V.; Hempstead, B.L. High Affinity Nerve Growth Factor Binding Displays a Faster Rate of Association than P140trk Binding. Implications for Multi-Subunit Polypeptide Receptors. J. Biol. Chem. 1994, 269, 6884–6891. [Google Scholar] [CrossRef]
- Selby, M.J.; Edwards, R.; Sharp, F.; Rutter, W.J. Mouse Nerve Growth Factor Gene: Structure and Expression. Mol. Cell. Biol. 1987, 7, 3057–3064. [Google Scholar] [CrossRef]
- Racke, M.M.; Mason, P.J.; Johnson, M.P.; Brankamp, R.G.; Linnik, M.D. Demonstration of a Second Pharmacologically Active Promoter Region in the NGF Gene That Induces Transcription at Exon 3. Mol. Brain Res. 1996, 41, 192–199. [Google Scholar] [CrossRef]
- Darling, T.L.J.; Petrides, P.E.; Beguin, P.; Frey, P.; Shooter, E.M.; Selby, M.; Rutter, W.J. The Biosynthesis and Processing of Proteins in the Mouse 7S Nerve Growth Factor Complex. Cold Spring Harb. Symp. Quant. Biol. 1983, 48, 427–434. [Google Scholar] [CrossRef]
- Ullrich, A.; Gray, A.; Berman, C.; Dull, T.J. Human β -Nerve Growth Factor Gene Sequence Highly Homologous to That of Mouse. Nature 1983, 303, 821–825. [Google Scholar] [CrossRef]
- Edwards, R.H.; Selby, M.J.; Rutter, W.J. Differential RNA Splicing Predicts Two Distinct Nerve Growth Factor Precursors. Nature 1986, 319, 784–787. [Google Scholar] [CrossRef]
- Edwards, R.H.; Selby, M.J.; Mobley, W.C.; Weinrich, S.L.; Hruby, D.E.; Rutter, W.J. Processing and Secretion of Nerve Growth Factor: Expression in Mammalian Cells with a Vaccinia Virus Vector. Mol. Cell. Biol. 1988, 8, 2456–2464. [Google Scholar] [CrossRef] [PubMed]
- Fahnestock, M. Structure and Biosynthesis of Nerve Growth Factor. Curr. Top. Microbiol. Immunol. 1991, 165, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Lee, R. Regulation of Cell Survival by Secreted Proneurotrophins. Science 2001, 294, 1945–1948. [Google Scholar] [CrossRef] [PubMed]
- Seidah, N.G.; Benjannet, S.; Pareek, S.; Savaria, D.; Hamelin, J.; Goulet, B.; Laliberté, J.; Lazure, C.; Chrétien, M.; Murphy, R.A. Cellular Processing of the Nerve Growth Factor Precursor by the Mammalian Pro-Protein Convertases. Biochem. J. 1996, 314, 951–960. [Google Scholar] [CrossRef]
- Bruno, M.A.; Cuello, A.C. Activity-Dependent Release of Precursor Nerve Growth Factor, Conversion to Mature Nerve Growth Factor, and Its Degradation by a Protease Cascade. Proc. Nat. Acad. Sci. USA 2006, 103, 6735–6740. [Google Scholar] [CrossRef] [PubMed]
- Fahnestock, M.; Michalski, B.; Xu, B.; Coughlin, M. The Precursor Pro-Nerve Growth Factor Is the Predominant Form of Nerve Growth Factor in Brain and Is Increased in Alzheimer’s Disease. Mol. Cell. Neurosci. 2001. [Google Scholar] [CrossRef] [PubMed]
- Fahnestock, M.; Yu, G.; Michalski, B.; Mathew, S.; Colquhoun, A.; Ross, G.M.; Coughlin, M.D. The Nerve Growth Factor Precursor ProNGF Exhibits Neurotrophic Activity but Is Less Active than Mature Nerve Growth Factor. J. Neurochem. 2004, 89, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Fahnestock, M.; Yu, G.; Coughlin, M.D. ProNGF: A neurotrophic or an apoptotic molecule? In Progress in Brain Research. NGF and Related Molecules in Health and Disease; Elsevier: Amsterdam, The Netherlands, 2004; Volume 146, pp. 101–110. [Google Scholar]
- Clewes, O.; Fahey, M.S.; Tyler, S.J.; Watson, J.J.; Seok, H.; Catania, C.; Cho, K.; Dawbarn, D.; Allen, S.J. Human ProNGF: Biological Effects and Binding Profiles at TrkA, P75NTR and Sortilin. J. Neurochem. 2008, 107, 1124–1135. [Google Scholar] [CrossRef]
- Masoudi, R.; Ioannou, M.S.; Coughlin, M.D.; Pagadala, P.; Neet, K.E.; Clewes, O.; Allen, S.J.; Dawbarn, D.; Fahnestock, M. Biological Activity of Nerve Growth Factor Precursor Is Dependent upon Relative Levels of Its Receptors. J. Biol. Chem. 2009, 284, 18424–18433. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, M.S.; Fahnestock, M. ProNGF, but Not NGF, Switches from Neurotrophic to Apoptotic Activity in Response to Reductions in TrkA Receptor Levels. Int. J. Mol. Sci. 2017, 18, 599. [Google Scholar] [CrossRef]
- Rattenholl, A.; Lilie, H.; Grossmann, A.; Stern, A.; Schwarz, E.; Rudolph, R. The Pro-Sequence Facilitates Folding of Human Nerve Growth Factor from Escherichia Coli Inclusion Bodies. Eur. J. Biochem. 2001, 268, 3296–3303. [Google Scholar] [CrossRef]
- Buttigieg, H.; Kawaja, M.D.; Fahnestock, M. Neurotrophic Activity of ProNGF in Vivo. Exp. Neurol. 2007, 204, 832–835. [Google Scholar] [CrossRef]
- Saboori, A.M.; Young, M. Nerve Growth Factor: Biosynthetic Products of the Mouse Salivary Glands. Characterization of Stable High Molecular Weight and 32,000-Dalton Nerve Growth Factors. Biochemistry 1986, 25, 5565–5571. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Dicou, E.; Djakiew, D. Characterization of Nerve Growth Factor Precursor Protein Expression in Rat Round Spermatids and the Trophic Effects of Nerve Growth Factor in the Maintenance of Sertoli Cell Viability. Mol. Cell. Endocrinol. 1997, 127, 129–136. [Google Scholar] [CrossRef]
- Nykjaer, A.; Lee, R.; Teng, K.K.; Jansen, P.; Madsen, P.; Nielsen, M.S.; Jacobsen, C.; Kliemannel, M.; Schwarz, E.; Willnow, T.E.; et al. Sortilin Is Essential for ProNGF-Induced Neuronal Cell Death. Nature 2004, 427, 843–848. [Google Scholar] [CrossRef] [PubMed]
- Fahnestock, M.; Shekari, A. ProNGF and Neurodegeneration in Alzheimer’s Disease. Front. Neurosci. 2019, 13. [Google Scholar] [CrossRef]
- Knopman, D.S.; Amieva, H.; Petersen, R.C.; Chételat, G.; Holtzman, D.M.; Hyman, B.T.; Nixon, R.A.; Jones, D.T. Alzheimer Disease. Nat. Rev. Dis. Primers 2021, 7, 33. [Google Scholar] [CrossRef]
- Schmitz, T.W.; Nathan Spreng, R. Basal Forebrain Degeneration Precedes and Predicts the Cortical Spread of Alzheimer’s Pathology. Nat. Commun. 2016, 7, 13249. [Google Scholar] [CrossRef] [PubMed]
- Colom, L.V. Septal Networks: Relevance to Theta Rhythm, Epilepsy and Alzheimer’s Disease. J. Neurochem. 2006, 96, 609–623. [Google Scholar] [CrossRef]
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer’s Disease: Targeting the Cholinergic System. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef]
- Ypsilanti, A.R.; Girão da Cruz, M.T.; Burgess, A.; Aubert, I. The Length of Hippocampal Cholinergic Fibers Is Reduced in the Aging Brain. Neurobiol. Aging 2008, 29, 1666–1679. [Google Scholar] [CrossRef]
- Baxter, M.G.; Chiba, A.A. Cognitive Functions of the Basal Forebrain. Curr. Opin. Neurobiol. 1999, 9, 178–183. [Google Scholar] [CrossRef]
- Sarter, M.; Parikh, V. Choline Transporters, Cholinergic Transmission and Cognition. Nat. Rev. Neurosci. 2005, 6, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.M.; Yates, P.O.; Marcyniuk, B.; Ravindra, C.R. Pathological Evidence for Neurotransmitter Deficits in Down’s Syndrome of Middle Age. J. Ment. Defic. Res. 1985, 29, 125–135. [Google Scholar] [CrossRef]
- Wisniewski, K.E.; Dalton, A.J.; McLachlan, D.R.C.; Wen, G.Y.; Wisniewski, H.M. Alzheimer’s Disease in Down’s Syndrome: Clinicopathologic Studies. Neurology 1985, 35, 957. [Google Scholar] [CrossRef] [PubMed]
- Hartikka, J.; Hefti, F. Development of Septal Cholinergic Neurons in Culture: Plating Density and Glial Cells Modulate Effects of NGF on Survival, Fiber Growth, and Expression of Transmitter-Specific Enzymes. J. Neurosci. 1988, 8, 2967–2985. [Google Scholar] [CrossRef] [PubMed]
- Hatanaka, H.; Tsukui, H.; Nihonmatsu, I. Developmental Change in the Nerve Growth Factor Action from Induction of Choline Acetyltransferase to Promotion of Cell Survival in Cultured Basal Forebrain Cholinergic Neurons from Postnatal Rats. Dev. Brain Res. 1988, 39, 85–95. [Google Scholar] [CrossRef]
- Friedman, W.J.; Ibáñez, C.F.; Hallböök, F.; Persson, H.; Cain, L.D.; Dreyfus, C.F.; Black, I.B. Differential Actions of Neurotrophins in the Locus Coeruleus and Basal Forebrain. Exp. Neurol. 1993, 119, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Hefti, F. Nerve Growth Factor Promotes Survival of Septal Cholinergic Neurons after Fimbrial Transections. J. Neurosci. 1986, 6, 2155–2162. [Google Scholar] [CrossRef]
- Lapchak, P.A.; Hefti, F. Effect of Recombinant Human Nerve Growth Factor on Presynaptic Cholinergic Function in Rat Hippocampal Slices Following Partial Septohippocampal Lesions: Measures of [3H]Acetylcholine Synthesis, [3H]Acetylcholine Release and Choline Acetyltransferase Activity. Neuroscience 1991, 42, 639–649. [Google Scholar] [CrossRef]
- Koliatsos, V.E.; Price, D.L.; Gouras, G.K.; Cayouette, M.H.; Burton, L.E.; Winslow, J.W. Highly Selective Effects of Nerve Growth Factor, Brain-Derived Neurotrophic Factor, and Neurotrophin-3 on Intact and Injured Basal Forebrain Magnocellular Neurons. J. Comp. Neurol. 1994, 343, 247–262. [Google Scholar] [CrossRef]
- Williams, L.R.; Varon, S.; Peterson, G.M.; Wictorin, K.; Fischer, W.; Bjorklund, A.; Gage, F.H. Continuous Infusion of Nerve Growth Factor Prevents Basal Forebrain Neuronal Death after Fimbria Fornix Transection. Proc. Nat. Acad. Sci. USA 1986, 83, 9231–9235. [Google Scholar] [CrossRef]
- Lauterborn, J.C.; Isackson, P.J.; Gall, C.M. Nerve Growth Factor MRNA-Containing Cells Are Distributed within Regions of Cholinergic Neurons in the Rat Basal Forebrain. J. Comp. Neurol. 1991, 306, 439–446. [Google Scholar] [CrossRef]
- Korsching, S.; Auburger, G.; Heumann, R.; Scott, J.; Thoenen, H. Levels of Nerve Growth Factor and Its MRNA in the Central Nervous System of the Rat Correlate with Cholinergic Innervation. EMBO J. 1985, 4, 1389–1393. [Google Scholar] [CrossRef]
- Lauterborn, J.C.; Bizon, J.L.; Tran, T.M.; Gall, C.M. NGF MRNA Is Expressed by GABAergic but Not Cholinergic Neurons in Rat Basal Forebrain. J. Comp. Neurol. 1995, 360, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Francis-Turner, L.; Valoušková, V. Nerve Growth Factor and Nootropic Drug Cerebrolysin but Not Fibroblast Growth Factor Can Reduce Spatial Memory Impairment Elicited by Fimbria-Fornix Transection: Short-Term Study. Neurosci. Lett. 1996, 202, 193–196. [Google Scholar] [CrossRef]
- Seiler, M.; Schwab, M.E. Specific Retrograde Transport of Nerve Growth Factor (NGF) from Neocortex to Nucleus Basalis in the Rat. Brain Res. 1984, 300, 33–39. [Google Scholar] [CrossRef]
- Barford, K.; Deppmann, C.; Winckler, B. The Neurotrophin Receptor Signaling Endosome: Where Trafficking Meets Signaling. Dev. Neurobiol. 2017, 77, 405–418. [Google Scholar] [CrossRef] [PubMed]
- Delcroix, J.-D.; Valletta, J.S.; Wu, C.; Hunt, S.J.; Kowal, A.S.; Mobley, W.C. NGF Signaling in Sensory Neurons: Evidence That Early Endosomes Carry NGF Retrograde Signals. Neuron 2003, 39, 69–84. [Google Scholar] [CrossRef]
- Salehi, A.; Delcroix, J.-D.; Belichenko, P.V.; Zhan, K.; Wu, C.; Valletta, J.S.; Takimoto-Kimura, R.; Kleschevnikov, A.M.; Sambamurti, K.; Chung, P.P.; et al. Increased App Expression in a Mouse Model of Down’s Syndrome Disrupts NGF Transport and Causes Cholinergic Neuron Degeneration. Neuron 2006, 51, 29–42. [Google Scholar] [CrossRef]
- Cosker, K.E.; Pazyra-Murphy, M.F.; Fenstermacher, S.J.; Segal, R.A. Target-Derived Neurotrophins Coordinate Transcription and Transport of Bclw to Prevent Axonal Degeneration. J. Neurosci. 2013, 33, 5195–5207. [Google Scholar] [CrossRef]
- Di Matteo, P.; Calvello, M.; Luin, S.; Marchetti, L.; Cattaneo, A. An Optimized Procedure for the Site-Directed Labeling of NGF and ProNGF for Imaging Purposes. Front. Mol. Biosci. 2017, 4. [Google Scholar] [CrossRef]
- De Nadai, T.; Marchetti, L.; Di Rienzo, C.; Calvello, M.; Signore, G.; Di Matteo, P.; Gobbo, F.; Turturro, S.; Meucci, S.; Viegi, A.; et al. Precursor and Mature NGF Live Tracking: One versus Many at a Time in the Axons. Sci. Rep. 2016, 6, 20272. [Google Scholar] [CrossRef]
- Shekari, A.; Fahnestock, M. Retrograde Axonal Transport of BDNF and ProNGF Diminishes with Age in Basal Forebrain Cholinergic Neurons. Neurobiol. Aging 2019, 84, 131–140. [Google Scholar] [CrossRef]
- Cooper, J.D.; Lindholm, D.; Sofroniew, M.V. Reduced Transport of [125I]Nerve Growth Factor by Cholinergic Neurons and down-Regulated Trka Expression in the Medial Septum of Aged Rats. Neuroscience 1994, 62, 625–629. [Google Scholar] [CrossRef]
- Bearer, E.L.; Manifold-Wheeler, B.C.; Medina, C.S.; Gonzales, A.G.; Chaves, F.L.; Jacobs, R.E. Alterations of Functional Circuitry in Aging Brain and the Impact of Mutated APP Expression. Neurobiol. Aging 2018, 70, 276–290. [Google Scholar] [CrossRef]
- Niewiadomska, G.; Baksalerska-Pazera, M. Age-Dependent Changes in Axonal Transport and Cellular Distribution of Tau 1 in the Rat Basal Forebrain Neurons. NeuroReport 2003, 14, 1701–1706. [Google Scholar] [CrossRef] [PubMed]
- Narisawa-Saito, M.; Wakabayashi, K.; Tsuji, S.; Takahashi, H.; Nawa, H. Regional Specificity of Alterations in NGF, BDNF, and NT-3 Levels in Alzheimer’s Disease. NeuroReport 1996, 7, 2925–2928. [Google Scholar] [CrossRef] [PubMed]
- Crutcher, K.A.; Scott, S.A.; Liang, S.; Everson, W.V.; Weingartner, J. Detection of NGF-like Activity in Human Brain Tissue: Increased Levels in Alzheimer’s Disease. J. Neurosci. 1993, 13, 2540–2550. [Google Scholar] [CrossRef]
- Fahnestock, M.; Scott, S.A.; Jetté, N.; Weingartner, J.A.; Crutcher, K.A. Nerve Growth Factor MRNA and Protein Levels Measured in the Same Tissue from Normal and Alzheimer’s Disease Parietal Cortex. Mol. Brain Res. 1996, 42, 175–178. [Google Scholar] [CrossRef]
- Scott, S.A.; Mufson, E.J.; Weingartner, J.A.; Skau, K.A.; Crutcher, K.A. Nerve Growth Factor in Alzheimer’s Disease: Increased Levels throughout the Brain Coupled with Declines in Nucleus Basalis. J. Neurosci. 1995, 15, 6213–6221. [Google Scholar] [CrossRef]
- Mufson, E.J.; Conner, J.M.; Kordower, J.H. Nerve Growth Factor in Alzheimer’s Disease: Defective Retrograde Transport to Nucleus Basalis. NeuroReport 1995, 6, 1063–1066. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Wuu, J.; Mufson, E.J.; Fahnestock, M. Increased ProNGF Levels in Subjects with Mild Cognitive Impairment and Mild Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2004, 63, 641–649. [Google Scholar] [CrossRef]
- Perez, S.E.; He, B.; Muhammad, N.; Oh, K.-J.; Fahnestock, M.; Ikonomovic, M.D.; Mufson, E.J. Cholinotrophic Basal Forebrain System Alterations in 3xTg-AD Transgenic Mice. Neurobiol. Dis. 2011, 41, 338–352. [Google Scholar] [CrossRef]
- Iulita, M.F.; Bistué Millón, M.B.; Pentz, R.; Aguilar, L.F.; Do Carmo, S.; Allard, S.; Michalski, B.; Wilson, E.N.; Ducatenzeiler, A.; Bruno, M.A.; et al. Differential Deregulation of NGF and BDNF Neurotrophins in a Transgenic Rat Model of Alzheimer’s Disease. Neurobiol. Dis. 2017, 108, 307–323. [Google Scholar] [CrossRef]
- Iulita, M.F.; Do Carmo, S.; Ower, A.K.; Fortress, A.M.; Aguilar, L.F.; Hanna, M.; Wisniewski, T.; Granholm, A.-C.; Buhusi, M.; Busciglio, J.; et al. Nerve Growth Factor Metabolic Dysfunction in Down’s Syndrome Brains. Brain 2014, 137, 860–872. [Google Scholar] [CrossRef]
- Pentz, R.; Iulita, M.F.; Ducatenzeiler, A.; Bennett, D.A.; Cuello, A.C. The Human Brain NGF Metabolic Pathway Is Impaired in the Pre-Clinical and Clinical Continuum of Alzheimers Disease. Mol. Psychiatry 2020, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Pentz, R.; Iulita, M.F.; Mikutra-Cencora, M.; Ducatenzeiler, A.; Bennett, D.A.; Cuello, A.C. A New Role for Matrix Metalloproteinase-3 in the NGF Metabolic Pathway: Proteolysis of Mature NGF and Sex-Specific Differences in the Continuum of Alzheimer’s Pathology. Neurobiol. Dis. 2021, 148, 105150. [Google Scholar] [CrossRef] [PubMed]
- Jetté, N.; Cole, M.S.; Fahnestock, M. NGF MRNA Is Not Decreased in Frontal Cortex from Alzheimer’s Disease Patients. Mol. Brain Res. 1994, 25, 242–250. [Google Scholar] [CrossRef]
- Goedert, M.; Fine, A.; Hunt, S.P.; Ullrich, A. Nerve Growth Factor MRNA in Peripheral and Central Rat Tissues and in the Human Central Nervous System: Lesion Effects in the Rat Brain and Levels in Alzheimer’s Disease. Mol. Brain Res. 1986, 1, 85–92. [Google Scholar] [CrossRef]
- Allard, S.; Leon, W.C.; Pakavathkumar, P.; Bruno, M.A.; Ribeiro-da-Silva, A.; Cuello, A.C. Impact of the NGF Maturation and Degradation Pathway on the Cortical Cholinergic System Phenotype. J. Neurosci. 2012, 32, 2002–2012. [Google Scholar] [CrossRef]
- Allard, S.; Jacobs, M.L.; Do Carmo, S.; Cuello, A.C. Compromise of Cortical ProNGF Maturation Causes Selective Retrograde Atrophy in Cholinergic Nucleus Basalis Neurons. Neurobiol. Aging 2018, 67, 10–20. [Google Scholar] [CrossRef]
- Hock, C.; Heese, K.; Müller-Spahn, F.; Hulette, C.; Rosenberg, C.; Otten, U. Decreased TrkA Neurotrophin Receptor Expression in the Parietal Cortex of Patients with Alzheimer’s Disease. Neurosci. Lett. 1998, 241, 151–154. [Google Scholar] [CrossRef]
- Mufson, E.J.; Li, J.M.; Sobreviela, T.; Kordower, J.H. Decreased TrkA Gene Expression within Basal Forebrain Neurons in Alzheimer’s Disease. NeuroReport 1996, 8, 25–29. [Google Scholar] [CrossRef]
- Mufson, E.J.; Lavine, N.; Jaffar, S.; Kordower, J.H.; Quirion, R.; Saragovi, H.U. Reduction in P140-TrkA Receptor Protein within the Nucleus Basalis and Cortex in Alzheimer’s Disease. Exp. Neurol. 1997, 146, 91–103. [Google Scholar] [CrossRef]
- Salehi, A.; Verhaagen, J.; Dijkhuizen, P.A.; Swaab, D.F. Co-Localization of High-Affinity Neurotrophin Receptors in Nucleus Basalis of Meynert Neurons and Their Differential Reduction in Alzheimer’s Disease. Neuroscience 1996, 75, 373–387. [Google Scholar] [CrossRef]
- Podlesniy, P.; Kichev, A.; Pedraza, C.; Saurat, J.; Encinas, M.; Perez, B.; Ferrer, I.; Espinet, C. Pro-NGF from Alzheimer’s Disease and Normal Human Brain Displays Distinctive Abilities to Induce Processing and Nuclear Translocation of Intracellular Domain of P75NTR and Apoptosis. Am. J. Pathol. 2006, 169, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-K.; Kumar, P.; Fu, Q.; Rosen, K.M.; Querfurth, H.W. The Insulin/Akt Signaling Pathway Is Targeted by Intracellular β-Amyloid. Mol. Biol. Cell 2009, 20, 1533–1544. [Google Scholar] [CrossRef] [PubMed]
- Ernfors, P.; Lindefors, N.; Chan-Palay, V.; Persson, H. Cholinergic Neurons of the Nucleus Basalis Express Elevated Levels of Nerve Growth Factor Receptor MRNA in Senile Dementia of the Alzheimer Type. Dement. Geriatr. Cogn. Disord. 1990, 1, 138–145. [Google Scholar] [CrossRef]
- Granholm, A.-C.E.; Sanders, L.A.; Crnic, L.S. Loss of Cholinergic Phenotype in Basal Forebrain Coincides with Cognitive Decline in a Mouse Model of Down’s Syndrome. Exp. Neurol. 2000, 161, 647–663. [Google Scholar] [CrossRef]
- Ozcan, A.; Ogun, M. Biochemistry of Reactive Oxygen and Nitrogen Species; IntechOpen: London, UK, 2015; ISBN 978-953-51-2200-5. [Google Scholar]
- Cobb, C.A.; Cole, M.P. Oxidative and Nitrative Stress in Neurodegeneration. Neurobiol. Dis. 2015, 84, 4–21. [Google Scholar] [CrossRef]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The Role of Oxidative Stress in Neurodegenerative Diseases. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef]
- Markesbery, W.R. Oxidative Stress Hypothesis in Alzheimer’s Disease. Free Radic. Biol. Med. 1997, 23, 134–147. [Google Scholar] [CrossRef]
- Kim, T.-S.; Pae, C.-U.; Yoon, S.-J.; Jang, W.-Y.; Lee, N.J.; Kim, J.-J.; Lee, S.-J.; Lee, C.; Paik, I.-H.; Lee, C.-U. Decreased Plasma Antioxidants in Patients with Alzheimer’s Disease. Int. J. Geriatr. Psychiatry 2006, 21, 344–348. [Google Scholar] [CrossRef]
- Hensley, K.; Maidt, M.L.; Yu, Z.; Sang, H.; Markesbery, W.R.; Floyd, R.A. Electrochemical Analysis of Protein Nitrotyrosine and Dityrosine in the Alzheimer Brain Indicates Region-Specific Accumulation. J. Neurosci. 1998, 18, 8126–8132. [Google Scholar] [CrossRef]
- Mecocci, P.; Beal, M.F.; Cecchetti, R.; Polidori, M.C.; Cherubini, A.; Chionne, F.; Avellini, L.; Romano, G.; Senin, U. Mitochondrial Membrane Fluidity and Oxidative Damage to Mitochondrial DNA in Aged and AD Human Brain. Mol. Chem. Neuropathol. 1997, 31, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Lyras, L.; Cairns, N.J.; Jenner, A.; Jenner, P.; Halliwell, B. An Assessment of Oxidative Damage to Proteins, Lipids, and DNA in Brain from Patients with Alzheimer’s Disease. J. Neurochem. 1997, 68, 2061–2069. [Google Scholar] [CrossRef] [PubMed]
- Lovell, M.A.; Ehmann, W.D.; Butler, S.M.; Markesbery, W.R. Elevated Thiobarbituric Acid-Reactive Substances and Antioxidant Enzyme Activity in the Brain in Alzheimer’s Disease. Neurology 1995, 45, 1594–1601. [Google Scholar] [CrossRef] [PubMed]
- Subbarao, K.V.; Richardson, J.S.; Ang, L.C. Autopsy Samples of Alzheimer’s Cortex Show Increased Peroxidation In Vitro. J. Neurochem. 1990, 55, 342–345. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.D.; Carney, J.M.; Starke-Reed, P.E.; Oliver, C.N.; Stadtman, E.R.; Floyd, R.A.; Markesbery, W.R. Excess Brain Protein Oxidation and Enzyme Dysfunction in Normal Aging and in Alzheimer Disease. Proc. Nat. Acad. Sci. USA 1991, 88, 10540–10543. [Google Scholar] [CrossRef] [PubMed]
- Praticò, D.; Uryu, K.; Leight, S.; Trojanoswki, J.Q.; Lee, V.M.-Y. Increased Lipid Peroxidation Precedes Amyloid Plaque Formation in an Animal Model of Alzheimer Amyloidosis. J. Neurosci. 2001, 21, 4183–4187. [Google Scholar] [CrossRef]
- Yan, S.D.; Chen, X.; Schmidt, A.M.; Brett, J.; Godman, G.; Zou, Y.S.; Scott, C.W.; Caputo, C.; Frappier, T.; Smith, M.A. Glycated Tau Protein in Alzheimer Disease: A Mechanism for Induction of Oxidant Stress. Proc. Nat. Acad. Sci. USA 1994, 91, 7787–7791. [Google Scholar] [CrossRef]
- Good, P.F.; Werner, P.; Hsu, A.; Olanow, C.W.; Perl, D.P. Evidence of Neuronal Oxidative Damage in Alzheimer’s Disease. Am. J. Pathol. 1996, 149, 21–28. [Google Scholar]
- Smith, M.A.; Taneda, S.; Richey, P.L.; Miyata, S.; Yan, S.D.; Stern, D.; Sayre, L.M.; Monnier, V.M.; Perry, G. Advanced Maillard Reaction End Products Are Associated with Alzheimer Disease Pathology. Proc. Nat. Acad. Sci. USA 1994, 91, 5710–5714. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhao, B. Oxidative Stress and the Pathogenesis of Alzheimer’s Disease. Oxid. Med. Cell. Longev. 2013, 2013, 316523. [Google Scholar] [CrossRef]
- Mattson, M.P.; Pedersen, W.A. Effects of Amyloid Precursor Protein Derivatives and Oxidative Stress on Basal Forebrain Cholinergic Systems in Alzheimer’s Disease. Int. J. Dev. Neurosci. 1998, 16, 737–753. [Google Scholar] [CrossRef]
- Bruno, M.A.; Cuello, A.C. Cortical Peroxynitration of Nerve Growth Factor in Aged and Cognitively Impaired Rats. Neurobiol. Aging 2012, 33, 1927–1937. [Google Scholar] [CrossRef] [PubMed]
- Berr, C. Cognitive Impairment and Oxidative Stress in the Elderly: Results of Epidemiological Studies. BioFactors 2000, 13, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Perkins, A.J.; Hendrie, H.C.; Callahan, C.M.; Gao, S.; Unverzagt, F.W.; Xu, Y.; Hall, K.S.; Hui, S.L. Association of Antioxidants with Memory in a Multiethnic Elderly Sample Using the Third National Health and Nutrition Examination Survey. Am. J. Epidemiol. 1999, 150, 37–44. [Google Scholar] [CrossRef]
- Perrig, W.J.; Perrig, P.; Stähelin, H.B. The Relation between Antioxidants and Memory Performance in the Old and Very Old. J. Am. Geriatr. Soc. 1997, 45, 718–724. [Google Scholar] [CrossRef]
- Rinaldi, P.; Polidori, M.C.; Metastasio, A.; Mariani, E.; Mattioli, P.; Cherubini, A.; Catani, M.; Cecchetti, R.; Senin, U.; Mecocci, P. Plasma Antioxidants Are Similarly Depleted in Mild Cognitive Impairment and in Alzheimer’s Disease. Neurobiol. Aging 2003, 24, 915–919. [Google Scholar] [CrossRef]
- Carney, J.M.; Starke-Reed, P.E.; Oliver, C.N.; Landum, R.W.; Cheng, M.S.; Wu, J.F.; Floyd, R.A. Reversal of Age-Related Increase in Brain Protein Oxidation, Decrease in Enzyme Activity, and Loss in Temporal and Spatial Memory by Chronic Administration of the Spin-Trapping Compound N-Tert-Butyl-Alpha-Phenylnitrone. Proc. Nat. Acad. Sci. USA 1991, 88, 3633–3636. [Google Scholar] [CrossRef]
- Tran, M.H.; Yamada, K.; Nakajima, A.; Mizuno, M.; He, J.; Kamei, H.; Nabeshima, T. Tyrosine Nitration of a Synaptic Protein Synaptophysin Contributes to Amyloid β -Peptide-Induced Cholinergic Dysfunction. Mol. Psychiatry 2003, 8, 407–412. [Google Scholar] [CrossRef]
- Guermonprez, L.; Ducrocq, C.; Gaudry-Talarmain, Y.M. Inhibition of Acetylcholine Synthesis and Tyrosine Nitration Induced by Peroxynitrite Are Differentially Prevented by Antioxidants. Mol. Pharmacol. 2001, 60, 838–846. [Google Scholar]
- Lockrow, J.; Prakasam, A.; Huang, P.; Bimonte-Nelson, H.; Sambamurti, K.; Granholm, A.-C. Cholinergic Degeneration and Memory Loss Delayed by Vitamin E in a Down Syndrome Mouse Model. Exp. Neurol. 2009, 216, 278–289. [Google Scholar] [CrossRef]
- Bruno, M.A.; Leon, W.C.; Fragoso, G.; Mushynski, W.E.; Almazan, G.; Cuello, A.C. Amyloid β-Induced Nerve Growth Factor Dysmetabolism in Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2009, 68, 857–869. [Google Scholar] [CrossRef] [PubMed]
- Lan, Z.; Chen, L.; Fu, Q.; Ji, W.; Wang, S.; Liang, Z.; Qu, R.; Kong, L.; Ma, S. Paeoniflorin Attenuates Amyloid-Beta Peptide-Induced Neurotoxicity by Ameliorating Oxidative Stress and Regulating the NGF-Mediated Signaling in Rats. Brain Res. 2013, 1498, 9–19. [Google Scholar] [CrossRef]
- Lim, S.; Moon, M.; Oh, H.; Kim, H.G.; Kim, S.Y.; Oh, M.S. Ginger Improves Cognitive Function via NGF-Induced ERK/CREB Activation in the Hippocampus of the Mouse. J. Nutr. Biochem. 2014, 25, 1058–1065. [Google Scholar] [CrossRef]
- Jackson, G.R.; Apffel, L.; Werrbach-Perez, K.; Perez-Polo, J.R. Role of Nerve Growth Factor in Oxidant-Antioxidant Balance and Neuronal Injury. I. Stimulation of Hydrogen Peroxide Resistance. J. Neurosci. Res. 1990, 25, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Perez-Polo, R. Role of Nerve Growth Factor in Oxidanl Homeostasis: Glutathione Metabolism. J. Neurochem. 1993, 61, 1713–1721. [Google Scholar] [CrossRef] [PubMed]
- Amara, F.; Berbenni, M.; Fragni, M.; Leoni, G.; Viggiani, S.; Ippolito, V.M.; Larocca, M.; Rossano, R.; Alberghina, L.; Riccio, P.; et al. Neuroprotection by Cocktails of Dietary Antioxidants under Conditions of Nerve Growth Factor Deprivation. Oxid. Med. Cell. Longev. 2015, 2015, 217258. [Google Scholar] [CrossRef]
- Spear, N.; Estévez, A.G.; Barbeito, L.; Beckman, J.S.; Johnson, G.V.W. Nerve Growth Factor Protects PC12 Cells Against Peroxynitrite-Induced Apoptosis via a Mechanism Dependent on Phosphatidylinositol 3-Kinase. J. Neurochem. 1997, 69, 53–59. [Google Scholar] [CrossRef]
- Wang, W.; Dow, K.E.; Riopelle, R.J.; Ross, G.M. The Common Neurotrophin Receptor P75NTR Enhances the Ability of PC12 Cells to Resist Oxidative Stress by a TrkA-Dependent Mechanism. Neurotox. Res. 2001, 3, 485–499. [Google Scholar] [CrossRef]
- Tang, L.-L.; Wang, R.; Tang, X.-C. Huperzine A Protects SHSY5Y Neuroblastoma Cells against Oxidative Stress Damage via Nerve Growth Factor Production. Eur. J. Pharmacol. 2005, 519, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.; King, M.A.; Heaton, M.B.; Walker, D.W. The Effects of Chronic Ethanol Consumption on Neurotrophins and Their Receptors in the Rat Hippocampus and Basal Forebrain. Brain Res. 2002, 950, 137–147. [Google Scholar] [CrossRef]
- Song, K.; Na, J.-Y.; Kim, S.; Kwon, J. Rutin Upregulates Neurotrophic Factors Resulting in Attenuation of Ethanol-Induced Oxidative Stress in HT22 Hippocampal Neuronal Cells. J. Sci. Food Agric. 2015, 95, 2117–2123. [Google Scholar] [CrossRef]
- Guo, C.; Sun, L.; Liu, G.; Zhao, S.; Liu, W.; Zhao, Y. Protective Effect of Nicotine on the Cultured Rat Basal Forebrain Neurons Damaged by B-Amyloid (Ab)25-35 Protein Cytotoxicity. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 2964–2972. [Google Scholar]
- Olivieri, G.; Otten, U.; Meier, F.; Baysang, G.; Dimitriades-Schmutz, B.; Müller-Spahn, F.; Savaskan, E. Oxidative Stress Modulates Tyrosine Kinase Receptor A and P75 Receptor (Low-Affinity Nerve Growth Factor Receptor) Expression in SHSY5Y Neuroblastoma Cells. Neurol. Clin. Neurophysiol. 2002, 2002, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Gupta, A.K.; Shukla, R.K.; Tripathi, V.K.; Jahan, S.; Pandey, A.; Srivastava, A.; Agrawal, M.; Yadav, S.; Khanna, V.K.; et al. Molecular Mechanism of Switching of TrkA/P75 NTR Signaling in Monocrotophos Induced Neurotoxicity. Sci. Rep. 2015, 5, 14038. [Google Scholar] [CrossRef]
- Ali, T.K.; Matragoon, S.; Pillai, B.A.; Liou, G.I.; El-Remessy, A.B. Peroxynitrite Mediates Retinal Neurodegeneration by Inhibiting Nerve Growth Factor Survival Signaling in Experimental and Human Diabetes. Diabetes 2008, 57, 10. [Google Scholar] [CrossRef]
- del Pino, J.; Moyano, P.; Anadon, M.J.; García, J.M.; Díaz, M.J.; Gómez, G.; García, J.; Frejo, M.T. SN56 Basal Forebrain Cholinergic Neuronal Loss after Acute and Long-Term Chlorpyrifos Exposure through Oxidative Stress Generation; P75NTR and A7-NAChRs Alterations Mediated Partially by AChE Variants Disruption. Toxicology 2016, 353–354, 48–57. [Google Scholar] [CrossRef]
- Kim, S.; Kwon, J. [6]-Shogaol Attenuates Neuronal Apoptosis in Hydrogen Peroxide-Treated Astrocytes through the Up-Regulation of Neurotrophic Factors. Phytother. Res. 2013, 27, 1795–1799. [Google Scholar] [CrossRef]
- Martindale, J.L.; Holbrook, N.J. Cellular Response to Oxidative Stress: Signaling for Suicide and Survival. J. Cell. Physiol. 2002, 192, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.-L.; Yen, G.-C. Effect of Hesperetin against Oxidative Stress via ER- and TrkA-Mediated Actions in PC12 Cells. J. Agric. Food Chem. 2011, 59, 5779–5785. [Google Scholar] [CrossRef] [PubMed]
- Hamano, T.; Mutoh, T.; Tabira, T.; Araki, W.; Kuriyama, M.; Mihara, T.; Yano, S.; Yamamoto, H. Abnormal Intracellular Trafficking of High Affinity Nerve Growth Factor Receptor, Trk, in Stable Transfectants Expressing Presenilin 1 Protein. Mol. Brain Res. 2005, 137, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Jonnala, R.R.; Buccafusco, J.J. Inhibition of Nerve Growth Factor Signaling by Peroxynitrite. J. Neurosci. Res. 2001, 63, 27–34. [Google Scholar] [CrossRef]
- Iannilli, F.; Sodero, A.O.; Ledesma, M.D.; Dotti, C.G. Oxidative Stress Activates the Pro-Survival TrkA Pathway through Membrane Cholesterol Loss. Neurobiol. Aging 2011, 32, 1033–1042. [Google Scholar] [CrossRef]
- Zhang, L.; Jope, R.S. Oxidative Stress Differentially Modulates Phosphorylation of ERK, P38 and CREB Induced by NGF or EGF in PC12 Cells. Neurobiol. Aging 1999, 20, 271–278. [Google Scholar] [CrossRef]
- Scott-Solomon, E.; Kuruvilla, R. Mechanisms of Neurotrophin Trafficking via Trk Receptors. Mol. Cell. Neurosci. 2018, 91, 25–33. [Google Scholar] [CrossRef]
- Heerssen, H.M.; Pazyra, M.F.; Segal, R.A. Dynein Motors Transport Activated Trks to Promote Survival of Target-Dependent Neurons. Nat. Neurosci. 2004, 7, 596–604. [Google Scholar] [CrossRef]
- King, S.M. The Dynein Microtubule Motor. Biochim. Biophys. Acta. Mol. Cell Res. 2000, 1496, 60–75. [Google Scholar] [CrossRef]
- Ogawa, K.; Takai, H.; Ogiwara, A.; Yokota, E.; Shimizu, T.; Inaba, K.; Mohri, H. Is Outer Arm Dynein Intermediate Chain 1 Multifunctional? Mol. Biol. Cell 1996, 7, 1895–1907. [Google Scholar] [CrossRef]
- Patel-King, R.S.; Benashski, S.E.; Harrison, A.; King, S.M. Two Functional Thioredoxins Containing Redox-Sensitive Vicinal Dithiols from the Chlamydomonas Outer Dynein Arm (∗). J. Biol. Chem. 1996, 271, 6283–6291. [Google Scholar] [CrossRef]
- Hashemy, S.I.; Holmgren, A. Regulation of the Catalytic Activity and Structure of Human Thioredoxin 1 via Oxidation and S-Nitrosylation of Cysteine Residues. J. Biol. Chem. 2008, 283, 21890–21898. [Google Scholar] [CrossRef]
- Chaves, R.; Quevedo Melo, T.; D’Unhao, A.; Farizatto, K.; Ferrari, M.F.R. Dynein C1h1, Dynactin and Syntaphilin Expression in Brain Areas Related to Neurodegenerative Diseases Following Exposure to Rotenone. Acta Neurobiol. Exp. 2013, 73, 541–556. [Google Scholar]
- Eiserich, J.P.; Estévez, A.G.; Bamberg, T.V.; Ye, Y.Z.; Chumley, P.H.; Beckman, J.S.; Freeman, B.A. Microtubule Dysfunction by Posttranslational Nitrotyrosination of α-Tubulin: A Nitric Oxide-Dependent Mechanism of Cellular Injury. Proc. Nat. Acad. Sci. USA 1999, 96, 6365–6370. [Google Scholar] [CrossRef] [PubMed]
- Chou, A.P.; Li, S.; Fitzmaurice, A.G.; Bronstein, J.M. Mechanisms of Rotenone-Induced Proteasome Inhibition. NeuroToxicology 2010, 31, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Sherer, T.B.; Betarbet, R.; Testa, C.M.; Seo, B.B.; Richardson, J.R.; Kim, J.H.; Miller, G.W.; Yagi, T.; Matsuno-Yagi, A.; Greenamyre, J.T. Mechanism of Toxicity in Rotenone Models of Parkinson’s Disease. J. Neurosci. 2003, 23, 10756–10764. [Google Scholar] [CrossRef]
- Melo, T.Q.; D’unhao, A.M.; Martins, S.A.; Farizatto, K.L.G.; Chaves, R.S.; Ferrari, M.F.R. Rotenone-Dependent Changes of Anterograde Motor Protein Expression and Mitochondrial Mobility in Brain Areas Related to Neurodegenerative Diseases. Cell. Mol. Neurobiol. 2013, 33, 327–335. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Xu, Y.F.; Chen, X.Q.; Wang, X.C.; Wang, J.-Z. Nitration and Oligomerization of Tau Induced by Peroxynitrite Inhibit Its Microtubule-Binding Activity. FEBS Lett. 2005, 579, 2421–2427. [Google Scholar] [CrossRef]
- Fang, C.; Bourdette, D.; Banker, G. Oxidative Stress Inhibits Axonal Transport: Implications for Neurodegenerative Diseases. Mol. Neurodegener. 2012, 7, 29. [Google Scholar] [CrossRef]
- Olmsted, J.B.; Borisy, G.G. Microtubules. Annu. Rev. Biochem. 1973, 42, 507–540. [Google Scholar] [CrossRef]
- Matsuyama, S.S.; Jarvik, L.F. Hypothesis: Microtubules, a Key to Alzheimer Disease. Proc. Nat. Acad. Sci. USA 1989, 86, 8152–8156. [Google Scholar] [CrossRef] [PubMed]
- Stagi, M. Breakdown of Axonal Synaptic Vesicle Precursor Transport by Microglial Nitric Oxide. J. Neurosci. 2005, 25, 352–362. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hirokawa, N.; Noda, Y.; Tanaka, Y.; Niwa, S. Kinesin Superfamily Motor Proteins and Intracellular Transport. Nat. Rev. Mol. Cell Biol. 2009, 10, 682–696. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Niwa, S.; Dong, M.; Farkhondeh, A.; Wang, L.; Zhou, R.; Hirokawa, N. The Molecular Motor KIF1A Transports the TrkA Neurotrophin Receptor and Is Essential for Sensory Neuron Survival and Function. Neuron 2016, 90, 1215–1229. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, N.; Joshi, R.; Zhang, S.; Zhang, Z.-Y.; Kuruvilla, R. Phospho-Regulation of Soma-to-Axon Transcytosis of Neurotrophin Receptors. Dev. Cell 2017, 42, 626.e5–639.e5. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kropf, E.; Fahnestock, M. Effects of Reactive Oxygen and Nitrogen Species on TrkA Expression and Signalling: Implications for proNGF in Aging and Alzheimer’s Disease. Cells 2021, 10, 1983. https://doi.org/10.3390/cells10081983
Kropf E, Fahnestock M. Effects of Reactive Oxygen and Nitrogen Species on TrkA Expression and Signalling: Implications for proNGF in Aging and Alzheimer’s Disease. Cells. 2021; 10(8):1983. https://doi.org/10.3390/cells10081983
Chicago/Turabian StyleKropf, Erika, and Margaret Fahnestock. 2021. "Effects of Reactive Oxygen and Nitrogen Species on TrkA Expression and Signalling: Implications for proNGF in Aging and Alzheimer’s Disease" Cells 10, no. 8: 1983. https://doi.org/10.3390/cells10081983
APA StyleKropf, E., & Fahnestock, M. (2021). Effects of Reactive Oxygen and Nitrogen Species on TrkA Expression and Signalling: Implications for proNGF in Aging and Alzheimer’s Disease. Cells, 10(8), 1983. https://doi.org/10.3390/cells10081983