
Amyloid-Beta Peptides and Activated Astroglia Impairs Proliferation of Nerve Growth Factor Releasing Cells In Vitro: Implication for Encapsulated Cell Biodelivery-Mediated AD Therapy

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Plasmid Preparation and Generation of NGC0211 Cells
2.2. Two-Dimensional Cell Cultures
2.3. Aβ Peptide Preparation and Cell Culture Exposure
2.4. Astrocyte-Conditioned Media (ACM)
2.5. ECB Device Preparation and Treatment Exposure
2.6. Biochemical Measurements
2.7. Immunocytochemistry and Image Analysis
2.8. Cell Death Assay
2.9. Measurements of hmNGF by ELISA
2.10. Human CSF Collection and Cell Exposure
2.11. Statistical Analysis
3. Results
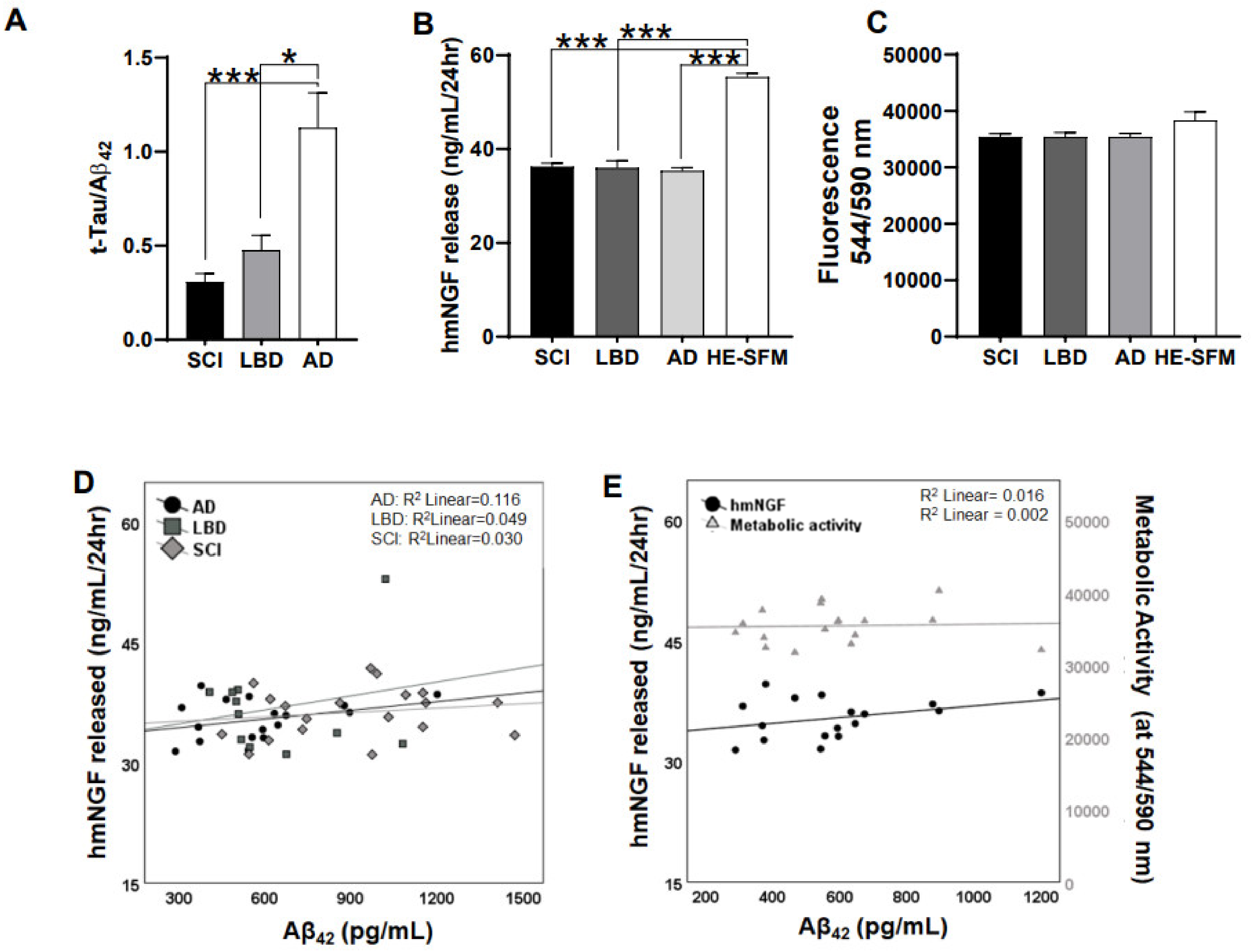
3.1. Release of hmNGF Is Altered by Exposure to Human Patient CSF (SCI, LBD, AD)
3.2. Direct Exposure of Aβ40/42 Peptides Marginally Affected Cell Death, hmNGF Release, and Stress Response
3.3. Severe Anti-Proliferative Impact of Aβ40/42 Peptides on NGC0211 Cells
3.4. Astrocyte-Conditioned Media (ACM) Shows Minimal Impact on NGC0211 Cell Death, hmNGF Release, and Stress Response
3.5. ACM’s Showed Significant Anti-Proliferative Effects on NGC0211 Cells
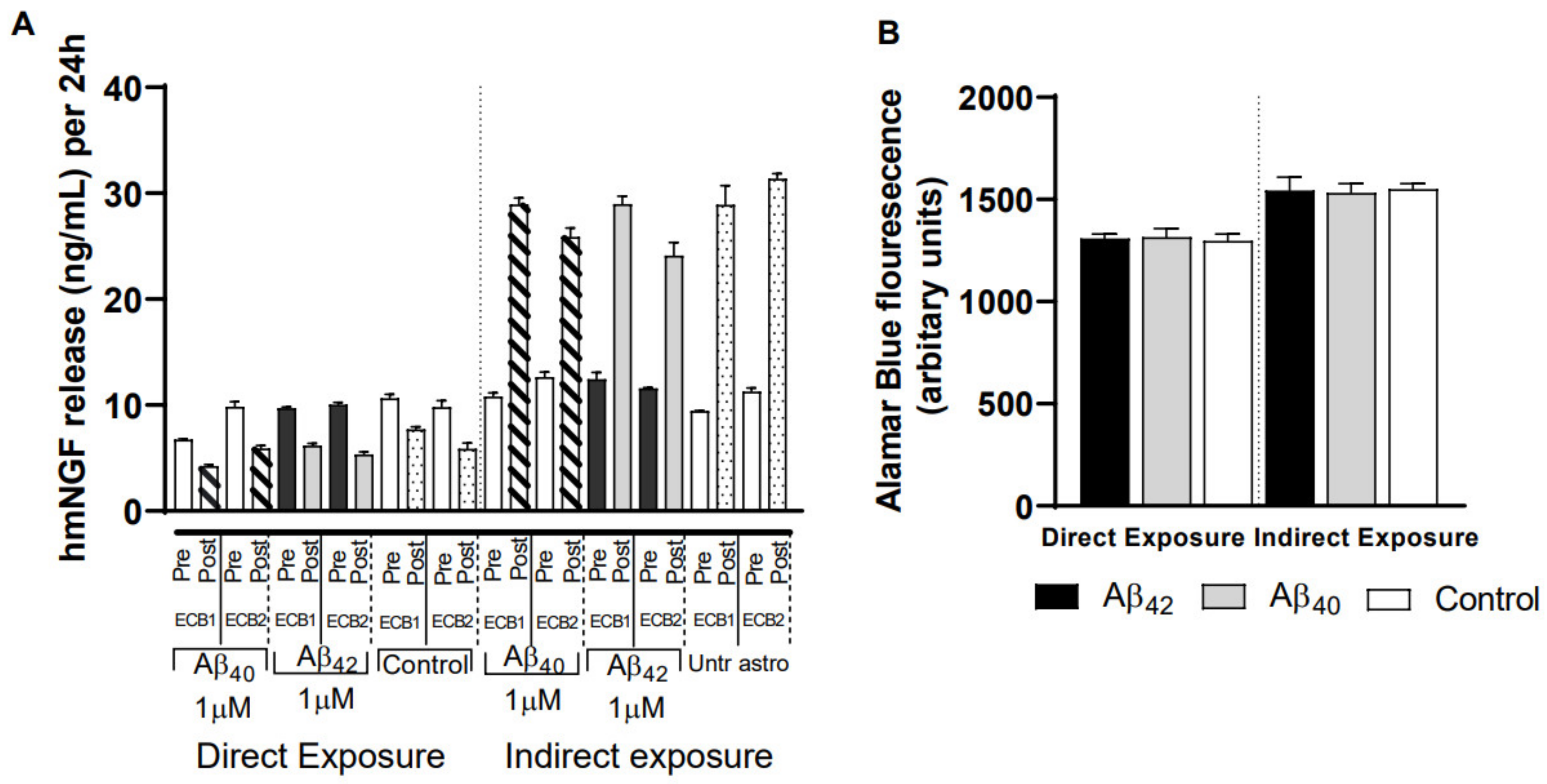
3.6. Impact of Different Exposure Regimes on the ECB-NGF Device
4. Discussion
5. Limitations
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s Disease |
| SCI | Subjective cognitive impairment |
| LBD | Lewy body dementia |
| CSF | Cerebrospinal fluid |
| Aβ | Amyloid beta peptide |
| ECB | Encapsulated cell biodelivery |
| NGF | Nerve Growth Factor |
| ECB-NGF | Encapsulated cell biodelivery-NGF device |
| RPE | Retinal pigment epithelial |
| AMD | Age-related macular degeneration |
| mNGF | mature NGF |
| hmNGF | human mature NGF |
| proNGF | Precursor form of NGF |
| IL-1β | Interleukin-1beta |
| TNFα | Tumour necrosis factor alpha |
| BFCN | Basal forebrain cholinergic neurons |
| TrkA | Tropomyosin receptor kinase A |
| PCR | Polymerase chain reaction |
| ACM | Astrocyte conditioned media |
| V/PI | Annexin-V/Propidium Iodide |
References
- Cuello, A.C.; Pentz, R.; Hall, H. The brain NGF metabolic pathway in health and in Alzheimer’s pathology. Front. Neurosci. 2019, 13, 62. [Google Scholar] [CrossRef]
- Teipel, S.J.; Cavedo, E.; Hampel, H.; Grothe, M.J. Basal forebrain volume, but not hippocampal volume, is a predictor of global cognitive decline in patients with Alzheimer’s disease treated with cholinesterase inhibitors. Front. Neurol. 2018, 9, 642. [Google Scholar] [CrossRef] [PubMed]
- Aloe, L.; Rocco, M.L.; Balzamino, B.O.; Micera, A. Nerve growth factor: A focus on neuroscience and therapy. Curr. Neuropharmacol. 2015, 13, 294–303. [Google Scholar] [CrossRef]
- Cuello, A.C.; Bruno, M.A.; Allard, S.; Leon, W.; Iulita, M.F. Cholinergic involvement in Alzheimer’s disease. A link with NGF maturation and degradation. J. Mol. Neurosci. 2010, 40, 230–235. [Google Scholar] [CrossRef]
- Peng, S.; Wuu, J.; Mufson, E.J.; Fahnestock, M. Increased proNGF levels in subjects with mild cognitive impairment and mild Alzheimer disease. J. Neuropathol. Exp. Neurol. 2004, 63, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Volosin, M.; Cragnolini, A.B.; Hempstead, B.L.; Friedman, W.J. ProNGF induces PTEN via p75NTR to suppress Trk-mediated survival signaling in brain neurons. J. Neurosci. 2010, 30, 15608–15615. [Google Scholar] [CrossRef] [PubMed]
- Mufson, E.J.; Li, J.M.; Sobreviela, T.; Kordower, J.H. Decreased trkA gene expression within basal forebrain neurons in Alzheimer’s disease. Neuroreport 1996, 8, 25–29. [Google Scholar] [CrossRef]
- Boissiere, F.; Hunot, S.; Faucheux, B.; Hersh, L.B.; Agid, Y.; Hirsch, E.C. Trk neurotrophin receptors in cholinergic neurons of patients with Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 1997, 8, 1–8. [Google Scholar] [CrossRef]
- Ginsberg, S.D.; Che, S.; Wuu, J.; Counts, S.E.; Mufson, E.J. Down regulation of trk but not p75NTR gene expression in single cholinergic basal forebrain neurons mark the progression of Alzheimer’s disease. J. Neurochem. 2006, 97, 475–487. [Google Scholar] [CrossRef]
- Sanchez-Ortiz, E.; Yui, D.; Song, D.; Li, Y.; Rubenstein, J.L.; Reichardt, L.F.; Parada, L.F. TrkA gene ablation in basal forebrain results in dysfunction of the cholinergic circuitry. J. Neurosci. 2012, 32, 4065–4079. [Google Scholar] [CrossRef]
- Haam, J.; Yakel, J.L. Cholinergic modulation of the hippocampal region and memory function. J. Neurochem. 2017, 142 (Suppl. 2), 111–121. [Google Scholar] [CrossRef]
- Ballinger, E.C.; Ananth, M.; Talmage, D.A.; Role, L.W. Basal forebrain cholinergic circuits and signaling in cognition and cognitive decline. Neuron 2016, 91, 1199–1218. [Google Scholar] [CrossRef]
- Hasselmo, M.E.; Sarter, M. Modes and models of forebrain cholinergic neuromodulation of cognition. Neuropsychopharmacology 2011, 36, 52–73. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.; Behbahani, H.; Eriksdotter, M. Innovative therapy for Alzheimer’s disease-with focus on biodelivery of NGF. Front. Neurosci. 2019, 13, 38. [Google Scholar] [CrossRef]
- Wahlberg, L.U.; Lind, G.; Almqvist, P.M.; Kusk, P.; Tornoe, J.; Juliusson, B.; Soderman, M.; Sellden, E.; Seiger, A.; Eriksdotter-Jonhagen, M.; et al. Targeted delivery of nerve growth factor via encapsulated cell biodelivery in Alzheimer disease: A technology platform for restorative neurosurgery. J. Neurosurg. 2012, 117, 340–347. [Google Scholar] [CrossRef]
- Eriksdotter-Jonhagen, M.; Linderoth, B.; Lind, G.; Aladellie, L.; Almkvist, O.; Andreasen, N.; Blennow, K.; Bogdanovic, N.; Jelic, V.; Kadir, A.; et al. Encapsulated cell biodelivery of nerve growth factor to the Basal forebrain in patients with Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2012, 33, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Eyjolfsdottir, H.; Eriksdotter, M.; Linderoth, B.; Lind, G.; Juliusson, B.; Kusk, P.; Almkvist, O.; Andreasen, N.; Blennow, K.; Ferreira, D.; et al. Targeted delivery of nerve growth factor to the cholinergic basal forebrain of Alzheimer’s disease patients: Application of a second-generation encapsulated cell biodelivery device. Alzheimers Res. Ther. 2016, 8, 30. [Google Scholar] [CrossRef] [PubMed]
- Fjord-Larsen, L.; Kusk, P.; Emerich, D.F.; Thanos, C.; Torp, M.; Bintz, B.; Tornoe, J.; Johnsen, A.H.; Wahlberg, L.U. Increased encapsulated cell biodelivery of nerve growth factor in the brain by transposon-mediated gene transfer. Gene. Ther. 2012, 19, 1010–1017. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kauppinen, A.; Paterno, J.J.; Blasiak, J.; Salminen, A.; Kaarniranta, K. Inflammation and its role in age-related macular degeneration. Cell Mol. Life Sci. 2016, 73, 1765–1786. [Google Scholar] [CrossRef]
- Saxena, S.; Maze, M. Impact on the brain of the inflammatory response to surgery. Presse. Med. 2018, 47, e73–e81. [Google Scholar] [CrossRef]
- Eriksdotter, M.; Navarro-Oviedo, M.; Mitra, S.; Wahlberg, L.; Linderoth, B.; Tjernberg, L.O.; Behbahani, H. Cerebrospinal fluid from Alzheimer patients affects cell-mediated nerve growth factor production and cell survival in vitro. Exp. Cell Res. 2018, 371, 175–184. [Google Scholar] [CrossRef]
- Stine, W.B.; Jungbauer, L.; Yu, C.; LaDu, M.J. Preparing synthetic Abeta in different aggregation states. Methods Mol. Biol. 2011, 670, 13–32. [Google Scholar] [CrossRef]
- Wicklund, L.; Leao, R.N.; Stromberg, A.M.; Mousavi, M.; Hovatta, O.; Nordberg, A.; Marutle, A. Beta-amyloid 1-42 oligomers impair function of human embryonic stem cell-derived forebrain cholinergic neurons. PLoS ONE 2010, 5, e15600. [Google Scholar] [CrossRef] [PubMed]
- Darreh-Shori, T.; Rezaeianyazdi, S.; Lana, E.; Mitra, S.; Gellerbring, A.; Karami, A.; Bogdanovic, N.; Lithner, C.U.; Winblad, B.; Behbahani, H. Increased active OMI/HTRA2 serine protease displays a positive correlation with cholinergic alterations in the Alzheimer’s disease brain. Mol. Neurobiol. 2019, 56, 4601–4619. [Google Scholar] [CrossRef] [PubMed]
- Hoitzing, H.; Johnston, I.G.; Jones, N.S. What is the function of mitochondrial networks? A theoretical assessment of hypotheses and proposal for future research. Bioessays 2015, 37, 687–700. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.Y.; McLaurin, J. Mechanisms of amyloid-Beta peptide uptake by neurons: The role of lipid rafts and lipid raft-associated proteins. Int. J. Alzheimer’s Dis. 2010, 2011, 548380. [Google Scholar] [CrossRef]
- Roher, A.E.; Esh, C.L.; Kokjohn, T.A.; Castano, E.M.; Van Vickle, G.D.; Kalback, W.M.; Patton, R.L.; Luehrs, D.C.; Daugs, I.D.; Kuo, Y.M.; et al. Amyloid beta peptides in human plasma and tissues and their significance for Alzheimer’s disease. Alzheimer’s Dement. 2009, 5, 18–29. [Google Scholar] [CrossRef]
- Collins-Praino, L.E.; Francis, Y.I.; Griffith, E.Y.; Wiegman, A.F.; Urbach, J.; Lawton, A.; Honig, L.S.; Cortes, E.; Vonsattel, J.P.; Canoll, P.D.; et al. Soluble amyloid beta levels are elevated in the white matter of Alzheimer’s patients, independent of cortical plaque severity. Acta Neuropathol. Commun. 2014, 2, 83. [Google Scholar] [CrossRef]
- Takahashi, R.H.; Nam, E.E.; Edgar, M.; Gouras, G.K. Alzheimer beta-amyloid peptides: Normal and abnormal localization. Histol. Histopathol. 2002, 17, 239–246. [Google Scholar] [CrossRef]
- Chen, J.X.; Yan, S.D. Amyloid-beta-induced mitochondrial dysfunction. J. Alzheimer’s Dis. 2007, 12, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Masuda, N.; Tsujinaka, H.; Hirai, H.; Yamashita, M.; Ueda, T.; Ogata, N. Effects of concentration of amyloid beta (Abeta) on viability of cultured retinal pigment epithelial cells. BMC Ophthalmol. 2019, 19, 70. [Google Scholar] [CrossRef]
- Do, K.V.; Kautzmann, M.I.; Jun, B.; Gordon, W.C.; Nshimiyimana, R.; Yang, R.; Petasis, N.A.; Bazan, N.G. Elovanoids counteract oligomeric beta-amyloid-induced gene expression and protect photoreceptors. Proc. Natl. Acad. Sci. USA 2019, 116, 24317–24325. [Google Scholar] [CrossRef]
- Liu, R.T.; Gao, J.; Cao, S.; Sandhu, N.; Cui, J.Z.; Chou, C.L.; Fang, E.; Matsubara, J.A. Inflammatory mediators induced by amyloid-beta in the retina and RPE in vivo: Implications for inflammasome activation in age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 2013, 54, 2225–2237. [Google Scholar] [CrossRef]
- Mitra, S.; Prasad, P.; Chakraborty, S. A unified view of assessing the pro-oxidant versus antioxidant nature of amyloid beta conformers. Chembiochem 2018, 19, 2360–2371. [Google Scholar] [CrossRef] [PubMed]
- Zou, K.; Gong, J.S.; Yanagisawa, K.; Michikawa, M. A novel function of monomeric amyloid beta-protein serving as an antioxidant molecule against metal-induced oxidative damage. J. Neurosci. 2002, 22, 4833–4841. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Akama, K.T.; Krafft, G.A.; Chromy, B.A.; Van Eldik, L.J. Amyloid-beta peptide activates cultured astrocytes: Morphological alterations, cytokine induction and nitric oxide release. Brain Res. 1998, 785, 195–206. [Google Scholar] [CrossRef]
- Hu, J.; Van Eldik, L.J. Glial-derived proteins activate cultured astrocytes and enhance beta amyloid-induced glial activation. Brain Res. 1999, 842, 46–54. [Google Scholar] [CrossRef]
- Lian, H.; Yang, L.; Cole, A.; Sun, L.; Chiang, A.C.; Fowler, S.W.; Shim, D.J.; Rodriguez-Rivera, J.; Taglialatela, G.; Jankowsky, J.L.; et al. NFkappaB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer’s disease. Neuron 2015, 85, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Vecino, E.; Rodriguez, F.D.; Ruzafa, N.; Pereiro, X.; Sharma, S.C. Glia-neuron interactions in the mammalian retina. Prog. Retin. Eye Res. 2016, 51, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Walker, G.B.; Wang, X.; Cui, J.Z.; Matsubara, J.A. Altered cytokine profiles of human retinal pigment epithelium: Oxidant injury and replicative senescence. Mol. Vis. 2013, 19, 718–728. [Google Scholar]
- Li, Y.; Frei, A.W.; Yang, E.Y.; Labrada-Miravet, I.; Sun, C.; Rong, Y.; Samojlik, M.M.; Bayer, A.L.; Stabler, C.L. In vitro platform establishes antigen-specific CD8(+) T cell cytotoxicity to encapsulated cells via indirect antigen recognition. Biomaterials 2020, 256, 120182. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.G.; Hulseberg, P.; Ling, C.; Karman, J.; Clarkson, B.D.; Harding, J.S.; Zhang, M.; Sandor, A.; Christensen, K.; Nagy, A.; et al. Immune privilege of the CNS is not the consequence of limited antigen sampling. Sci. Rep. 2014, 4, 4422. [Google Scholar] [CrossRef] [PubMed]
- Szatmari-Toth, M.; Kristof, E.; Vereb, Z.; Akhtar, S.; Facsko, A.; Fesus, L.; Kauppinen, A.; Kaarniranta, K.; Petrovski, G. Clearance of autophagy-associated dying retinal pigment epithelial cells—A possible source for inflammation in age-related macular degeneration. Cell Death Dis. 2016, 7, e2367. [Google Scholar] [CrossRef]
- Inana, G.; Murat, C.; An, W.; Yao, X.; Harris, I.R.; Cao, J. RPE phagocytic function declines in age-related macular degeneration and is rescued by human umbilical tissue derived cells. J. Transl. Med. 2018, 16, 63. [Google Scholar] [CrossRef]
- Ishii, M.; Rohrer, B. Mechanisms of bystander effects in retinal pigment epithelium cell networks. Cell Death Dis. 2017, 8, e3061. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Bergmann, A. Apoptosis-induced compensatory proliferation. The Cell is dead. Long live the Cell! Trends Cell Biol. 2008, 18, 467–473. [Google Scholar] [CrossRef]
- Goss, J.R.; O’Malley, M.E.; Zou, L.; Styren, S.D.; Kochanek, P.M.; DeKosky, S.T. Astrocytes are the major source of nerve growth factor upregulation following traumatic brain injury in the rat. Exp. Neurol. 1998, 149, 301–309. [Google Scholar] [CrossRef]
- Paolone, G.; Falcicchia, C.; Lovisari, F.; Kokaia, M.; Bell, W.J.; Fradet, T.; Barbieri, M.; Wahlberg, L.U.; Emerich, D.F.; Simonato, M. Long-term, targeted delivery of GDNF from encapsulated cells is neuroprotective and reduces seizures in the pilocarpine model of epilepsy. J. Neurosci. 2019, 39, 2144–2156. [Google Scholar] [CrossRef]
- Falcicchia, C.; Paolone, G.; Emerich, D.F.; Lovisari, F.; Bell, W.J.; Fradet, T.; Wahlberg, L.U.; Simonato, M. Seizure-suppressant and neuroprotective effects of encapsulated bdnf-producing cells in a rat model of temporal lobe epilepsy. Mol. Ther. Methods Clin. Dev. 2018, 9, 211–224. [Google Scholar] [CrossRef]
- Jorgensen, J.R.; Xu, X.J.; Arnold, H.M.; Munro, G.; Hao, J.X.; Pepinsky, B.; Huang, C.; Gong, B.J.; Wiesenfeld-Hallin, Z.; Wahlberg, L.U.; et al. Meteorin reverses hypersensitivity in rat models of neuropathic pain. Exp. Neurol. 2012, 237, 260–266. [Google Scholar] [CrossRef]
- Jorgensen, J.R.; Fransson, A.; Fjord-Larsen, L.; Thompson, L.H.; Houchins, J.P.; Andrade, N.; Torp, M.; Kalkkinen, N.; Andersson, E.; Lindvall, O.; et al. Cometin is a novel neurotrophic factor that promotes neurite outgrowth and neuroblast migration in vitro and supports survival of spiral ganglion neurons in vivo. Exp. Neurol. 2012, 233, 172–181. [Google Scholar] [CrossRef]
- Yoshida, T.; Ohno-Matsui, K.; Ichinose, S.; Sato, T.; Iwata, N.; Saido, T.C.; Hisatomi, T.; Mochizuki, M.; Morita, I. The potential role of amyloid beta in the pathogenesis of age-related macular degeneration. J. Clin. Investig. 2005, 115, 2793–2800. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Reyes, R.E.; Nava-Mesa, M.O.; Vargas-Sanchez, K.; Ariza-Salamanca, D.; Mora-Munoz, L. Involvement of astrocytes in Alzheimer’s disease from a neuroinflammatory and oxidative stress perspective. Front. Mol. Neurosci. 2017, 10, 427. [Google Scholar] [CrossRef] [PubMed]
- Wyssenbach, A.; Quintela, T.; Llavero, F.; Zugaza, J.L.; Matute, C.; Alberdi, E. Amyloid beta-induced astrogliosis is mediated by beta1-integrin via NADPH oxidase 2 in Alzheimer’s disease. Aging Cell 2016, 15, 1140–1152. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mitra, S.; Turchetto, S.; Van Os, W.; Wahlberg, L.U.; Linderoth, B.; Behbahani, H.; Eriksdotter, M. Amyloid-Beta Peptides and Activated Astroglia Impairs Proliferation of Nerve Growth Factor Releasing Cells In Vitro: Implication for Encapsulated Cell Biodelivery-Mediated AD Therapy. Cells 2021, 10, 2834. https://doi.org/10.3390/cells10112834
Mitra S, Turchetto S, Van Os W, Wahlberg LU, Linderoth B, Behbahani H, Eriksdotter M. Amyloid-Beta Peptides and Activated Astroglia Impairs Proliferation of Nerve Growth Factor Releasing Cells In Vitro: Implication for Encapsulated Cell Biodelivery-Mediated AD Therapy. Cells. 2021; 10(11):2834. https://doi.org/10.3390/cells10112834
Chicago/Turabian StyleMitra, Sumonto, Silvia Turchetto, Winant Van Os, Lars U. Wahlberg, Bengt Linderoth, Homira Behbahani, and Maria Eriksdotter. 2021. "Amyloid-Beta Peptides and Activated Astroglia Impairs Proliferation of Nerve Growth Factor Releasing Cells In Vitro: Implication for Encapsulated Cell Biodelivery-Mediated AD Therapy" Cells 10, no. 11: 2834. https://doi.org/10.3390/cells10112834
APA StyleMitra, S., Turchetto, S., Van Os, W., Wahlberg, L. U., Linderoth, B., Behbahani, H., & Eriksdotter, M. (2021). Amyloid-Beta Peptides and Activated Astroglia Impairs Proliferation of Nerve Growth Factor Releasing Cells In Vitro: Implication for Encapsulated Cell Biodelivery-Mediated AD Therapy. Cells, 10(11), 2834. https://doi.org/10.3390/cells10112834