
Identification of Novel Cathepsin B Inhibitors with Implications in Alzheimer’s Disease: Computational Refining and Biochemical Evaluation

 , , , ,
, , , ,  , ,
, , 
Abstract
1. Introduction
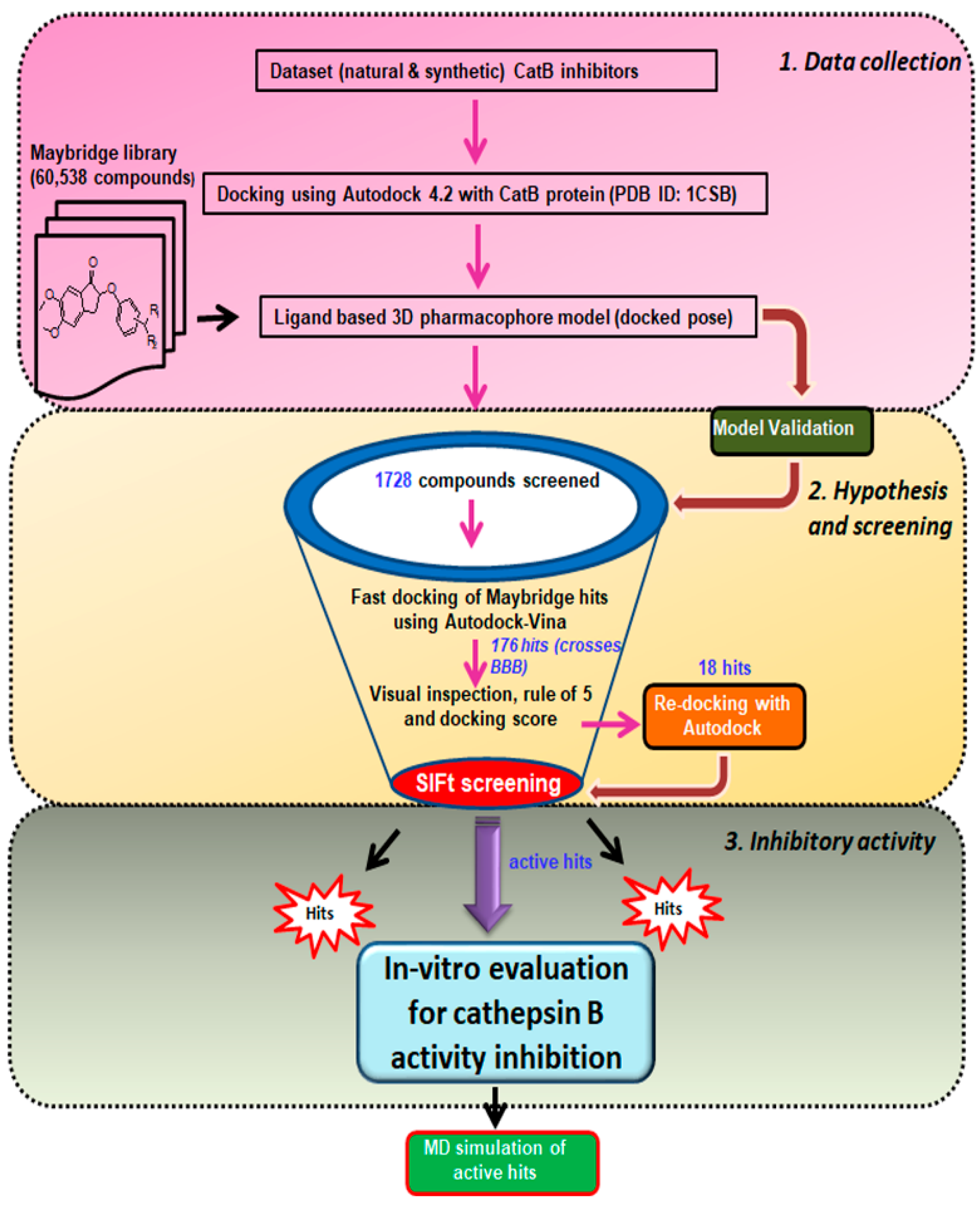
2. Materials and Methods
2.1. Selection and Preparation of Macromolecule
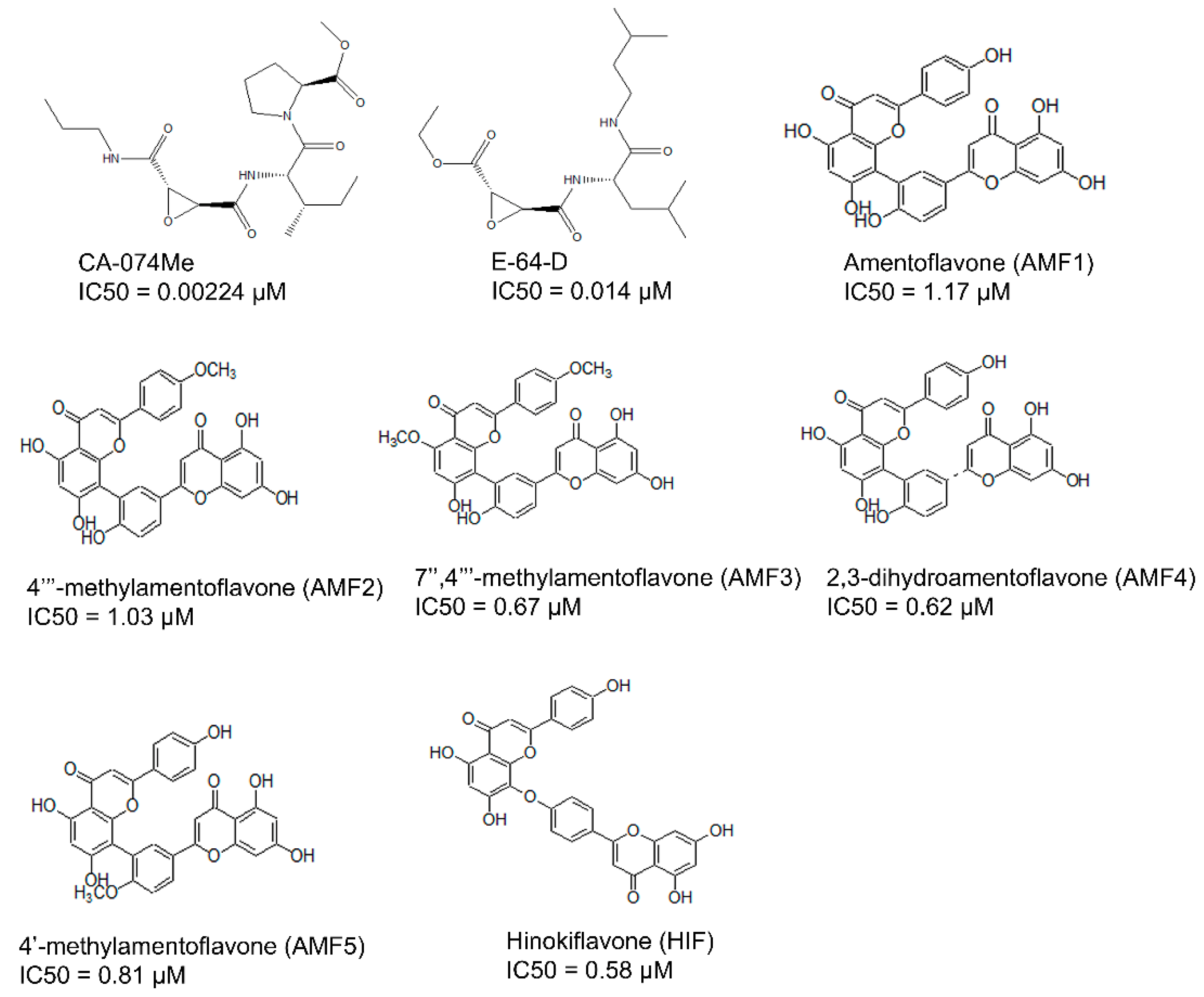
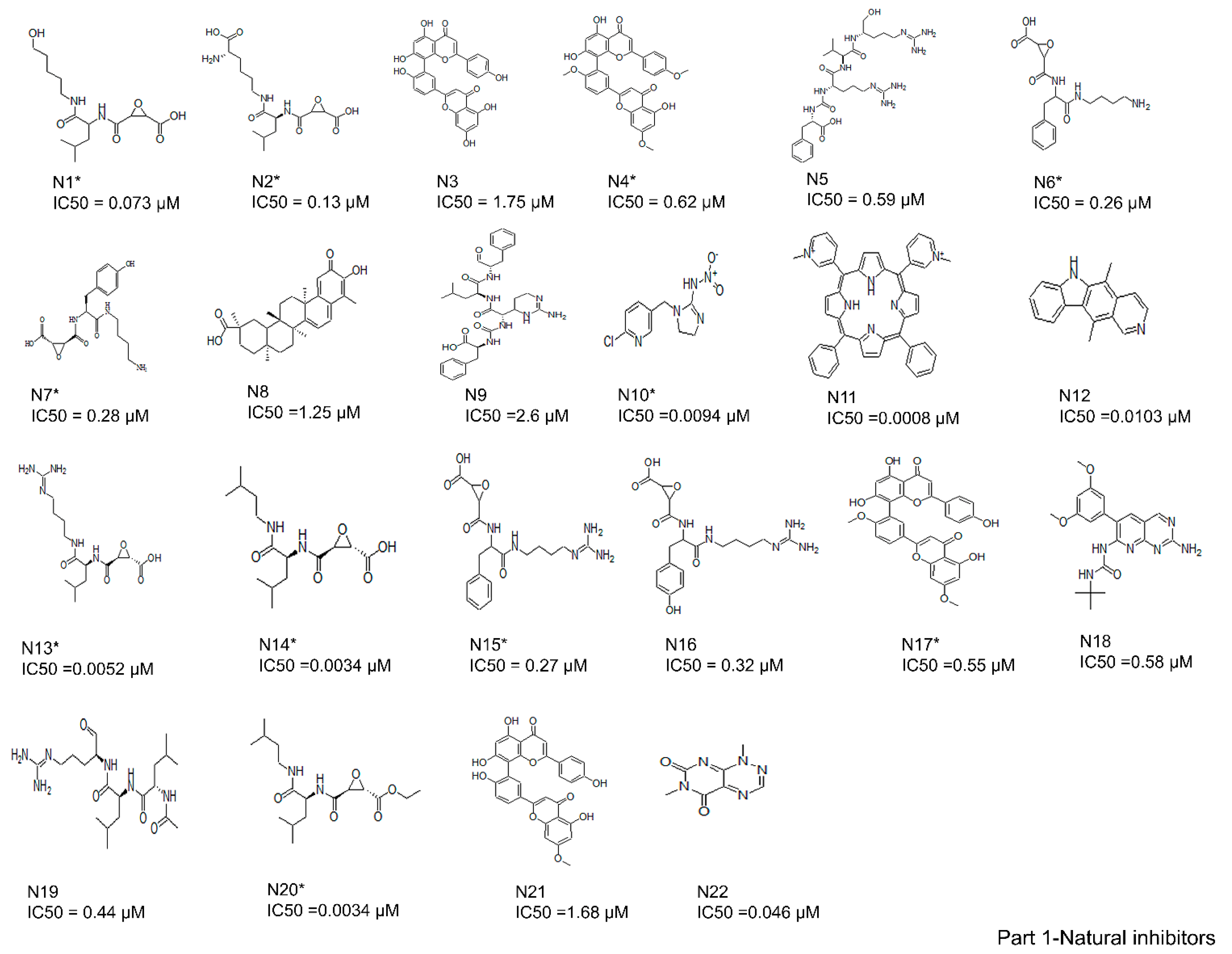
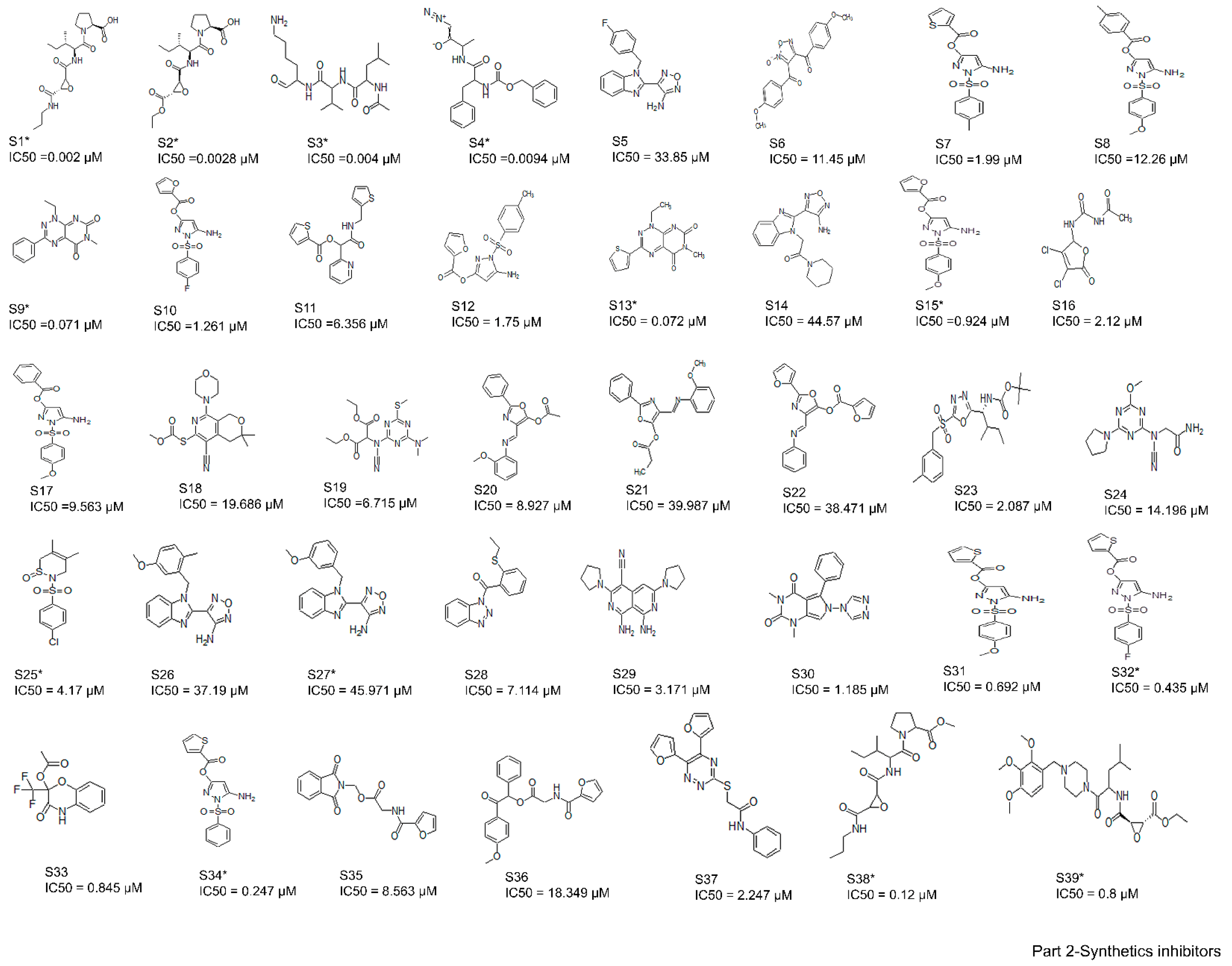
2.2. Collection and Preparation of Ligand Dataset
2.3. Molecular Docking and Interaction Studies
2.4. Ligand-Based Three-Dimensional Pharmacophore Search and Virtual Screening
2.5. Screening of Maybridge Database and Fast Docking Using AutoDock Vina
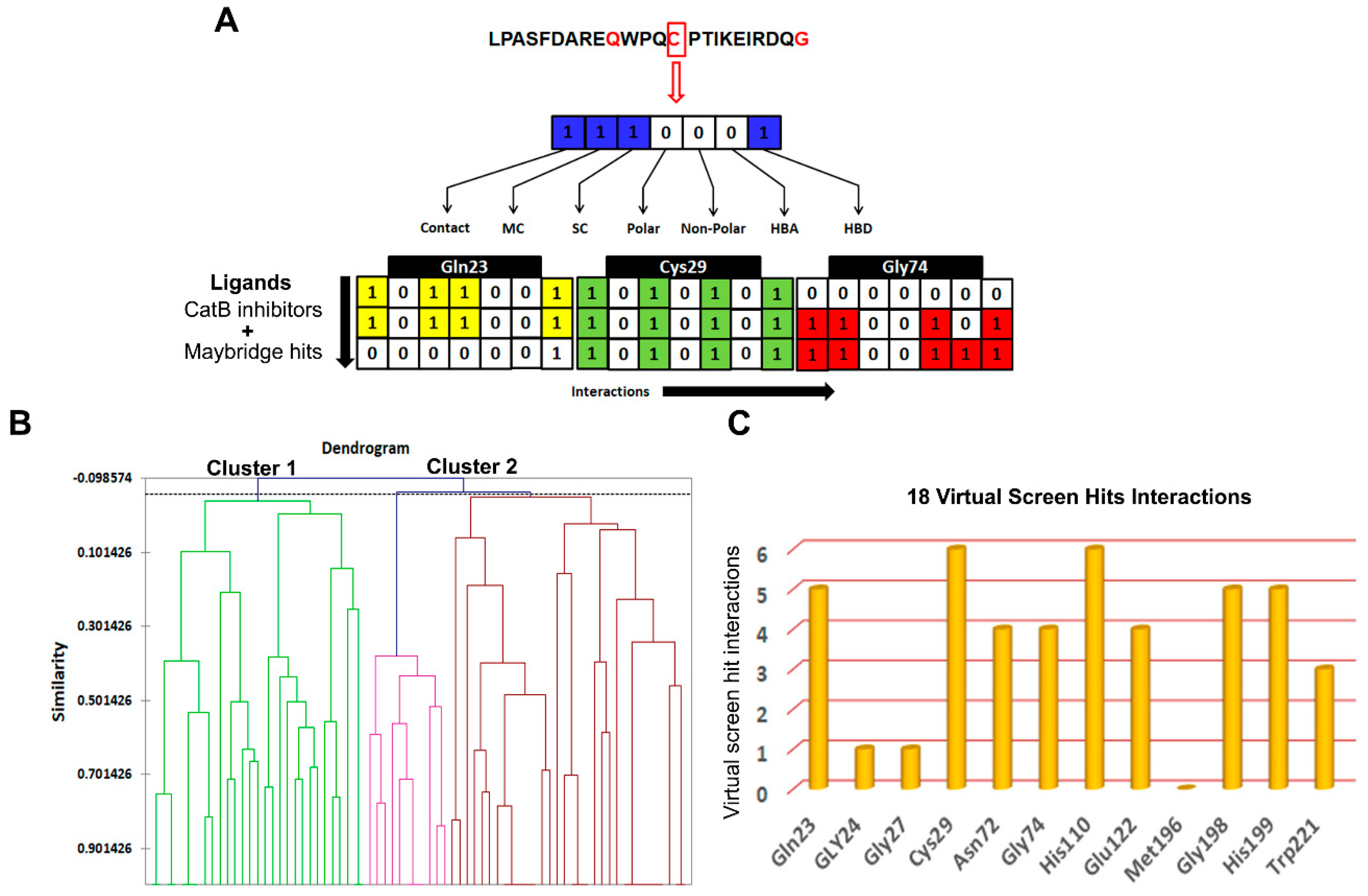
2.6. Construction of Structural Interaction Fingerprints (SIFts), Similarity Analysis, and Hierarchical Clustering
2.7. Molecular Dynamics Simulation and Binding Free Energy Calculations
2.8. Cathepsin B Inhibitory Assay
3. Results
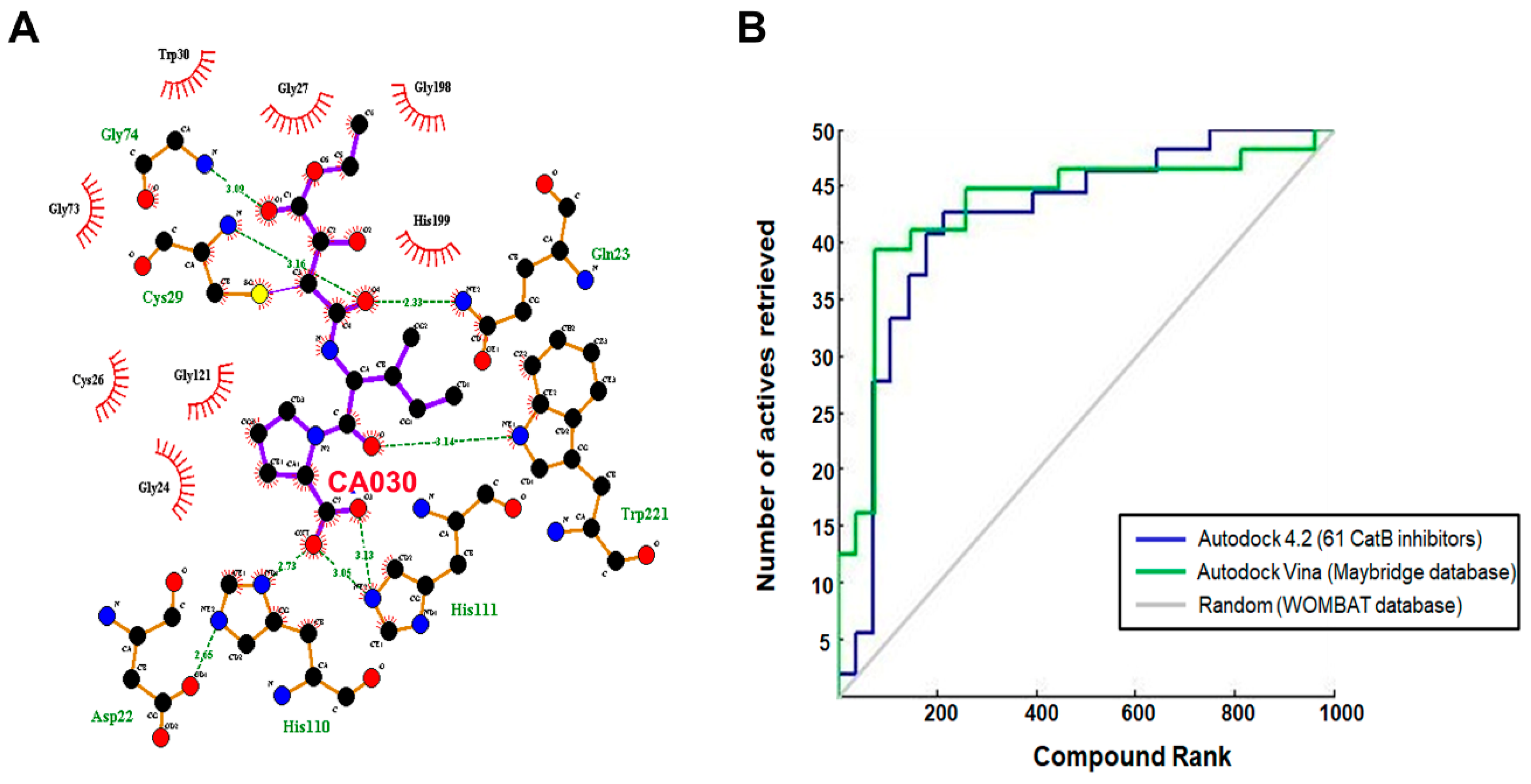
3.1. Binding Modes of the 61 Active Cathepsin B Inhibitors Using the AutoDock v4.2 Docking Program
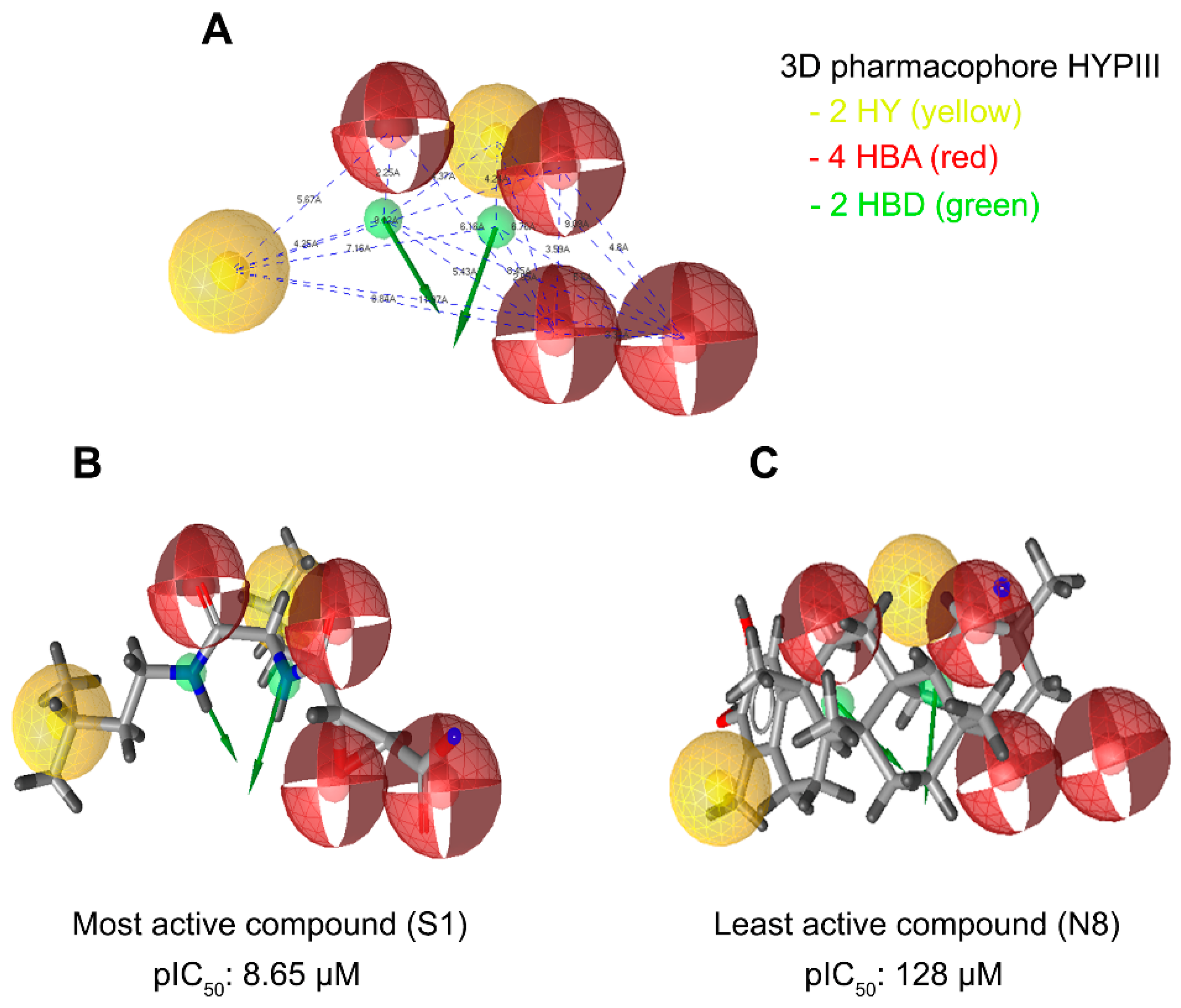
3.2. Generation and Validation of Mixed-Feature Ligand-Based Pharmacophore Models Using LigandScout 3.1
3.3. Virtual Screening and Hit Filtration
3.3.1. Maybridge Database Screen and Fast Docking of Maybridge Molecular Library Hits Using AutoDock-Vina
3.3.2. Structure Interaction Fingerprint Based Clustering
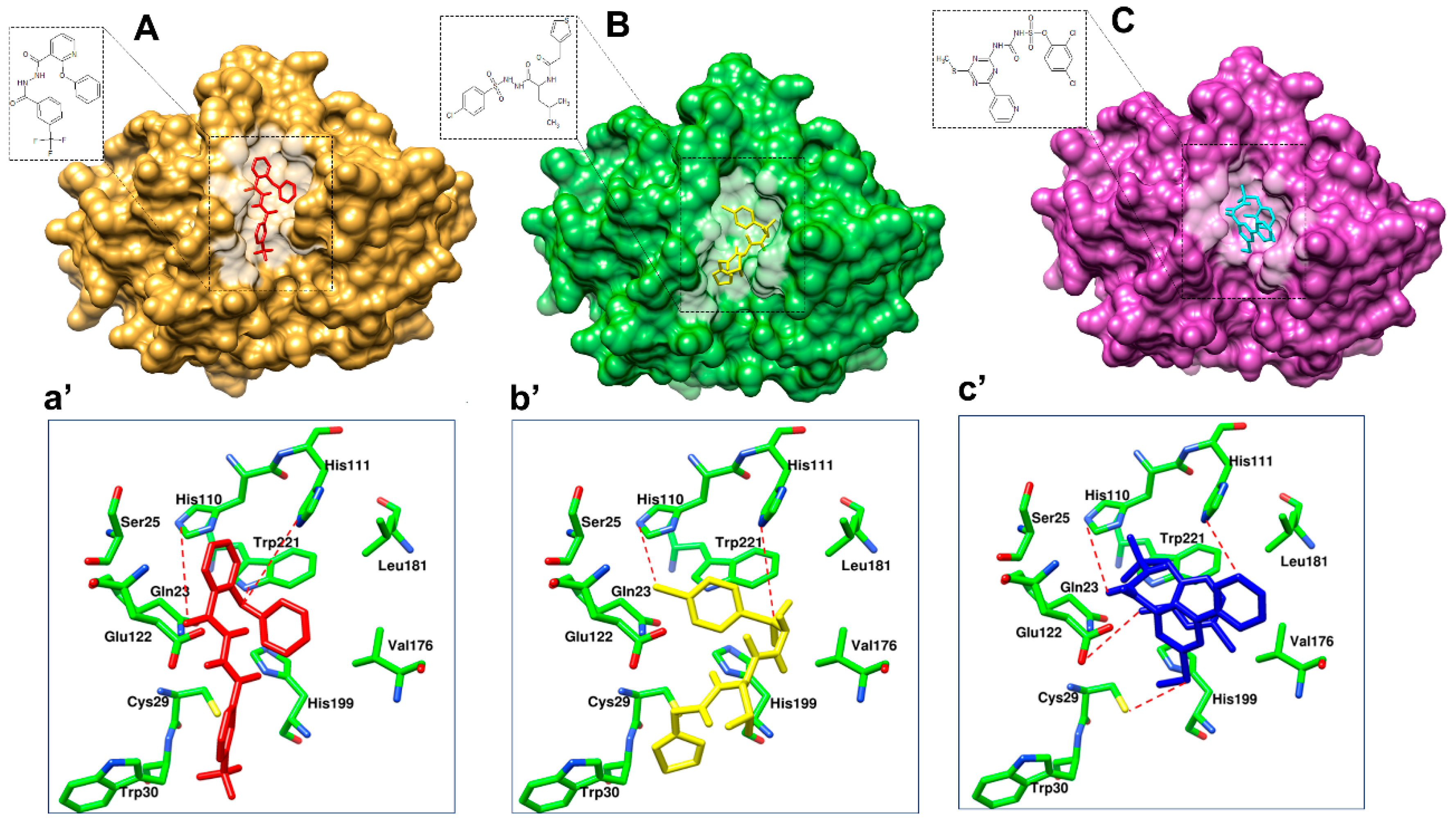
3.4. Prioritization and Binding Modes of Hit Molecules
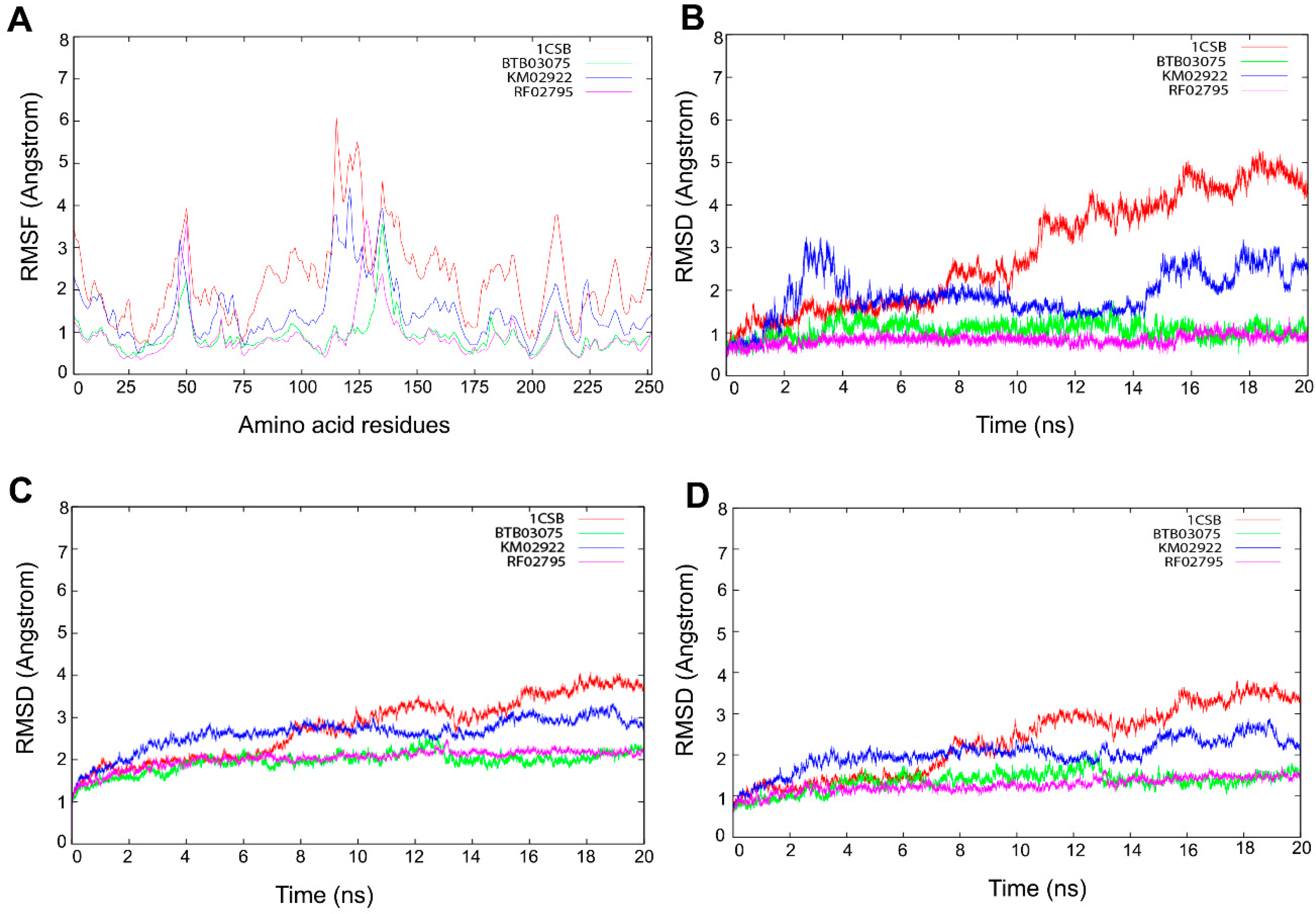
3.5. Molecular Dynamics Simulations
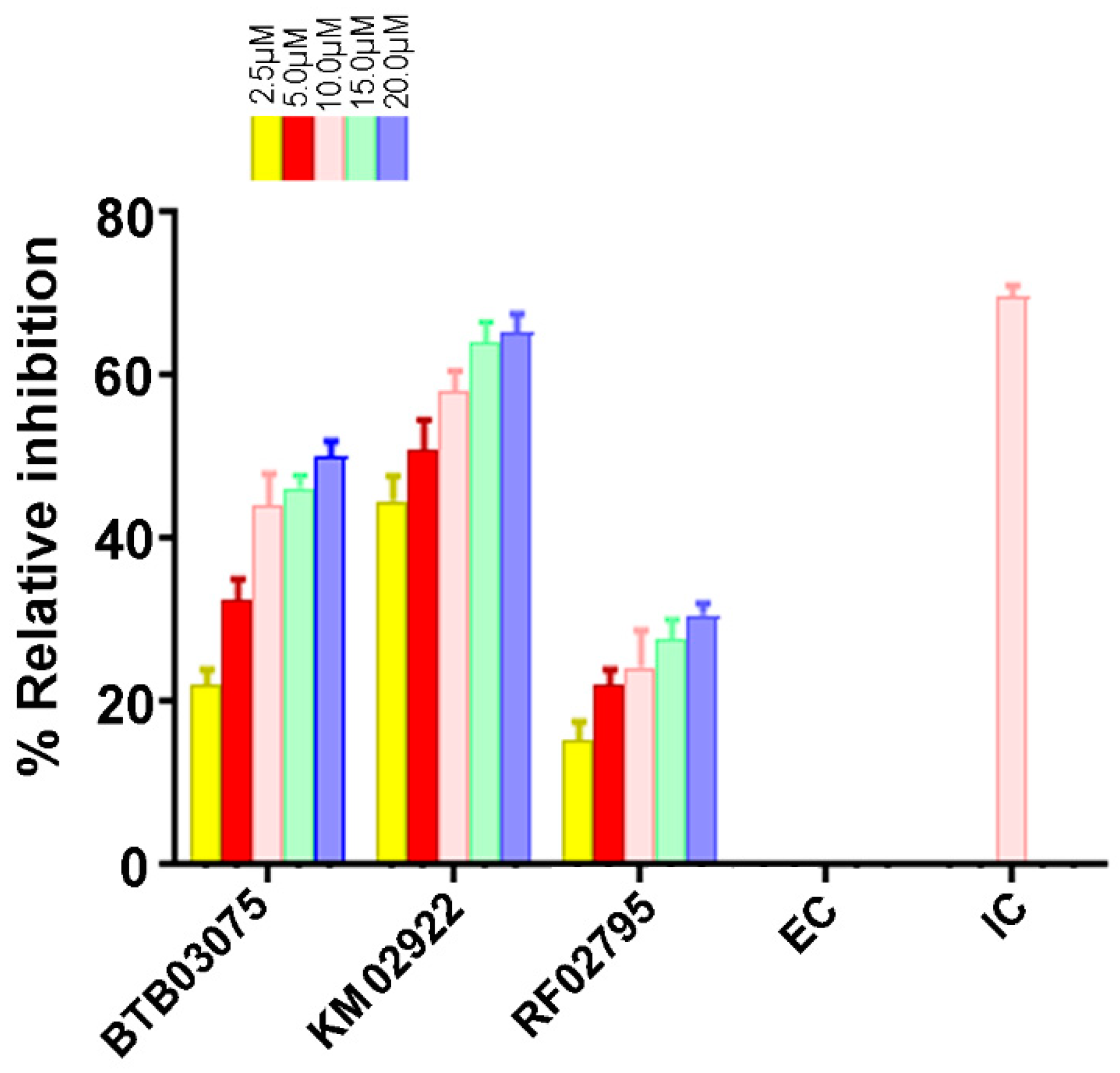
3.6. Cathepsin B Inhibition Activity of Virtually Screened Hit Compounds
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Bogdanovic, N.; Hansson, O.; Zetterberg, H.; Basun, H.; Ingelsson, M.; Lannfelt, L.; Blennow, K. Alzheimer’s disease—The most common cause of dementia. Lakartidningen 2020, 117. [Google Scholar]
- Marelli, C.; Hourregue, C.; Gutierrez, L.A.; Paquet, C.; de Menjot Champfleur, N.; De Verbizier, D.; Jacob, M.; Dubois, J.; Maleska, A.M.; Hirtz, C.; et al. Cerebrospinal Fluid and Plasma Biomarkers do not Differ in the Presenile and Late-Onset Behavioral Variants of Frontotemporal Dementia. J. Alzheimers Dis. 2020, 74, 903–911. [Google Scholar] [CrossRef]
- Wilson, R.S.; Segawa, E.; Boyle, P.A.; Anagnos, S.E.; Hizel, L.P.; Bennett, D.A. The natural history of cognitive decline in Alzheimer’s disease. Psychol. Aging 2012, 27, 1008–1017. [Google Scholar] [CrossRef] [PubMed]
- Gaugler, J.; James, B.; Johnson, T.; Marin, A.; Weuve, J.; Assoc, A.S. 2019 Alzheimer’s disease facts and figures. Alzheimers Dement. 2019, 15, 321–387. [Google Scholar] [CrossRef]
- Gupta, V.; Gupta, V.B.; Chitranshi, N.; Gangoda, S.; Vander Wall, R.; Abbasi, M.; Golzan, M.; Dheer, Y.; Shah, T.; Avolio, A.; et al. One protein, multiple pathologies: Multifaceted involvement of amyloid beta in neurodegenerative disorders of the brain and retina. Cell Mol. Life Sci. 2016, 73, 4279–4297. [Google Scholar] [CrossRef]
- Gupta, V.K.; Chitranshi, N.; Gupta, V.B.; Golzan, M.; Dheer, Y.; Wall, R.V.; Georgevsky, D.; King, A.E.; Vickers, J.C.; Chung, R.; et al. Amyloid beta accumulation and inner retinal degenerative changes in Alzheimer’s disease transgenic mouse. Neurosci. Lett. 2016, 623, 52–56. [Google Scholar] [CrossRef]
- Zhang, X.; Fu, Z.; Meng, L.; He, M.; Zhang, Z. The Early Events That Initiate beta-Amyloid Aggregation in Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 359. [Google Scholar] [CrossRef] [PubMed]
- Giorgetti, S.; Greco, C.; Tortora, P.; Aprile, F.A. Targeting Amyloid Aggregation: An Overview of Strategies and Mechanisms. Int. J. Mol. Sci. 2018, 19, 2677. [Google Scholar] [CrossRef]
- Wang, Q.; Yu, X.; Li, L.; Zheng, J. Inhibition of amyloid-beta aggregation in Alzheimer’s disease. Curr. Pharm. Des. 2014, 20, 1223–1243. [Google Scholar] [CrossRef]
- Cavallo-Medved, D.; Moin, K.; Sloane, B. Cathepsin B: Basis Sequence: Mouse. AFCS Nat. Mol. Pages 2011, 2011, A000508. [Google Scholar]
- Linebaugh, B.E.; Sameni, M.; Day, N.A.; Sloane, B.F.; Keppler, D. Exocytosis of active cathepsin B enzyme activity at pH 7.0, inhibition and molecular mass. Eur. J. Biochem. 1999, 264, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.L.; Guo, J.; Zhang, X.; Sukhova, G.K.; Libby, P.; Shi, G.P. Cysteine protease cathepsins in cardiovascular disease: From basic research to clinical trials. Nat. Rev. Cardiol. 2018, 15, 351–370. [Google Scholar] [CrossRef]
- Gondi, C.S.; Rao, J.S. Cathepsin B as a cancer target. Expert Opin. Ther. Targets 2013, 17, 281–291. [Google Scholar] [CrossRef]
- Sendler, M.; Maertin, S.; John, D.; Persike, M.; Weiss, F.U.; Kruger, B.; Wartmann, T.; Wagh, P.; Halangk, W.; Schaschke, N.; et al. Cathepsin B Activity Initiates Apoptosis via Digestive Protease Activation in Pancreatic Acinar Cells and Experimental Pancreatitis. J. Biol. Chem. 2016, 291, 14717–14731. [Google Scholar] [CrossRef]
- Bernstein, H.G.; Keilhoff, G. Putative roles of cathepsin B in Alzheimer’s disease pathology: The good, the bad, and the ugly in one? Neural. Regen. Res. 2018, 13, 2100–2101. [Google Scholar] [CrossRef]
- Dewachter, I.; Van Leuven, F. Secretases as targets for the treatment of Alzheimer’s disease: The prospects. Lancet Neurol. 2002, 1, 409–416. [Google Scholar] [CrossRef]
- Yan, R.; Bienkowski, M.J.; Shuck, M.E.; Miao, H.; Tory, M.C.; Pauley, A.M.; Brashier, J.R.; Stratman, N.C.; Mathews, W.R.; Buhl, A.E.; et al. Membrane-anchored aspartyl protease with Alzheimer’s disease beta-secretase activity. Nature 1999, 402, 533–537. [Google Scholar] [CrossRef]
- Lin, X.; Koelsch, G.; Wu, S.; Downs, D.; Dashti, A.; Tang, J. Human aspartic protease memapsin 2 cleaves the beta-secretase site of beta-amyloid precursor protein. Proc. Natl. Acad. Sci. USA 2000, 97, 1456–1460. [Google Scholar] [CrossRef]
- Vassar, R.; Bennett, B.D.; Babu-Khan, S.; Kahn, S.; Mendiaz, E.A.; Denis, P.; Teplow, D.B.; Ross, S.; Amarante, P.; Loeloff, R.; et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999, 286, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, M.; Pushpitha, K.; Deng, L.; Chitranshi, N.; Gupta, V.; Rajput, R.; Mangani, A.B.; Dheer, Y.; Godinez, A.; McKay, M.J.; et al. Upregulation of Proteolytic Pathways and Altered Protein Biosynthesis Underlie Retinal Pathology in a Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2019, 56, 6017–6034. [Google Scholar] [CrossRef]
- Chitranshi, N.; Tiwari, A.K.; Somvanshi, P.; Tripathi, P.K.; Seth, P.K. Investigating the function of single nucleotide polymorphisms in the CTSB gene: A computational approach. Futur. Neurol. 2013, 8, 469–483. [Google Scholar] [CrossRef]
- Sosic, I.; Mitrovic, A.; Curic, H.; Knez, D.; Brodnik Zugelj, H.; Stefane, B.; Kos, J.; Gobec, S. Cathepsin B inhibitors: Further exploration of the nitroxoline core. Bioorg. Med. Chem. Lett. 2018, 28, 1239–1247. [Google Scholar] [CrossRef]
- Greenspan, P.D.; Clark, K.L.; Tommasi, R.A.; Cowen, S.D.; McQuire, L.W.; Farley, D.L.; van Duzer, J.H.; Goldberg, R.L.; Zhou, H.; Du, Z.; et al. Identification of dipeptidyl nitriles as potent and selective inhibitors of cathepsin B through structure-based drug design. J. Med. Chem. 2001, 44, 4524–4534. [Google Scholar] [CrossRef]
- Rasnick, D. Synthesis of peptide fluoromethyl ketones and the inhibition of human cathepsin B. Anal. Biochem. 1985, 149, 461–465. [Google Scholar] [CrossRef]
- Jilkova, A.; Rezacova, P.; Lepsik, M.; Horn, M.; Vachova, J.; Fanfrlik, J.; Brynda, J.; McKerrow, J.H.; Caffrey, C.R.; Mares, M. Structural basis for inhibition of cathepsin B drug target from the human blood fluke, Schistosoma mansoni. J. Biol. Chem. 2011, 286, 35770–35781. [Google Scholar] [CrossRef]
- Zhou, Z.; Wang, Y.; Bryant, S.H. Computational analysis of the cathepsin B inhibitors activities through LR-MMPBSA binding affinity calculation based on docked complex. J. Comput. Chem. 2009, 30, 2165–2175. [Google Scholar] [CrossRef]
- Perlman, N.; Hazan, M.; Shokhen, M.; Albeck, A. Peptidyl epoxides extended in the P’ direction as cysteine protease inhibitors: Effect on affinity and mechanism of inhibition. Bioorg. Med. Chem. 2008, 16, 9032–9039. [Google Scholar] [CrossRef]
- Siklos, M.; BenAissa, M.; Thatcher, G.R. Cysteine proteases as therapeutic targets: Does selectivity matter? A systematic review of calpain and cathepsin inhibitors. Acta Pharm. Sin. B 2015, 5, 506–519. [Google Scholar] [CrossRef]
- Walker, B.; McCarthy, N.; Healy, A.; Ye, T.; McKervey, M.A. Peptide glyoxals: A novel class of inhibitor for serine and cysteine proteinases. Biochem. J. 1993, 293, 321–323. [Google Scholar] [CrossRef] [PubMed]
- Schaschke, N.; Deluca, D.; Assfalg-Machleidt, I.; Hohneke, C.; Sommerhoff, C.P.; Machleidt, W. Epoxysuccinyl peptide-derived cathepsin B inhibitors: Modulating membrane permeability by conjugation with the C-terminal heptapeptide segment of penetratin. Biol. Chem. 2002, 383, 849–852. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, J.; Li, T.; Bartz, U.; Gutschow, M. Cathepsin B Inhibitors: Combining Dipeptide Nitriles with an Occluding Loop Recognition Element by Click Chemistry. ACS Med. Chem. Lett. 2016, 7, 211–216. [Google Scholar] [CrossRef]
- Pan, X.; Tan, N.; Zeng, G.; Zhang, Y.; Jia, R. Amentoflavone and its derivatives as novel natural inhibitors of human Cathepsin B. Bioorg. Med. Chem. 2005, 13, 5819–5825. [Google Scholar] [CrossRef]
- Turk, D.; Podobnik, M.; Popovic, T.; Katunuma, N.; Bode, W.; Huber, R.; Turk, V. Crystal structure of cathepsin B inhibited with CA030 at 2.0-A resolution: A basis for the design of specific epoxysuccinyl inhibitors. Biochemistry 1995, 34, 4791–4797. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic. Acids. Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Chitranshi, N.; Gupta, V.K.; Rajput, R.; Godinez, A.; Pushpitha, K.; Shen, T.; Mirzaei, M.; You, Y.; Basavarajappa, D.; Gupta, V.; et al. Evolving geographic diversity in SARS-CoV2 and in silico analysis of replicating enzyme 3CL(pro) targeting repurposed drug candidates. J. Transl. Med. 2020, 18, 278. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic. Acids. Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef]
- Zeng, G.Z.; Pan, X.L.; Tan, N.H.; Xiong, J.; Zhang, Y.M. Natural biflavones as novel inhibitors of cathepsin B and K. Eur. J. Med. Chem. 2006, 41, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Powers, J.C.; Asgian, J.L.; Ekici, O.D.; James, K.E. Irreversible inhibitors of serine, cysteine, and threonine proteases. Chem. Rev. 2002, 102, 4639–4750. [Google Scholar] [CrossRef]
- Sarabia, F.; Sanchez-Ruiz, A.; Chammaa, S. Stereoselective synthesis of E-64 and related cysteine proteases inhibitors from 2,3-epoxyamides. Bioorg. Med. Chem. 2005, 13, 1691–1705. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Chitranshi, N.; Gupta, S.; Tripathi, P.K.; Seth, P.K. New molecular scaffolds for the design of Alzheimer’s acetylcholinesterase inhibitors identified using ligand- and receptor-based virtual screening. Med. Chem. Res. 2013, 22, 2328–2345. [Google Scholar] [CrossRef]
- Chitranshi, N.; Gupta, V.; Dheer, Y.; Gupta, V.; Vander Wall, R.; Graham, S. Molecular determinants and interaction data of cyclic peptide inhibitor with the extracellular domain of TrkB receptor. Data Brief. 2016, 6, 776–782. [Google Scholar] [CrossRef]
- Xiao, J.; Zhang, S.; Luo, M.; Zou, Y.; Zhang, Y.; Lai, Y. Effective virtual screening strategy focusing on the identification of novel Bruton’s tyrosine kinase inhibitors. J. Mol. Graph. Model. 2015, 60, 142–154. [Google Scholar] [CrossRef]
- Wolber, G.; Langer, T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Truchon, J.F.; Bayly, C.I. Evaluating virtual screening methods: Good and bad metrics for the “early recognition” problem. J. Chem. Inf. Model. 2007, 47, 488–508. [Google Scholar] [CrossRef]
- Benet, L.Z.; Hosey, C.M.; Ursu, O.; Oprea, T.I. BDDCS, the Rule of 5 and drugability. Adv. Drug Deliv. Rev. 2016, 101, 89–98. [Google Scholar] [CrossRef]
- Gupta, S.; Misra, G.; Chandra Pant, M.; Kishore Seth, P. Targeting the epidermal growth factor receptor: Exploring the potential of novel inhibitor N-(3-ethynylphenyl)-6, 7-bis (2-methoxyethoxy) quinolin- 4-amine using docking and molecular dynamics simulation. Protein Pept. Lett. 2012, 19, 955–968. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Siddiqi, M.I. Virtual screening against Mycobacterium tuberculosis dihydrofolate reductase: Suggested workflow for compound prioritization using structure interaction fingerprints. J. Mol. Graph. Model. 2008, 27, 476–488. [Google Scholar] [CrossRef] [PubMed]
- Velazquez-Libera, J.L.; Murillo-Lopez, J.A.; de la Torre, A.F.; Caballero, J. Structural Requirements of N-alpha-Mercaptoacetyl Dipeptide (NAMdP) Inhibitors of Pseudomonas Aeruginosa Virulence Factor LasB: 3D-QSAR, Molecular Docking, and Interaction Fingerprint Studies. Int. J. Mol. Sci. 2019, 20, 6133. [Google Scholar] [CrossRef] [PubMed]
- Racz, A.; Bajusz, D.; Heberger, K. Life beyond the Tanimoto coefficient: Similarity measures for interaction fingerprints. J. Cheminform. 2018, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bocker, A.; Derksen, S.; Schmidt, E.; Teckentrup, A.; Schneider, G. A hierarchical clustering approach for large compound libraries. J. Chem. Inf. Model. 2005, 45, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Ji, B.; Liu, S.; He, X.; Man, V.H.; Xie, X.Q.; Wang, J. Prediction of the Binding Affinities and Selectivity for CB1 and CB2 Ligands Using Homology Modeling, Molecular Docking, Molecular Dynamics Simulations, and MM-PBSA Binding Free Energy Calculations. ACS Chem. Neurosci. 2020, 11, 1139–1158. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug. Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Zhang, X.; Perez-Sanchez, H.; Lightstone, F.C. A Comprehensive Docking and MM/GBSA Rescoring Study of Ligand Recognition upon Binding Antithrombin. Curr. Top. Med. Chem. 2017, 17, 1631–1639. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, M.; Fujisaki, S.; Shirakura, M.; Watanabe, S.; Odagiri, T.; Ito, K.; Sato, H. Molecular Dynamics Simulation of the Influenza A(H3N2) Hemagglutinin Trimer Reveals the Structural Basis for Adaptive Evolution of the Recent Epidemic Clade 3C.2a. Front. Microbiol. 2017, 8, 584. [Google Scholar] [CrossRef]
- Chitranshi, N.; Dheer, Y.; Kumar, S.; Graham, S.L.; Gupta, V. Molecular docking, dynamics, and pharmacology studies on bexarotene as an agonist of ligand-activated transcription factors, retinoid X receptors. J. Cell Biochem. 2019, 120, 11745–11760. [Google Scholar] [CrossRef] [PubMed]
- Boateng, H.A. Periodic Coulomb Tree Method: An Alternative to Parallel Particle Mesh Ewald. J. Chem. Theory Comput. 2020, 16, 7–17. [Google Scholar] [CrossRef]
- Elber, R.; Ruymgaart, A.P.; Hess, B. SHAKE parallelization. Eur. Phys. J. Spec. Top. 2011, 200, 211–223. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E., 3rd; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Onufriev, A.V.; Case, D.A. Generalized Born Implicit Solvent Models for Biomolecules. Annu. Rev. Biophys. 2019, 48, 275–296. [Google Scholar] [CrossRef]
- Huang, H.; Simmerling, C. Fast Pairwise Approximation of Solvent Accessible Surface Area for Implicit Solvent Simulations of Proteins on CPUs and GPUs. J. Chem. Theory Comput. 2018, 14, 5797–5814. [Google Scholar] [CrossRef]
- Chitranshi, N.; Gupta, V.; Kumar, S.; Graham, S.L. Exploring the Molecular Interactions of 7,8-Dihydroxyflavone and Its Derivatives with TrkB and VEGFR2 Proteins. Int. J. Mol. Sci. 2015, 16, 21087–21108. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Chetter, B.A.; Kyriakis, E.; Barr, D.; Karra, A.G.; Katsidou, E.; Koulas, S.M.; Skamnaki, V.T.; Snape, T.J.; Psarra, A.G.; Leonidas, D.D.; et al. Synthetic flavonoid derivatives targeting the glycogen phosphorylase inhibitor site: QM/MM-PBSA motivated synthesis of substituted 5,7-dihydroxyflavones, crystallography, in vitro kinetics and ex-vivo cellular experiments reveal novel potent inhibitors. Bioorg. Chem. 2020, 102, 104003. [Google Scholar] [CrossRef]
- Ghosh, R.; Chakraborty, A.; Biswas, A.; Chowdhuri, S. Identification of polyphenols from Broussonetia papyrifera as SARS CoV-2 main protease inhibitors using in silico docking and molecular dynamics simulation approaches. J. Biomol. Struct. Dyn. 2020, 1–14. [Google Scholar] [CrossRef]
- El Khoury, L.; Santos-Martins, D.; Sasmal, S.; Eberhardt, J.; Bianco, G.; Ambrosio, F.A.; Solis-Vasquez, L.; Koch, A.; Forli, S.; Mobley, D.L. Comparison of affinity ranking using AutoDock-GPU and MM-GBSA scores for BACE-1 inhibitors in the D3R Grand Challenge 4. J. Comput. Aided. Mol. Des. 2019, 33, 1011–1020. [Google Scholar] [CrossRef]
- Wang, E.; Weng, G.; Sun, H.; Du, H.; Zhu, F.; Chen, F.; Wang, Z.; Hou, T. Assessing the performance of the MM/PBSA and MM/GBSA methods. 10. Impacts of enhanced sampling and variable dielectric model on protein-protein Interactions. Phys. Chem. Chem. Phys. 2019, 21, 18958–18969. [Google Scholar] [CrossRef]
- Joseph, C.; Mangani, A.S.; Gupta, V.; Chitranshi, N.; Shen, T.; Dheer, Y.; Kb, D.; Mirzaei, M.; You, Y.; Graham, S.L.; et al. Cell Cycle Deficits in Neurodegenerative Disorders: Uncovering Molecular Mechanisms to Drive Innovative Therapeutic development. Aging Dis. 2020, 11, 946–966. [Google Scholar] [CrossRef]
- Gupta, V.B.; Chitranshi, N.; Haan, J.D.; Mirzaei, M.; You, Y.; Lim, J.K.; Basavarajappa, D.; Godinez, A.; Di Angelantonio, S.; Sachdev, P.; et al. Retinal changes in Alzheimer’s disease- integrated prospects of imaging, functional and molecular advances. Prog. Retin. Eye. Res. 2020, 82, 100899. [Google Scholar] [CrossRef]
- Morris, G.P.; Clark, I.A.; Vissel, B. Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer’s disease. Acta Neuropathol. Commun. 2014, 2, 135. [Google Scholar] [CrossRef]
- Simic, G.; Babic Leko, M.; Wray, S.; Harrington, C.; Delalle, I.; Jovanov-Milosevic, N.; Bazadona, D.; Buee, L.; de Silva, R.; Di Giovanni, G.; et al. Tau Protein Hyperphosphorylation and Aggregation in Alzheimer’s Disease and Other Tauopathies, and Possible Neuroprotective Strategies. Biomolecules 2016, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Hook, G.; Hook, V.; Kindy, M. The cysteine protease inhibitor, E64d, reduces brain amyloid-beta and improves memory deficits in Alzheimer’s disease animal models by inhibiting cathepsin B, but not BACE1, beta-secretase activity. J. Alzheimers Dis. 2011, 26, 387–408. [Google Scholar] [CrossRef] [PubMed]
- Sakr, M.F.; Hassanein, T.I.; Zetti, G.M.; Van Thiel, D.H. FK 506 ameliorates the hepatic injury associated with ischemia. Life Sci. 1990, 47, 687–691. [Google Scholar] [CrossRef]
- Wang, C.; Sun, B.; Zhou, Y.; Grubb, A.; Gan, L. Cathepsin B degrades amyloid-beta in mice expressing wild-type human amyloid precursor protein. J. Biol. Chem. 2012, 287, 39834–39841. [Google Scholar] [CrossRef]
- Embury, C.M.; Dyavarshetty, B.; Lu, Y.; Wiederin, J.L.; Ciborowski, P.; Gendelman, H.E.; Kiyota, T. Cathepsin B Improves ss-Amyloidosis and Learning and Memory in Models of Alzheimer’s Disease. J Neuroimmune Pharm. 2017, 12, 340–352. [Google Scholar] [CrossRef]
- Smith, A.J.; Zhang, X.; Leach, A.G.; Houk, K.N. Beyond picomolar affinities: Quantitative aspects of noncovalent and covalent binding of drugs to proteins. J. Med. Chem. 2009, 52, 225–233. [Google Scholar] [CrossRef]
- Katritch, V.; Byrd, C.M.; Tseitin, V.; Dai, D.; Raush, E.; Totrov, M.; Abagyan, R.; Jordan, R.; Hruby, D.E. Discovery of small molecule inhibitors of ubiquitin-like poxvirus proteinase I7L using homology modeling and covalent docking approaches. J. Comput. Aided. Mol. Des. 2007, 21, 549–558. [Google Scholar] [CrossRef]
- Khan, M.S.; Mehmood, B.; Yousafi, Q.; Bibi, S.; Fazal, S.; Saleem, S.; Sajid, M.W.; Ihsan, A.; Azhar, M.; Kamal, M.A. Molecular Docking studies reveals Rhein from rhubarb (Rheum rhabarbarum) as a putative inhibitor of ATP-binding Cassette Super Family G member 2. Med. Chem. 2021, 17, 273–288. [Google Scholar] [CrossRef]
- Dhanjal, J.K.; Sharma, S.; Grover, A.; Das, A. Use of ligand-based pharmacophore modeling and docking approach to find novel acetylcholinesterase inhibitors for treating Alzheimer’s. Biomed. Pharm. 2015, 71, 146–152. [Google Scholar] [CrossRef]
- Gimeno, A.; Ojeda-Montes, M.J.; Tomas-Hernandez, S.; Cereto-Massague, A.; Beltran-Debon, R.; Mulero, M.; Pujadas, G.; Garcia-Vallve, S. The Light and Dark Sides of Virtual Screening: What is There to Know? Int. J. Mol. Sci. 2019, 20, 1375. [Google Scholar] [CrossRef]
- Kutlushina, A.; Khakimova, A.; Madzhidov, T.; Polishchuk, P. Ligand-Based Pharmacophore Modeling Using Novel 3D Pharmacophore Signatures. Molecules 2018, 23, 3094. [Google Scholar] [CrossRef]
- Chandra, N.; Bhagavat, R.; Sharma, E.; Sreekanthreddy, P.; Somasundaram, K. Virtual screening, identification and experimental testing of novel inhibitors of PBEF1/Visfatin/NMPRTase for glioma therapy. J. Clin. Bioinform. 2011, 1, 5. [Google Scholar] [CrossRef]
- Fu, Y.; Zhao, J.; Chen, Z. Insights into the Molecular Mechanisms of Protein-Ligand Interactions by Molecular Docking and Molecular Dynamics Simulation: A Case of Oligopeptide Binding Protein. Comput. Math. Methods Med. 2018, 2018, 3502514. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, L.; Lv, M.; Pei, R.; Li, P.; Pei, Z.; Wang, Y.; Su, W.; Xie, X.Q. AlzPlatform: An Alzheimer’s disease domain-specific chemogenomics knowledgebase for polypharmacology and target identification research. J. Chem. Inf. Model. 2014, 54, 1050–1060. [Google Scholar] [CrossRef] [PubMed]
- Nogara, P.A.; Saraiva Rde, A.; Caeran Bueno, D.; Lissner, L.J.; Lenz Dalla Corte, C.; Braga, M.M.; Rosemberg, D.B.; Rocha, J.B. Virtual screening of acetylcholinesterase inhibitors using the Lipinski’s rule of five and ZINC databank. Biomed. Res. Int. 2015, 2015, 870389. [Google Scholar] [CrossRef]
- Lionta, E.; Spyrou, G.; Vassilatis, D.K.; Cournia, Z. Structure-based virtual screening for drug discovery: Principles, applications and recent advances. Curr. Top. Med. Chem. 2014, 14, 1923–1938. [Google Scholar] [CrossRef]
- Fuhrmann, J.; Rurainski, A.; Lenhof, H.P.; Neumann, D. A new Lamarckian genetic algorithm for flexible ligand-receptor docking. J. Comput. Chem. 2010, 31, 1911–1918. [Google Scholar] [CrossRef]
- Ritschel, T.; Schirris, T.J.; Russel, F.G. KRIPO—A structure-based pharmacophores approach explains polypharmacological effects. J. Cheminform. 2014, 6, O26. [Google Scholar] [CrossRef]
- Loser, R.; Pietzsch, J. Cysteine cathepsins: Their role in tumor progression and recent trends in the development of imaging probes. Front. Chem. 2015, 3, 37. [Google Scholar] [CrossRef]
- Hospital, A.; Goni, J.R.; Orozco, M.; Gelpi, J.L. Molecular dynamics simulations: Advances and applications. Adv. Appl. Bioinform. Chem. 2015, 8, 37–47. [Google Scholar] [CrossRef]
- Durrant, J.D.; McCammon, J.A. Molecular dynamics simulations and drug discovery. BMC Biol. 2011, 9, 71. [Google Scholar] [CrossRef]
- Gohlke, H.; Klebe, G. Approaches to the description and prediction of the binding affinity of small-molecule ligands to macromolecular receptors. Angew. Chem. Int. Ed. Engl. 2002, 41, 2644–2676. [Google Scholar] [CrossRef]
- Wang, Y.Q.; Lin, W.W.; Wu, N.; Wang, S.Y.; Chen, M.Z.; Lin, Z.H.; Xie, X.Q.; Feng, Z.W. Structural insight into the serotonin (5-HT) receptor family by molecular docking, molecular dynamics simulation and systems pharmacology analysis. Acta Pharmacol. Sin. 2019, 40, 1138–1156. [Google Scholar] [CrossRef]
- Mazumder, M.; Ponnan, P.; Das, U.; Gourinath, S.; Khan, H.A.; Yang, J.; Sakharkar, M.K. Investigations on Binding Pattern of Kinase Inhibitors with PPARgamma: Molecular Docking, Molecular Dynamic Simulations, and Free Energy Calculation Studies. PPAR Res. 2017, 2017, 6397836. [Google Scholar] [CrossRef] [PubMed]
- Taddei, M.; Ferrini, S.; Giannotti, L.; Corsi, M.; Manetti, F.; Giannini, G.; Vesci, L.; Milazzo, F.M.; Alloatti, D.; Guglielmi, M.B.; et al. Synthesis and evaluation of new Hsp90 inhibitors based on a 1,4,5-trisubstituted 1,2,3-triazole scaffold. J. Med. Chem. 2014, 57, 2258–2274. [Google Scholar] [CrossRef]
- Roca, C.; Martinez-Gonzalez, L.; Daniel-Mozo, M.; Sastre, J.; Infantes, L.; Mansilla, A.; Chaves-Sanjuan, A.; Gonzalez-Rubio, J.M.; Gil, C.; Canada, F.J.; et al. Deciphering the Inhibition of the Neuronal Calcium Sensor 1 and the Guanine Exchange Factor Ric8a with a Small Phenothiazine Molecule for the Rational Generation of Therapeutic Synapse Function Regulators. J. Med. Chem. 2018, 61, 5910–5921. [Google Scholar] [CrossRef]
- Koukoulitsa, C.; Villalonga-Barber, C.; Csonka, R.; Alexi, X.; Leonis, G.; Dellis, D.; Hamelink, E.; Belda, O.; Steele, B.R.; Micha-Screttas, M.; et al. Biological and computational evaluation of resveratrol inhibitors against Alzheimer’s disease. J. Enzyme. Inhib. Med. Chem. 2016, 31, 67–77. [Google Scholar] [CrossRef]
- Sharma, T.; Harioudh, M.K.; Kuldeep, J.; Kumar, S.; Banerjee, D.; Ghosh, J.K.; Siddiqi, M.I. Identification of Potential Inhibitors of Cathepsin-B using Shape & Pharmacophore-based Virtual Screening, Molecular Docking and Explicit Water Thermodynamics. Mol. Inform. 2020, 39, e1900023. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S.No | C.Name | BEe (kcal/mol) | Ki (µM) | IMEe (kcal/mol) | Vdw-Hb-Ds (kcal/mol) | Ee (kcal/mol) | IEe (kcal/mol) | TFEe (kcal/mol) |
|---|---|---|---|---|---|---|---|---|
| 1 | CA030 | −5.89 | 48.10 | −6.32 | −6.77 | +0.44 | +0.16 | +0.27 |
| N1 * | −4.11 | 964.9 | −6.45 | −7.10 | +0.66 | −1.23 | +3.57 | |
| 2 | N2 * | −7.30 | 4.47 | −8.96 | −8.18 | −0.78 | −2.45 | +4.12 |
| 3 | N3 | −8.15 | 1.06 | −8.49 | −8.49 | +0.00 | −2.13 | +2.47 |
| 4 | N4 * | −10.28 | 29.17 | −9.54 | −9.31 | −0.22 | −2.94 | +2.20 |
| 5 | N5 | −5.37 | 115.2 | −8.85 | −7.81 | −1.05 | −2.01 | +5.49 |
| 6 | N6 * | −6.55 | 15.77 | −6.75 | −4.98 | −1.77 | −3.09 | +3.29 |
| 7 | N7 | −6.07 | 35.48 | −6.00 | −5.95 | −0.05 | −1.72 | +1.65 |
| 8 | N8 * | −7.11 | 6.17 | −8.15 | −8.96 | +0.81 | +0.22 | +0.82 |
| 9 | N9 | −6.61 | 14.25 | −8.28 | −7.61 | −0.67 | −2.72 | +4.39 |
| 10 | N10 * | −5.71 | 65.31 | −5.76 | −5.65 | −0.11 | −0.77 | +0.82 |
| 11 | N11 | −10.35 | 26.10 | −9.91 | −10.00 | +0.08 | −1.53 | +1.10 |
| 12 | N12 | −6.29 | 24.31 | −6.29 | −6.34 | +0.04 | +0.00 | +0.00 |
| 13 | N13 * | −6.26 | 25.83 | −8.34 | −7.26 | −1.08 | −1.21 | +3.29 |
| 14 | N14 * | −9.47 | 97.11 | −7.27 | −7.76 | +0.49 | −0.95 | +2.74 |
| 15 | N15 * | −6.20 | 28.60 | −7.08 | −6.94 | −0.15 | −2.41 | +3.29 |
| 16 | N16 | −5.30 | 130.1 | −7.19 | −6.13 | −1.05 | −1.68 | +3.57 |
| 17 | N17 * | −8.57 | 522.5 | −8.31 | −8.23 | −0.08 | −2.46 | +2.20 |
| 18 | N18 | −7.16 | 5.61 | −8.16 | −6.46 | −6.46 | −0.10 | +1.10 |
| 19 | N19 | −5.93 | 45.25 | −7.83 | −7.67 | −0.15 | −1.94 | +3.84 |
| 20 | N20 | −5.92 | 45.44 | −7.52 | −7.45 | −0.07 | −1.42 | +3.02 |
| 21 | N21 * | −9.35 | 0.134 | −9.59 | −9.40 | −0.19 | −2.24 | +2.47 |
| 22 | N22 | −5.03 | 205.2 | −5.03 | −4.95 | −0.08 | +0.00 | +0.00 |
| 23 | S1 * | −5.70 | 66.00 | −7.28 | −7.52 | +0.23 | −1.16 | +2.74 |
| 24 | S2 * | −6.42 | 19.52 | −8.01 | −8.01 | +0.16 | −1.16 | +2.74 |
| 25 | S3 | −6.65 | 13.30 | −8.07 | −6.52 | −1.55 | −2.43 | +3.84 |
| 26 | S4 * | −6.38 | 21.20 | −8.16 | −8.19 | +0.03 | −0.96 | +2.74 |
| 27 | S5 | −8.05 | 1.26 | −6.94 | −6.94 | −0.08 | −2.20 | +1.10 |
| 28 | S6 | −5.55 | 84.91 | −6.15 | −6.02 | −0.13 | −0.51 | +1.10 |
| 29 | S7 * | −6.26 | 25.76 | −8.32 | −8.30 | −0.03 | +0.42 | +1.65 |
| 30 | S8 | −7.56 | 2.89 | −8.65 | −8.50 | −0.15 | −0.83 | +1.92 |
| 31 | S9 * | −6.86 | 9.35 | −7.11 | −7.11 | −0.06 | −0.30 | −0.30 |
| 32 | S10 | −7.92 | 1.56 | −8.14 | −8.10 | −8.10 | −1.42 | +1.65 |
| 33 | S11 | −7.71 | 2.23 | −7.80 | −7.77 | −0.04 | −1.83 | +1.92 |
| 34 | S12 | −6.63 | 13.86 | −6.46 | −6.33 | −0.14 | −1.81 | +1.65 |
| 35 | S13 | −5.61 | 77.13 | −5.72 | −5.62 | −0.10 | −0.44 | +0.55 |
| 36 | S14 | −5.84 | 52.54 | −6.80 | −5.14 | −1.66 | −0.41 | +1.37 |
| 37 | S15 | −7.21 | 5.18 | −8.10 | −7.88 | −0.22 | −1.03 | +1.92 |
| 38 | S16 | −5.15 | 16.76 | −5.39 | −5.28 | −0.11 | −0.31 | +0.55 |
| 39 | S17 * | −9.34 | 0.14 | −9.77 | −9.55 | −0.23 | −1.48 | +1.92 |
| 40 | S18 * | −7.18 | 5.50 | −7.85 | −7.82 | −0.04 | −0.42 | +1.10 |
| 41 | S19 * | −8.07 | 1.21 | −9.05 | −6.28 | −2.77 | −1.22 | +2.20 |
| 42 | S20 | −7.19 | 5.36 | −8.17 | −7.99 | −0.18 | −0.67 | +1.65 |
| 43 | S21 | −6.95 | 8.05 | −7.99 | −8.02 | +0.03 | −0.88 | +1.92 |
| 44 | S22 * | −7.41 | 3.72 | −8.12 | −8.18 | +0.06 | −0.66 | +1.37 |
| 45 | S23 | −6.43 | 19.44 | −7.98 | −7.95 | −0.03 | −0.92 | +2.47 |
| 46 | S24 | −6.98 | 7.70 | −7.66 | −4.46 | −3.20 | −0.14 | +0.82 |
| 47 | S25 * | −7.20 | 5.27 | −7.01 | −6.92 | −0.09 | −0.74 | +0.55 |
| 48 | S26 | −7.86 | 1.73 | −8.21 | −8.08 | −0.13 | −1.02 | +1.37 |
| 49 | S27 * | −7.97 | 1.43 | −8.72 | −8.69 | −0.03 | −0.63 | +1.37 |
| 50 | S28 | −6.99 | 7.52 | −7.17 | −7.21 | +0.04 | −0.64 | +0.82 |
| 51 | S29 | −6.91 | 8.67 | −7.23 | −7.14 | −0.10 | −0.50 | +0.82 |
| 52 | S30 | −6.16 | 30.57 | −5.97 | −5.86 | −0.11 | −0.74 | +0.55 |
| 53 | S31 | −6.70 | 12.23 | −7.66 | −7.50 | −0.16 | −0.96 | +1.92 |
| 54 | S32 * | −6.59 | 14.68 | −8.48 | −8.28 | −0.21 | +0.24 | +1.65 |
| 55 | S33 | −5.19 | 155.8 | −6.13 | −6.05 | −0.08 | +0.11 | +0.82 |
| 56 | S34 * | −7.10 | 6.20 | −7.82 | −7.78 | −0.04 | −0.93 | +1.65 |
| 57 | S35 | −7.25 | 4.83 | −7.85 | −7.69 | −0.16 | −1.05 | +1.65 |
| 58 | S36 | −9.37 | 135.9 | −10.31 | −10.15 | −0.16 | −1.52 | +2.47 |
| 59 | S37 | −7.19 | 5.40 | −8.38 | −8.39 | +0.02 | −0.45 | +1.65 |
| 60 | S38 | −6.23 | 27.29 | −7.21 | −7.13 | −0.08 | −1.76 | +2.74 |
| 61 | S39 | −6.82 | 10.08 | −6.65 | −6.63 | −0.02 | −1.26 | +1.10 |
| Hypothesis Name | Features a | Matching Features | Pharmacohore-Fit Score b |
|---|---|---|---|
| Hyp I | HY, HBA, HBA, HBA, HBA, HBA, HBD | 7 | 74.81 |
| Hyp II | HY, HY, HBA, HBA, HBA, HBA, HBA, HBD, HBD | 9 | 75.24 |
| Hyp III | HY, HY, HBA, HBA, HBA, HBA, HBD, HBD | 8 | 75.98 |
| Hyp IV | HY, HBA, HBA, HBD, HBD, HBD | 6 | 74.22 |
| Hyp V | HY, HY, HBA, HBA, HBA, HBA | 6 | 74.17 |
| Hyp VI | HY, HBA, HBA, HBA, HBD, HBD | 6 | 73.96 |
| Hyp VII | HY, HY, HBA, HBA, HBA, HBA, HBA, HBD | 9 | 74.65 |
| Hyp VIII | HY, HBA, HBA, HBA, HBD, HBD, HBD | 7 | 74.57 |
| Hyp IX | HY, HY, HBA, HBA, HBD, HBD, HBD | 7 | 73.42 |
| Hyp X | HY, HY, HBA, HBA, HBA, HBA, HBA, HBD | 8 | 74.11 |
| No. | Parameter | Hypo I | Hypo II | Hypo III |
|---|---|---|---|---|
| 1 | Total number of molecules in the database (D) | 796 | 796 | 796 |
| 2 | Total number of actives in the database (A) | 37 | 37 | 37 |
| 3 | Total hits (Ht) | 121 | 60 | 45 |
| 4 | Active hits (Ha) | 28 | 30 | 35 |
| 5 | % Yield of actives (Ha/Ht × 100) | 23.15 | 50 | 77.78 |
| 6 | % Ratio of actives (Ha/A × 100) | 75.68 | 81.1 | 94.6 |
| 7 | Enrichment factor (E) a | 4.98 | 10.78 | 16.74 |
| 8 | False negatives (A − Ha) | 9 | 7 | 2 |
| 9 | False positives (Ht − Ha) | 93 | 30 | 10 |
| 10 | Goodness of hit score (GH) b | 0.32 | 0.56 | 0.81 |
| Compound | pIC50 | Error a | Fit Value b | Activity Scale c | ||
|---|---|---|---|---|---|---|
| Experimental | Estimated | Experimental | Estimated | |||
| * N1 | 7.14 | 6.22 | −1.1 | 74.21 | +++ | +++ |
| * N2 | 6.89 | 7.24 | +2.3 | 73.29 | +++ | ++ |
| N3 | 5.76 | 5.45 | +3.2 | 72.34 | ++ | ++ |
| * N4 | 6.21 | 6.33 | +0.9 | 71.91 | ++ | ++ |
| N5 | 6.23 | 6.1 | +2.9 | 70.14 | ++ | + |
| * N6 | 6.58 | 6.44 | −1.3 | 72.14 | +++ | +++ |
| N7 | 6.55 | 6.44 | −1.8 | 71.36 | +++ | +++ |
| * N8 | 3.91 | 4.23 | +2.4 | 69.15 | + | + |
| N9 | 5.59 | 6.27 | −1.9 | 71.85 | + | + |
| * N10 | 8.03 | 8.11 | +1.2 | 75.12 | +++ | +++ |
| N11 | 9.1 | 9.22 | +2.5 | 69.71 | +++ | ++ |
| N12 | 7.99 | 8.14 | +1.5 | 72.49 | +++ | +++ |
| * N13 | 8.28 | 8.22 | −0.6 | 75.16 | +++ | +++ |
| * N14 | 8.47 | 8.84 | +2.2 | 76.12 | +++ | +++ |
| * N15 | 6.57 | 8.53 | +3.2 | 73.11 | +++ | +++ |
| N16 | 6.49 | 6.78 | −1.5 | 72.44 | ++ | ++ |
| * N17 | 6.26 | 8.23 | +3.1 | 71.94 | ++ | +++ |
| N18 | 6.24 | 6.21 | +1.3 | 72.45 | ++ | ++ |
| N19 | 6.36 | 6.26 | +2.2 | 71.88 | ++ | ++ |
| N20 | 8.47 | 7.98 | +3.7 | 72.67 | +++ | ++ |
| * N21 | 5.77 | 5.72 | −0.5 | 71.26 | ++ | ++ |
| N22 | 7.34 | 7.31 | +2.8 | 72..12 | +++ | +++ |
| * S1 | 8.65 | 8.62 | −0.3 | 77.68 | +++ | +++ |
| * S2 | 8.55 | 8.53 | −0.4 | 76.23 | +++ | +++ |
| S3 | 8.4 | 8.34 | −0.5 | 70.11 | +++ | +++ |
| * S4 | 8.03 | 8.01 | −0.3 | 75.1 | +++ | +++ |
| S5 | 4.47 | 3.29 | +3.1 | 72.4 | + | + |
| S6 | 4.94 | 5.13 | −1.6 | 71.85 | + | + |
| * S7 | 5.7 | 4.91 | −1.8 | 71.22 | ++ | + |
| S8 | 4.91 | 4.9 | +0.4 | 69.36 | + | + |
| * S9 | 7.15 | 7.78 | +1.6 | 74.29 | +++ | +++ |
| S10 | 5.9 | 5.56 | +3.4 | 71.36 | ++ | + |
| S11 | 5.2 | 5.16 | −0.3 | 70.88 | + | + |
| S12 | 5.76 | 5.73 | +2.4 | 71.73 | ++ | ++ |
| S13 | 7.14 | 6.78 | +1.2 | 71.49 | +++ | ++ |
| S14 | 4.35 | 4.36 | −0.1 | 70.34 | + | + |
| S15 | 6.03 | 6.05 | +1.9 | 71.13 | ++ | ++ |
| S16 | 5.67 | 5.41 | +3.7 | 72.55 | ++ | + |
| * S17 | 5.02 | 6.01 | +1.4 | 70.63 | + | + |
| * S18 | 5.17 | 5.23 | +1.1 | 70.89 | + | + |
| * S19 | 4.71 | 4.67 | −1.6 | 70.25 | + | + |
| S20 | 5.05 | 5.03 | +0.4 | 71.61 | + | + |
| S21 | 4.39 | 4.38 | +0.6 | 70.78 | + | + |
| * S22 | 4.41 | 4.41 | −0.8 | 70.16 | + | + |
| S23 | 5.68 | 5.7 | +1.5 | 71.64 | ++ | ++ |
| S24 | 4.85 | 4.83 | +2.1 | 69.52 | + | + |
| * S25 | 5.38 | 5.96 | +1.7 | 71.13 | + | + |
| S26 | 4.43 | 4.42 | +1.4 | 71.47 | + | + |
| * S27 | 4.34 | 4.39 | +0.6 | 69.67 | + | + |
| S28 | 5.15 | 5.17 | −0.5 | 71.45 | + | + |
| S29 | 5.49 | 5.48 | +1.6 | 70.43 | + | + |
| S30 | 5.93 | 5.91 | +1.1 | 69.71 | ++ | ++ |
| S31 | 6.16 | 6.15 | +1.1 | 72.43 | ++ | ++ |
| * S32 | 6.36 | 7.63 | +1.9 | 71.98 | ++ | + |
| S33 | 6.07 | 6.08 | +1.3 | 69.11 | ++ | ++ |
| * S34 | 6.61 | 5.56 | −1.8 | 73.45 | +++ | +++ |
| S35 | 5.07 | 5.06 | −1.1 | 74.23 | + | + |
| S36 | 4.74 | 4.75 | +1.2 | 71.26 | + | + |
| S37 | 5.65 | 5.41 | +1.3 | 72.84 | ++ | + |
| S38 | 6.92 | 6.94 | +1.1 | 71.04 | +++ | +++ |
| S39 | 6.09 | 6.07 | −0.3 | 70.09 | ++ | ++ |
| Library | AUC | p-Value | BEDROC 20 | p-Value | |
|---|---|---|---|---|---|
| Cathepsin B dataset | Autodock v4.2 Adt-Vina | 0.74 | 0.00039 | 0.13 | 0.12 |
| 0.72 | 0.00086 | 0.18 | 0.03 | ||
| Maybridge dataset | Autodock v4.2 ADt-Vina | 0.63 | 0.00067 | 0.10 | 0.51 |
| 0.73 | 0.00088 | 0.17 | 0.0075 |
| S.No | C.Name | BEe (kcal/mol) | Ki (µM) | IMEe (kcal/mol) | Vdw-Hb-Ds (kcal/mol) | Ee (kcal/mol) | IEe (kcal/mol) | TFEe (kcal/mol) |
|---|---|---|---|---|---|---|---|---|
| 1 | AW01196 | −6.52 | 22.02 | −6.37 | −6.83 | +0.46 | −0.62 | +2.47 |
| 2 | BTB03075 | −8.17 | 1.02 | −8.45 | −8.42 | −0.03 | −1.37 | +1.65 |
| 3 | BTB11814 | −7.07 | 6.52 | −8.04 | −7.94 | −0.10 | −1.50 | +2.47 |
| 4 | HTS05162 | −7.65 | 2.46 | −7.88 | −7.77 | −0.11 | −0.87 | +1.10 |
| 5 | JFD02054 | −6.08 | 35.21 | −7.56 | −7.60 | +0.04 | −1.26 | +2.74 |
| 6 | JP00474 | −7.60 | 2.69 | −8.08 | −8.45 | +0.36 | −1.71 | +2.20 |
| 7 | KM02757 | −7.31 | 4.41 | −7.72 | −7.43 | −0.28 | −2.61 | +3.02 |
| 8 | KM02759 | −8.16 | 1.04 | −9.25 | −9.19 | −0.06 | −1.93 | +3.02 |
| 9 | KM02760 | −7.31 | 4.41 | −7.42 | −7.37 | −0.05 | −2.91 | +3.02 |
| 10 | KM02777 | −7.53 | 3.01 | −8.22 | −8.25 | +0.03 | −2.61 | +3.29 |
| 11 | KM02922 | −8.19 | 0.99 | −8.47 | −8.47 | −0.03 | −2.19 | +2.47 |
| 12 | RF02795 | −9.18 | 0.18 | −9.44 | −8.00 | −1.44 | −0.84 | +1.10 |
| 13 | RJC00586 | −6.29 | 24.43 | −7.84 | −7.81 | −0.03 | −1.20 | +2.74 |
| 14 | S12294 | −6.87 | 20.92 | −6.45 | −6.75 | +0.30 | −0.79 | +1.37 |
| 15 | SPB02418 | −7.15 | 5.75 | −7.91 | −7.62 | −0.29 | −0.61 | +1.37 |
| 16 | SPB03394 | −6.76 | 11.11 | −7.01 | −7.08 | +0.08 | −0.85 | +1.10 |
| 17 | SPB03600 | −8.01 | 1.35 | −8.27 | −8.15 | −0.12 | −0.83 | +1.10 |
| 18 | SPB08054 | −6.56 | 15.53 | −8.21 | −8.16 | −0.05 | −0.82 | +2.47 |
| Compounds | ΔEvdw | ΔEelec | ΔGMM | ΔGPBpolar solvation | ΔGPBnonpolar solvation | ΔGGBpolar solvation | ΔGGBnonpolar solvation | ΔGMM−PBSA binding | ΔGMM−GBSA binding |
|---|---|---|---|---|---|---|---|---|---|
| CA030 | −25.81 | −40.86 | −66.67 | 41.45 | −5.42 | 36.62 | −5.42 | −25.22 | −30.06 |
| BTB03075 | −19.43 | −39.35 | −58.78 | 39.53 | −5.94 | 31.97 | −5.64 | −19.25 | −26.81 |
| KM02922 | −29.50 | −51.66 | −81.66 | 46.48 | −6.61 | 38.96 | −6.61 | −34.68 | −42.20 |
| RF02795 | −17.39 | −58.86 | −76.25 | 44.32 | −6.38 | 37.24 | −6.38 | −31.93 | −39.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chitranshi, N.; Kumar, A.; Sheriff, S.; Gupta, V.; Godinez, A.; Saks, D.; Sarkar, S.; Shen, T.; Mirzaei, M.; Basavarajappa, D.; et al. Identification of Novel Cathepsin B Inhibitors with Implications in Alzheimer’s Disease: Computational Refining and Biochemical Evaluation. Cells 2021, 10, 1946. https://doi.org/10.3390/cells10081946
Chitranshi N, Kumar A, Sheriff S, Gupta V, Godinez A, Saks D, Sarkar S, Shen T, Mirzaei M, Basavarajappa D, et al. Identification of Novel Cathepsin B Inhibitors with Implications in Alzheimer’s Disease: Computational Refining and Biochemical Evaluation. Cells. 2021; 10(8):1946. https://doi.org/10.3390/cells10081946
Chicago/Turabian StyleChitranshi, Nitin, Ashutosh Kumar, Samran Sheriff, Veer Gupta, Angela Godinez, Danit Saks, Soumalya Sarkar, Ting Shen, Mehdi Mirzaei, Devaraj Basavarajappa, and et al. 2021. "Identification of Novel Cathepsin B Inhibitors with Implications in Alzheimer’s Disease: Computational Refining and Biochemical Evaluation" Cells 10, no. 8: 1946. https://doi.org/10.3390/cells10081946
APA StyleChitranshi, N., Kumar, A., Sheriff, S., Gupta, V., Godinez, A., Saks, D., Sarkar, S., Shen, T., Mirzaei, M., Basavarajappa, D., Abyadeh, M., Singh, S. K., Dua, K., Zhang, K. Y. J., Graham, S. L., & Gupta, V. (2021). Identification of Novel Cathepsin B Inhibitors with Implications in Alzheimer’s Disease: Computational Refining and Biochemical Evaluation. Cells, 10(8), 1946. https://doi.org/10.3390/cells10081946