
Age-Dependent Hippocampal Proteomics in the APP/PS1 Alzheimer Mouse Model: A Comparative Analysis with Classical SWATH/DIA and directDIA Approaches

 , , ,
, , , 
Abstract
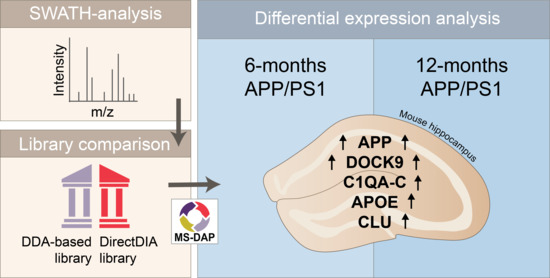
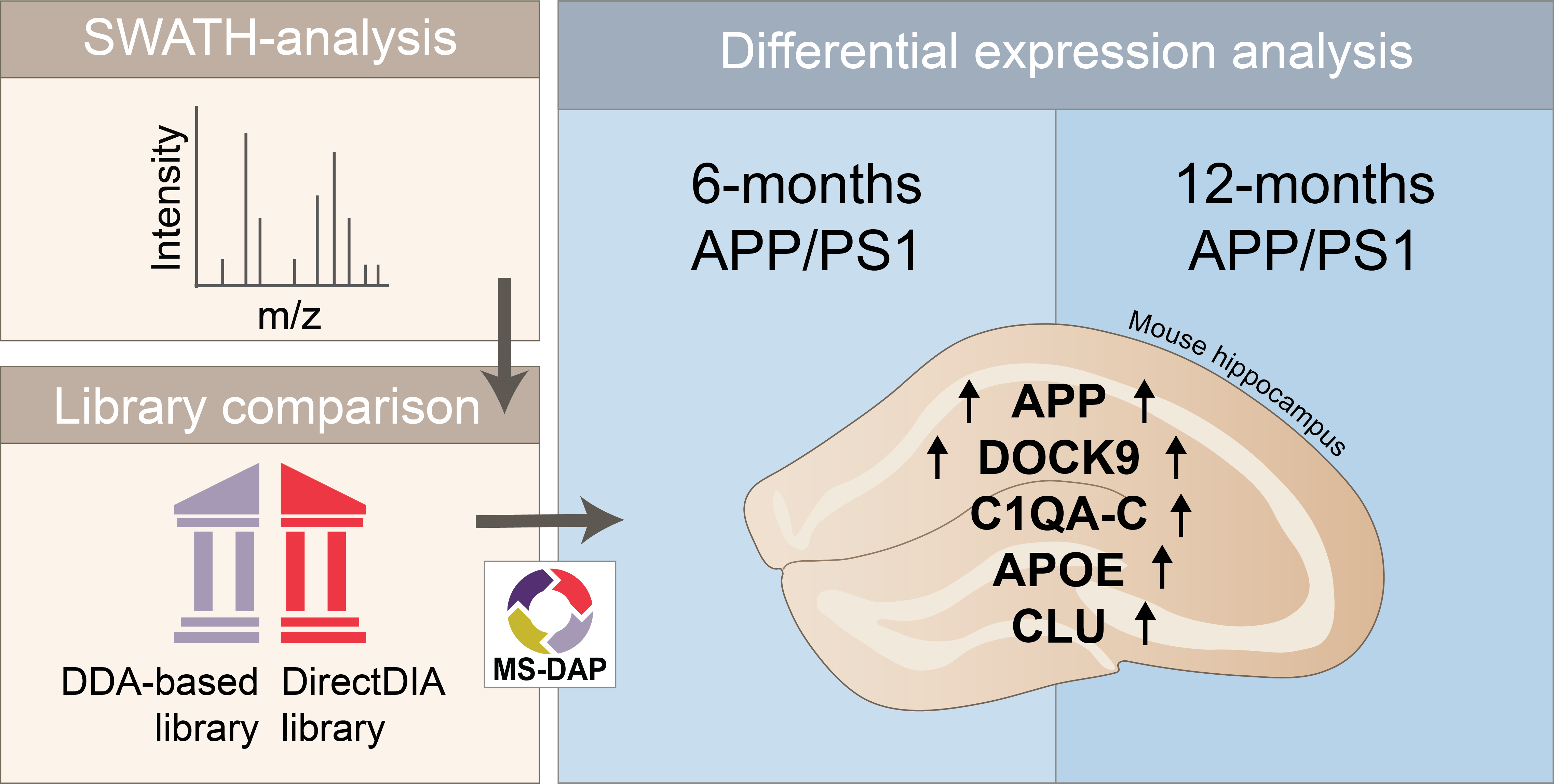{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Synaptosomal Enrichment
2.3. Filter-Aided Sample Preparation
2.4. Micro-LC and SWATH Mass Spectrometry
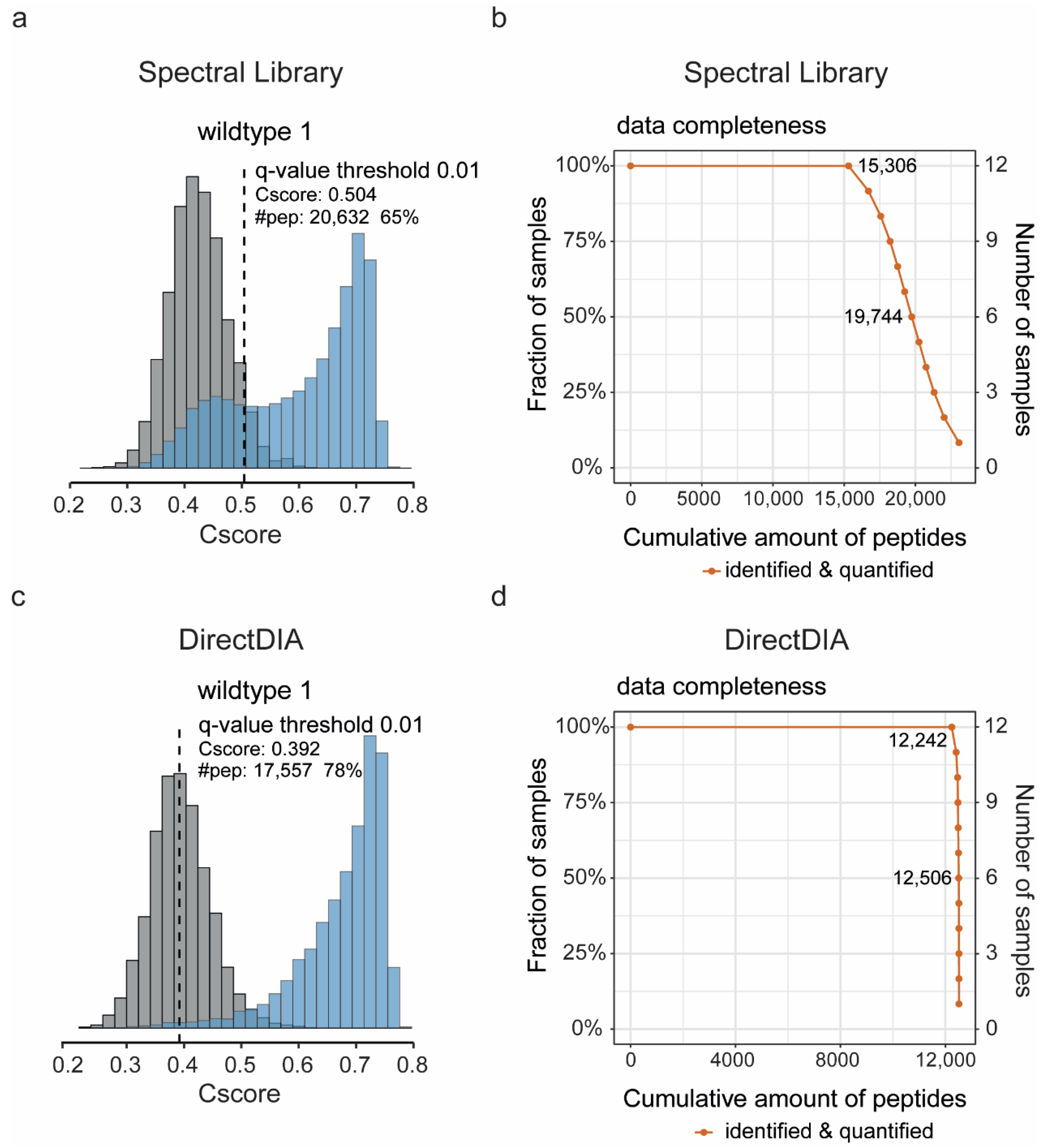
2.5. SWATH Data Analysis
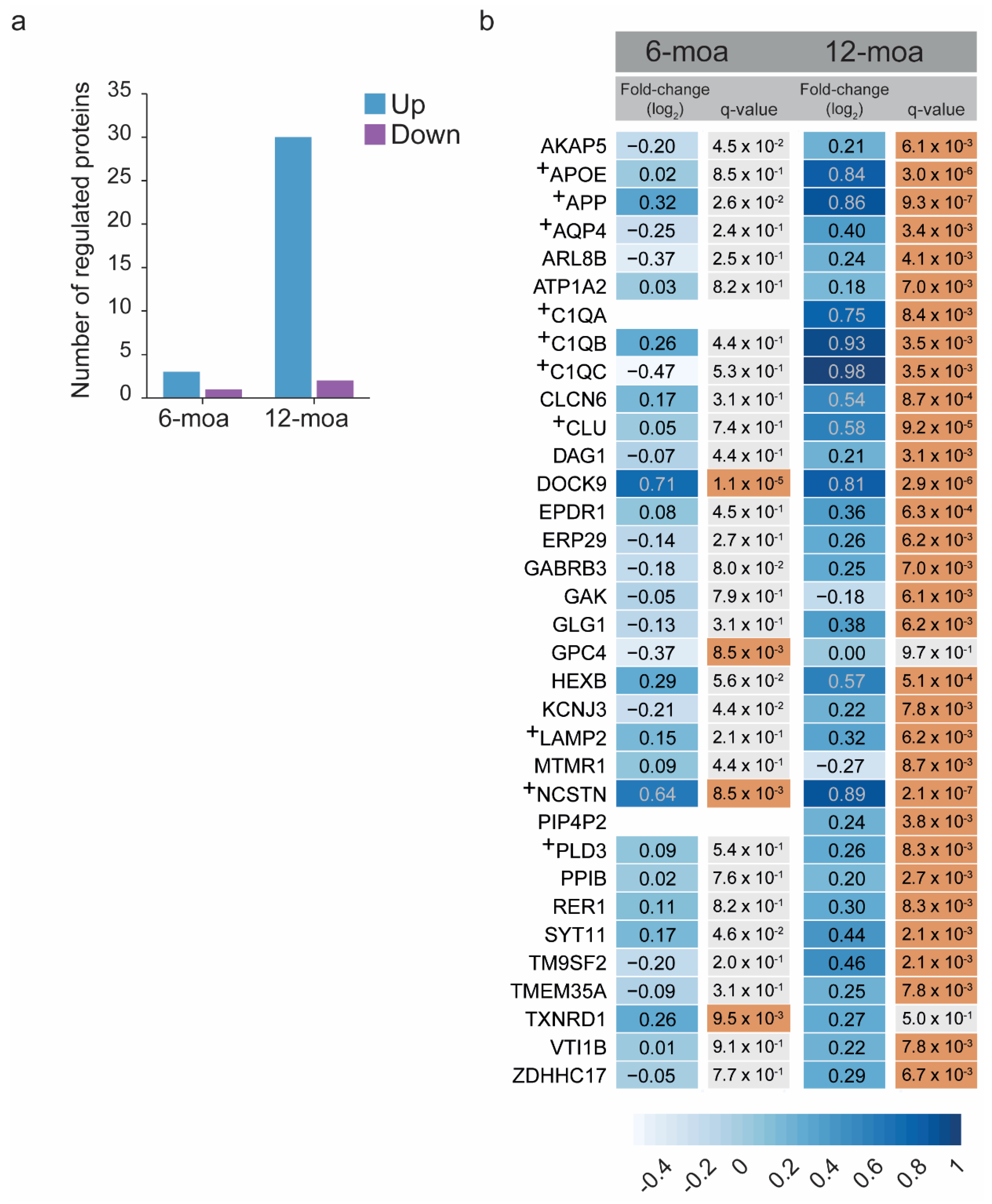
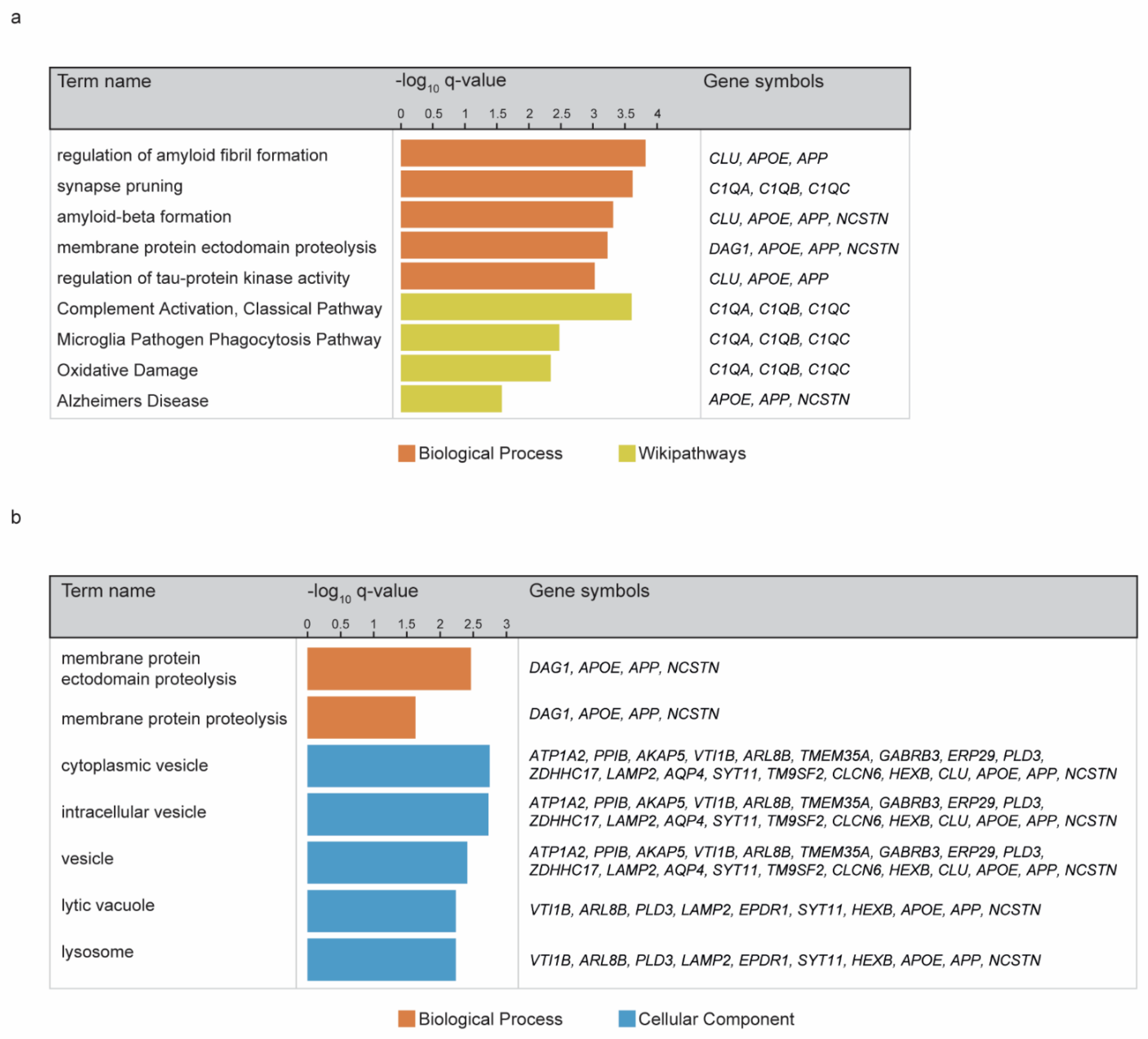
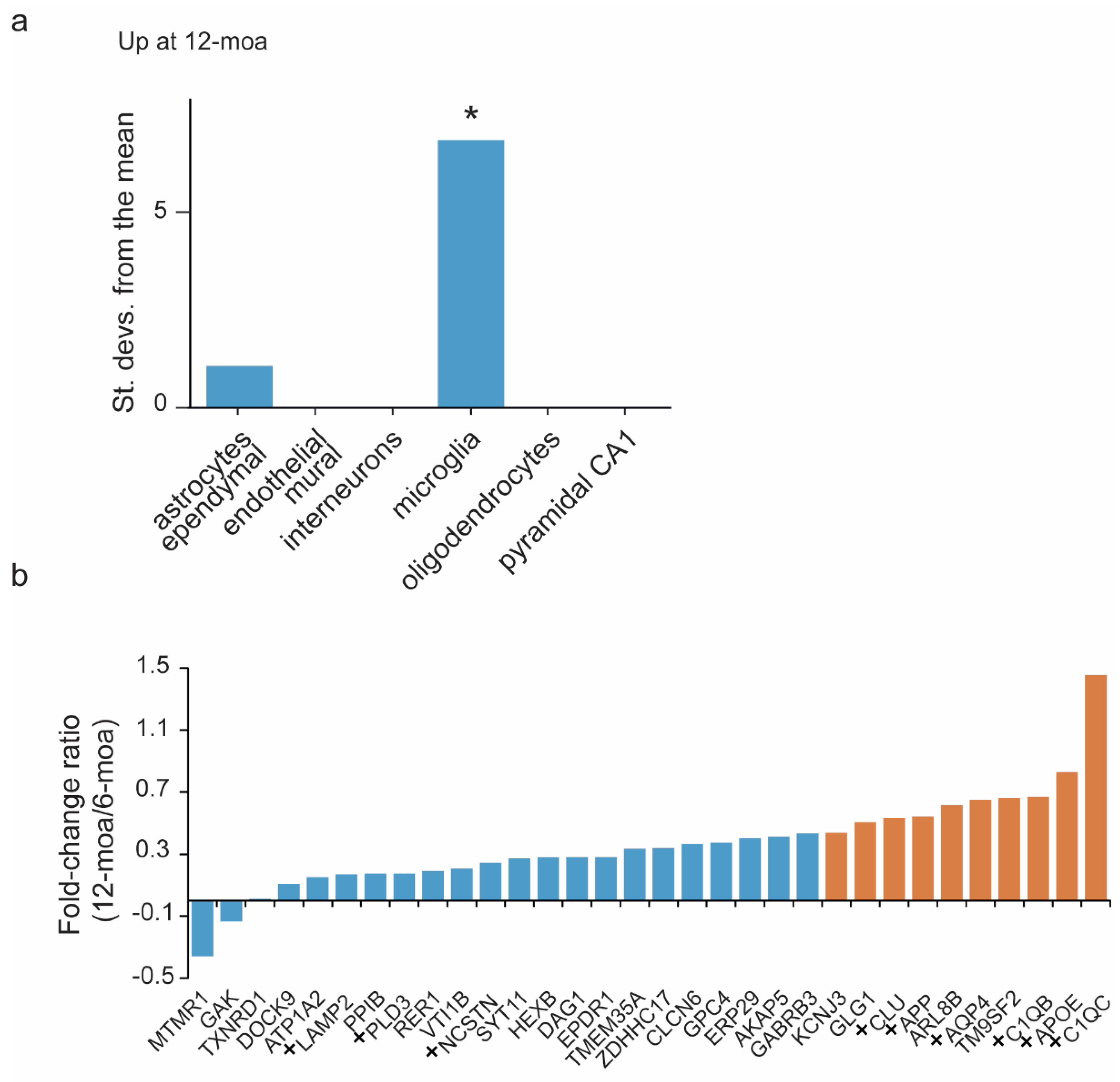
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| APP/PS1 | Amyloid Precursor Protein swe/ Presenilin 1 dE9 |
| Aβ | Amyloid-β |
| BP | Biological Process |
| CC | Cellular Component |
| Cscores | Confidence scores |
| DDA | Data-Dependent acquisition |
| DEA | Differential Expression Analysis |
| DIA | Data-independent acquisition |
| EWCE | Expression-Weighted Cell-type Enrichment |
| HPLC | High-Performance Liquid Chromatography |
| LC-MS/MS | Liquid Chromatography–Tandem Mass Spectrometry |
| Moa | Months of age |
| MS-DAP | Mass Spectrometry Downstream Analysis Pipeline |
| SWATH | Sequential Window Acquisition of all THeoretical fragment-ion |
| SynGO | Synaptic Gene Ontology |
| WP | WikiPathways |
References
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Li, S.; Selkoe, D.J. A mechanistic hypothesis for the impairment of synaptic plasticity by soluble Aβ oligomers from Alzheimer’s brain. J. Neurochem. 2020, 154, 583–597. [Google Scholar] [CrossRef] [PubMed]
- Raskin, J.; Cummings, J.; Hardy, J.; Schuh, K.; Dean, R.A. Neurobiology of Alzheimer’s Disease: Integrated Molecular, Physiological, Anatomical, Biomarker, and Cognitive Dimensions. Curr. Alzheimer Res. 2015, 12, 712–722. [Google Scholar] [CrossRef] [PubMed]
- Borchelt, D.R.; Ratovitski, T.; van Lare, J.; Lee, M.K.; Gonzales, V.; Jenkins, N.A.; Copeland, N.G.; Price, D.L.; Sisodia, S.S. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron 1997, 19, 939–945. [Google Scholar] [CrossRef]
- Sasaguri, H.; Nilsson, P.; Hashimoto, S.; Nagata, K.; Saito, T.; De Strooper, B.; Hardy, J.; Vassar, R.; Winblad, B.; Saido, T.C. APP mouse models for Alzheimer’s disease preclinical studies. EMBO J. 2017, 36, 2473–2487. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Nie, S.; Cao, M.; Marshall, C.; Gao, J.; Xiao, N.; Hu, G.; Xiao, M. Characterization of AD-like phenotype in aged APPSwe/PS1dE9 mice. Age 2016, 38, 303–322. [Google Scholar] [CrossRef]
- Oyelami, T.; De Bondt, A.; Van den Wyngaert, I.; Van Hoorde, K.; Hoskens, L.; Shaban, H.; Kemp, J.A.; Drinkenburg, W.H. Age-dependent concomitant changes in synaptic dysfunction and GABAergic pathway in the APP/PS1 mouse model. Acta Neurobiol. Exp. 2016, 76, 282–293. [Google Scholar] [CrossRef]
- Reinders, N.R.; Pao, Y.; Renner, M.C.; da Silva-Matos, C.M.; Lodder, T.R.; Malinow, R.; Kessels, H.W. Amyloid-β effects on synapses and memory require AMPA receptor subunit GluA3. Proc. Natl. Acad. Sci. USA 2016, 113, E6526–E6534. [Google Scholar] [CrossRef]
- Abbink, M.R.; Kotah, J.M.; Hoeijmakers, L.; Mak, A.; Yvon-Durocher, G.; van der Gaag, B.; Lucassen, P.J.; Korosi, A. Characterization of astrocytes throughout life in wildtype and APP/PS1 mice after early-life stress exposure. J. Neuroinflam. 2020, 17, 91. [Google Scholar] [CrossRef]
- Scheffler, K.; Stenzel, J.; Krohn, M.; Lange, C.; Hofrichter, J.; Schumacher, T.; Brüning, T.; Plath, A.-S.; Walker, L.; Pahnke, J. Determination of spatial and temporal distribution of microglia by 230 nm-high-resolution, high-throughput automated analysis reveals different amyloid plaque populations in an APP/PS1 mouse model of Alzheimer’s disease. Curr. Alzheimer Res. 2011, 8, 781–788. [Google Scholar] [CrossRef]
- Remmelink, E.; Smit, A.B.; Verhage, M.; Loos, M. Measuring discrimination- and reversal learning in mouse models within 4 days and without prior food deprivation. Learn. Mem. 2016, 23, 660–667. [Google Scholar] [CrossRef]
- Arendt, T. Synaptic degeneration in Alzheimer’s disease. Acta Neuropathol. 2009, 118, 167–179. [Google Scholar] [CrossRef]
- Schubert, O.T.; Gillet, L.C.; Collins, B.C.; Navarro, P.; Rosenberger, G.; Wolski, W.E.; Lam, H.; Amodei, D.; Mallick, P.; MacLean, B.; et al. Building high-quality assay libraries for targeted analysis of SWATH MS data. Nat. Protoc. 2015, 10, 426–441. [Google Scholar] [CrossRef]
- Koopmans, F.; Ho, J.T.C.; Smit, A.B.; Li, K.W. Comparative Analyses of Data Independent Acquisition Mass Spectrometric Approaches: DIA, WiSIM-DIA, and Untargeted DIA. Proteomics 2018, 18. [Google Scholar] [CrossRef]
- Tsou, C.-C.; Avtonomov, D.; Larsen, B.; Tucholska, M.; Choi, H.; Gingras, A.-C.; Nesvizhskii, A.I. DIA-Umpire: Comprehensive computational framework for data-independent acquisition proteomics. Nat. Methods 2015, 12, 258–264. [Google Scholar] [CrossRef]
- Koopmans, F.; Pandya, N.J.; Franke, S.K.; Phillippens, I.H.C.M.H.; Paliukhovich, I.; Li, K.W.; Smit, A.B. Comparative Hippocampal Synaptic Proteomes of Rodents and Primates: Differences in Neuroplasticity-Related Proteins. Front. Mol. Neurosci. 2018, 11, 364. [Google Scholar] [CrossRef]
- Wisniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- He, E.; Lozano, M.A.G.; Stringer, S.; Watanabe, K.; Sakamoto, K.; den Oudsten, F.; Koopmans, F.; Giamberardino, S.N.; Hammerschlag, A.; Cornelisse, L.N.; et al. MIR137 schizophrenia-associated locus controls synaptic function by regulating synaptogenesis, synapse maturation and synaptic transmission. Hum. Mol. Genet. 2018, 27, 1879–1891. [Google Scholar] [CrossRef] [PubMed]
- Rosato, M.; Stringer, S.; Gebuis, T.; Paliukhovich, I.; Li, K.W.; Posthuma, D.; Sullivan, P.F.; Smit, A.B.; van Kesteren, R.E. Combined cellomics and proteomics analysis reveals shared neuronal morphology and molecular pathway phenotypes for multiple schizophrenia risk genes. Mol. Psychiatry 2021, 26, 784–799. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucl. Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Lozano, M.A.; Klemmer, P.; Gebuis, T.; Hassan, C.; van Nierop, P.; van Kesteren, R.E.; Smit, A.B.; Li, K.W. Dynamics of the mouse brain cortical synaptic proteome during postnatal brain development. Sci. Rep. 2016, 6, 35456. [Google Scholar] [CrossRef]
- Klaassen, R.V.; Stroeder, J.; Coussen, F.; Petersen, J.D.; Renancio, C.; Lodder, J.C.; Diana, C.; Schmitz, L.J.M.; Rao-ruiz, P.; Spijker, S.; et al. Shisa6 traps AMPARs at postsynaptic sites and prevents their desensitization during high frequency synaptic stimulation. Nat. Commun. 2016, 1–12. [Google Scholar] [CrossRef]
- Wilhelm, B.G.; Mandad, S.; Truckenbrodt, S.; Kröhnert, K.; Schäfer, C.; Rammner, B.; Koo, S.J.; Claßen, G.A.; Krauss, M.; Haucke, V.; et al. Composition of isolated synaptic boutons reveals the amounts of vesicle trafficking proteins. Science 2014, 344, 1023–1028. [Google Scholar] [CrossRef] [PubMed]
- Pandya, N.J.; Koopmans, F.; Slotman, J.A.; Paliukhovich, I.; Houtsmuller, A.B.; Smit, A.B.; Li, K.W. Correlation profiling of brain sub-cellular proteomes reveals co-assembly of synaptic proteins and subcellular distribution. Sci. Rep. 2017, 7, 12107. [Google Scholar] [CrossRef] [PubMed]
- Koopmans, F.; van Nierop, P.; Andres-Alonso, M.; Byrnes, A.; Cijsouw, T.; Coba, M.P.; Cornelisse, L.N.; Farrell, R.J.; Goldschmidt, H.L.; Howrigan, D.P.; et al. SynGO: An Evidence-Based, Expert-Curated Knowledge Base for the Synapse. Neuron 2019, 103, 217–234.e4. [Google Scholar] [CrossRef] [PubMed]
- Skene, N.G.; Grant, S.G.N. Identification of Vulnerable Cell Types in Major Brain Disorders Using Single Cell Transcriptomes and Expression Weighted Cell Type Enrichment. Front. Neurosci. 2016, 10, 16. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, A.; Muñoz-Manchado, A.B.; Codeluppi, S.; Lönnerberg, P.; La Manno, G.; Juréus, A.; Marques, S.; Munguba, H.; He, L.; Betsholtz, C.; et al. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 2015, 347, 1138–1142. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef]
- Li, X.; Tsolis, K.C.; Koper, M.J.; Ronisz, A.; Ospitalieri, S.; von Arnim, C.A.F.; Vandenberghe, R.; Tousseyn, T.; Scheuerle, A.; Economou, A.; et al. Sequence of proteome profiles in preclinical and symptomatic Alzheimer’s disease. Alzheimers. Dement. 2021. [Google Scholar] [CrossRef]
- Végh, M.J.; Heldring, C.M.; Kamphuis, W.; Hijazi, S.; Timmerman, A.J.; Li, K.W.; van Nierop, P.; Mansvelder, H.D.; Hol, E.M.; Smit, A.B.; et al. Reducing hippocampal extracellular matrix reverses early memory deficits in a mouse model of Alzheimer’s disease. Acta Neuropathol. Commun. 2014, 2, 76. [Google Scholar] [CrossRef][Green Version]
- Wan, Y.-W.; Al-Ouran, R.; Mangleburg, C.G.; Perumal, T.M.; Lee, T.V.; Allison, K.; Swarup, V.; Funk, C.C.; Gaiteri, C.; Allen, M.; et al. Meta-Analysis of the Alzheimer’s Disease Human Brain Transcriptome and Functional Dissection in Mouse Models. Cell Rep. 2020, 32, 107908. [Google Scholar] [CrossRef]
- Sebastian Monasor, L.; Müller, S.A.; Colombo, A.V.; Tanrioever, G.; König, J.; Roth, S.; Liesz, A.; Berghofer, A.; Piechotta, A.; Prestel, M.; et al. Fibrillar Aβ triggers microglial proteome alterations and dysfunction in Alzheimer mouse models. Elife 2020, 9, e54083. [Google Scholar] [CrossRef]
- Parhizkar, S.; Arzberger, T.; Brendel, M.; Kleinberger, G.; Deussing, M.; Focke, C.; Nuscher, B.; Xiong, M.; Ghasemigharagoz, A.; Katzmarski, N.; et al. Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat. Neurosci. 2019, 22, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Sala Frigerio, C.; Wolfs, L.; Fattorelli, N.; Thrupp, N.; Voytyuk, I.; Schmidt, I.; Mancuso, R.; Chen, W.-T.; Woodbury, M.E.; Srivastava, G.; et al. The Major Risk Factors for Alzheimer’s Disease: Age, Sex, and Genes Modulate the Microglia Response to Aβ Plaques. Cell Rep. 2019, 27, 1293–1306.e6. [Google Scholar] [CrossRef]
- Saito, T.; Matsuba, Y.; Mihira, N.; Takano, J.; Nilsson, P.; Itohara, S.; Iwata, N.; Saido, T.C. Single App knock-in mouse models of Alzheimer’s disease. Nat. Neurosci. 2014, 17, 661–663. [Google Scholar] [CrossRef] [PubMed]
- Holtzman, D.M.; Herz, J.; Bu, G. Apolipoprotein E and apolipoprotein E receptors: Normal biology and roles in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006312. [Google Scholar] [CrossRef] [PubMed]
- Yeh, F.L.; Wang, Y.; Tom, I.; Gonzalez, L.C.; Sheng, M. TREM2 Binds to Apolipoproteins, Including APOE and CLU/APOJ, and Thereby Facilitates Uptake of Amyloid-Beta by Microglia. Neuron 2016, 91, 328–340. [Google Scholar] [CrossRef]
- Radde, R.; Bolmont, T.; Kaeser, S.A.; Coomaraswamy, J.; Lindau, D.; Stoltze, L.; Calhoun, M.E.; Jäggi, F.; Wolburg, H.; Gengler, S.; et al. Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 2006, 7, 940–946. [Google Scholar] [CrossRef]
- De Strooper, B. Aph-1, Pen-2, and Nicastrin with Presenilin generate an active gamma-Secretase complex. Neuron 2003, 38, 9–12. [Google Scholar] [CrossRef]
- Annaert, W.; Kaether, C. Bring it back, bring it back, don’t take it away from me—The sorting receptor RER1. J. Cell Sci. 2020, 133. [Google Scholar] [CrossRef]
- Kaether, C.; Scheuermann, J.; Fassler, M.; Zilow, S.; Shirotani, K.; Valkova, C.; Novak, B.; Kacmar, S.; Steiner, H.; Haass, C. Endoplasmic reticulum retention of the gamma-secretase complex component Pen2 by Rer1. EMBO Rep. 2007, 8, 743–748. [Google Scholar] [CrossRef]
- Spasic, D.; Raemaekers, T.; Dillen, K.; Declerck, I.; Baert, V.; Serneels, L.; Füllekrug, J.; Annaert, W. Rer1p competes with APH-1 for binding to nicastrin and regulates gamma-secretase complex assembly in the early secretory pathway. J. Cell Biol. 2007, 176, 629–640. [Google Scholar] [CrossRef]
- Park, H.-J.; Shabashvili, D.; Nekorchuk, M.D.; Shyqyriu, E.; Jung, J.I.; Ladd, T.B.; Moore, B.D.; Felsenstein, K.M.; Golde, T.E.; Kim, S.-H. Retention in endoplasmic reticulum 1 (RER1) modulates amyloid-β (Aβ) production by altering trafficking of γ-secretase and amyloid precursor protein (APP). J. Biol. Chem. 2012, 287, 40629–40640. [Google Scholar] [CrossRef]
- Jansen, I.E.; Savage, J.E.; Watanabe, K.; Bryois, J.; Williams, D.M.; Steinberg, S.; Sealock, J.; Karlsson, I.K.; Hägg, S.; Athanasiu, L.; et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 2019, 51, 404–413. [Google Scholar] [CrossRef]
- De Silva, H.V.; Stuart, W.D.; Duvic, C.R.; Wetterau, J.R.; Ray, M.J.; Ferguson, D.G.; Albers, H.W.; Smith, W.R.; Harmony, J.A. A 70-kDa apolipoprotein designated ApoJ is a marker for subclasses of human plasma high density lipoproteins. J. Biol. Chem. 1990, 265, 13240–13247. [Google Scholar] [CrossRef]
- Xie, Z.; Harris-White, M.E.; Wals, P.A.; Frautschy, S.A.; Finch, C.E.; Morgan, T.E. Apolipoprotein J (clusterin) activates rodent microglia in vivo and in vitro. J. Neurochem. 2005, 93, 1038–1046. [Google Scholar] [CrossRef] [PubMed]
- Kishore, U.; Reid, K.B. C1q: Structure, function, and receptors. Immunopharmacology 2000, 49, 159–170. [Google Scholar] [CrossRef]
- Afagh, A.; Cummings, B.J.; Cribbs, D.H.; Cotman, C.W.; Tenner, A.J. Localization and cell association of C1q in Alzheimer’s disease brain. Exp. Neurol. 1996, 138, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Reichwald, J.; Danner, S.; Wiederhold, K.-H.; Staufenbiel, M. Expression of complement system components during aging and amyloid deposition in APP transgenic mice. J. Neuroinflam. 2009, 6, 35. [Google Scholar] [CrossRef] [PubMed]
- Webster, S.; Glabe, C.; Rogers, J. Multivalent binding of complement protein C1Q to the amyloid beta-peptide (A beta) promotes the nucleation phase of A beta aggregation. Biochem. Biophys. Res. Commun. 1995, 217, 869–875. [Google Scholar] [CrossRef] [PubMed]
- Webster, S.; O’Barr, S.; Rogers, J. Enhanced aggregation and beta structure of amyloid beta peptide after coincubation with C1q. J. Neurosci. Res. 1994, 39, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Boyett, K.W.; DiCarlo, G.; Jantzen, P.T.; Jackson, J.; O’Leary, C.; Wilcock, D.; Morgan, D.; Gordon, M.N. Increased fibrillar beta-amyloid in response to human clq injections into hippocampus and cortex of APP+PS1 transgenic mice. Neurochem. Res. 2003, 28, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Webster, S.D.; Yang, A.J.; Margol, L.; Garzon-Rodriguez, W.; Glabe, C.G.; Tenner, A.J. Complement component C1q modulates the phagocytosis of Abeta by microglia. Exp. Neurol. 2000, 161, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Sanders, S.S.; Hou, J.; Sutton, L.M.; Garside, V.C.; Mui, K.K.N.; Singaraja, R.R.; Hayden, M.R.; Hoodless, P.A. Huntingtin interacting proteins 14 and 14-like are required for chorioallantoic fusion during early placental development. Dev. Biol. 2015, 397, 257–266. [Google Scholar] [CrossRef]
- Nalls, M.A.; Plagnol, V.; Hernandez, D.G.; Sharma, M.; Sheerin, U.-M.; Saad, M.; Simón-Sánchez, J.; Schulte, C.; Lesage, S.; Sveinbjörnsdóttir, S.; et al. Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet 2011, 377, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Sanghvi, H.; Singh, R.; Morrin, H.; Rajkumar, A.P. Systematic review of genetic association studies in people with Lewy body dementia. Int. J. Geriatr. Psychiatry 2020, 35, 436–448. [Google Scholar] [CrossRef]
- Rong, Z.; Cheng, B.; Zhong, L.; Ye, X.; Li, X.; Jia, L.; Li, Y.; Shue, F.; Wang, N.; Cheng, Y.; et al. Activation of FAK/Rac1/Cdc42-GTPase signaling ameliorates impaired microglial migration response to Aβ(42) in triggering receptor expressed on myeloid cells 2 loss-of-function murine models. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2020, 34, 10984–10997. [Google Scholar] [CrossRef]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.K.; Younkin, S.; et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef]
- Cimino, P.J.; Yang, Y.; Li, X.; Hemingway, J.F.; Cherne, M.K.; Khademi, S.B.; Fukui, Y.; Montine, K.S.; Montine, T.J.; Keene, C.D. Ablation of the microglial protein DOCK2 reduces amyloid burden in a mouse model of Alzheimer’s disease. Exp. Mol. Pathol. 2013, 94, 366–371. [Google Scholar] [CrossRef]
- Chen, Q.; Peto, C.A.; Shelton, G.D.; Mizisin, A.; Sawchenko, P.E.; Schubert, D. Loss of modifier of cell adhesion reveals a pathway leading to axonal degeneration. J. Neurosci. 2009, 29, 118–130. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van der Spek, S.J.F.; Gonzalez-Lozano, M.A.; Koopmans, F.; Miedema, S.S.M.; Paliukhovich, I.; Smit, A.B.; Li, K.W. Age-Dependent Hippocampal Proteomics in the APP/PS1 Alzheimer Mouse Model: A Comparative Analysis with Classical SWATH/DIA and directDIA Approaches. Cells 2021, 10, 1588. https://doi.org/10.3390/cells10071588
van der Spek SJF, Gonzalez-Lozano MA, Koopmans F, Miedema SSM, Paliukhovich I, Smit AB, Li KW. Age-Dependent Hippocampal Proteomics in the APP/PS1 Alzheimer Mouse Model: A Comparative Analysis with Classical SWATH/DIA and directDIA Approaches. Cells. 2021; 10(7):1588. https://doi.org/10.3390/cells10071588
Chicago/Turabian Stylevan der Spek, Sophie J. F., Miguel A. Gonzalez-Lozano, Frank Koopmans, Suzanne S. M. Miedema, Iryna Paliukhovich, August B. Smit, and Ka Wan Li. 2021. "Age-Dependent Hippocampal Proteomics in the APP/PS1 Alzheimer Mouse Model: A Comparative Analysis with Classical SWATH/DIA and directDIA Approaches" Cells 10, no. 7: 1588. https://doi.org/10.3390/cells10071588
APA Stylevan der Spek, S. J. F., Gonzalez-Lozano, M. A., Koopmans, F., Miedema, S. S. M., Paliukhovich, I., Smit, A. B., & Li, K. W. (2021). Age-Dependent Hippocampal Proteomics in the APP/PS1 Alzheimer Mouse Model: A Comparative Analysis with Classical SWATH/DIA and directDIA Approaches. Cells, 10(7), 1588. https://doi.org/10.3390/cells10071588