
Understanding Cervical Cancer through Proteomics

 ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Proteomics
2.1. Two-Dimensional Gels Electrophoresis
2.2. Mass Spectrometry
3. Proteomics and Cervical Cancer
3.1. Biological Samples in Proteomic Studies of CC
3.1.1. Cervical Cancer Cell Line
3.1.2. Cervical-Vaginal Fluid
3.1.3. Cervical Cancer Tissue
3.1.4. Blood
4. Other Omics Studies in Cervical Cancer
4.1. Genomics
4.2. Metagenomic
4.3. Transcriptomic
4.4. Metabolomics
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sankaranarayanan, R. Cervical cancer in developing countries. Trans. R. Soc. Trop. 2002, 96, 580–585. [Google Scholar] [CrossRef]
- Gargano, J.; Meites, E.; Watson, M.; Unger, E.; Markowitz, L.; Background, I. Manual for the Surveillance of Vac-cine-Preventable Diseases; Chapter 5: Human Papillomavirus; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2014.
- Walboomers, J.M.; Jacobs, M.V.; Manos, M.M.; Bosch, F.X.; Kummer, J.A.; Shah, K.V.; Snijders, P.J.; Peto, J.; Meijer, C.J.; Muñoz, N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide–PubMed. J. Pathol. 1999, 1, 9–12. [Google Scholar]
- Muñoz, N.; Bosch, F.X.; de Sanjosé, S.; Herrero, R.; Castellsagué, X.; Shah, K.V.; Snijders, P.J.F.; Meijer, C.J.L.M. Epidemiologic Classification of Human Papillomavirus Types Associated with Cervical Cancer. N. Engl. J. Med. 2003, 348, 518–527. [Google Scholar] [CrossRef] [Green Version]
- De Sanjose, S.; Quint, W.G.V.; Alemany, L.; Geraets, D.T.; Klaustermeier, J.E.; Lloveras, B.; Tous, S.; Felix, A.; Bravo, L.E.; Shin, H.R.; et al. Human papillomavirus genotype attribution in invasive cervical cancer: A retrospective cross-sectional worldwide study. Lancet Oncol. 2010, 11, 1048–1056. [Google Scholar] [CrossRef]
- Mendoza-Almanza, G.; Burciaga-Hernández, L.; Maldonado, V.; Melendez-Zajgla, J.; Olmos, J. Role of platelets and breast cancer stem cells in metastasis. World J. Stem Cells 2020, 12, 1237–1254. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 340, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [Green Version]
- Kontomanolis, E.N.; Koutras, A.; Syllaios, A.; Schizas, D.; Mastoraki, A.; Garmpis, N.; Diakosavvas, M.; Angelou, K.; Tsatsaris, G.; Pagkalos, A.; et al. Role of oncogenes and tumor-suppressor genes in carcinogenesis: A review. Anticancer Res. 2020, 40, 6009–6015. [Google Scholar] [CrossRef]
- Wang, L.H.; Wu, C.F.; Rajasekaran, N.; Shin, Y.K. Loss of tumor suppressor gene function in human cancer: An overview. Cell. Physiol. Biochem. 2019, 51, 2647–2693. [Google Scholar] [CrossRef]
- Chow, A.Y. Cell Cycle Control, Oncogenes, Tumor Suppressors | Learn Science at Scitable. Nat. Sci. Educ. 2010, 3, 7. [Google Scholar]
- Moreno-Layseca, P.; Streuli, C.H. Signalling pathways linking integrins with cell cycle progression. Matrix Biol. 2014, 34, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Petsalaki, E.; Zachos, G. DNA damage response proteins regulating mitotic cell division: Double agents preserving genome stability. FEBS J. 2020, 287, 1700–1721. [Google Scholar] [CrossRef]
- Yang, J.; Chen, L.; Kong, X.; Huang, T.; Cai, Y.D. Analysis of tumor suppressor genes based on gene ontology and the KEGG pathway. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Mishra, L.; Deng, C.X. The role of TGF-β/SMAD4 signaling in cancer. Int. J. Biol. Sci. 2018, 14, 111–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, P.-H.; Lin, Y.-W.; Huang, R.-L.; Liao, Y.-P.; Lee, H.-Y.; Wang, H.-C.; Chao, T.-K.; Chen, C.-K.; Chan, M.; Chu, T.-Y.; et al. Epigenetic silencing of PTPRR activates MAPK signaling, promotes metastasis and serves as a biomarker of invasive cervical cancer. Oncogene 2012, 32, 15–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, A.A.; Howitt, B.E.; Myers, A.P.; Dahlberg, S.E.; Palescandolo, E.; Van Hummelen, P.; Macconaill, L.E.; Shoni, M.; Wagle, N.; Jones, R.T.; et al. Oncogenic mutations in cervical cancer: Genomic differences between adenocarcinomas and squamous cell carcinomas of the cervix. Cancer 2013, 119, 3776–3783. [Google Scholar] [CrossRef]
- He, C.; Mao, D.; Hua, G.; Lv, X.; Chen, X.; Angeletti, P.C.; Dong, J.; Remmenga, S.W.; Rodabaugh, K.J.; Zhou, J.; et al. The Hippo/ YAP pathway interacts with EGFR signaling and HPV oncoproteins to regulate cervical cancer progression. EMBO Mol. Med. 2015, 7, 1426–1449. [Google Scholar] [CrossRef]
- Mendoza-Almanza, G.; Ortíz-Sánchez, E.; Rocha-Zavaleta, L.; Rivas-Santiago, C.; Esparza-Ibarra, E.; Olmos, J. Cervical cancer stem cells and other leading factors associated with cervical cancer development (Review). Oncol. Lett. 2019, 18, 3423–3432. [Google Scholar] [CrossRef] [PubMed]
- Kabakov, A.; Yakimova, A.; Matchuk, O. Molecular Chaperones in Cancer Stem Cells: Determinants of Stemness and Potential Targets for Antitumor Therapy. Cells 2020, 9, 892. [Google Scholar] [CrossRef] [Green Version]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [Green Version]
- Lu, W.; Kang, Y. Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Dev. Cell 2019, 49, 361–374. [Google Scholar] [CrossRef]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal. Transduct. Target. 2020, 5, 1–17. [Google Scholar] [CrossRef]
- Wang, W.; Jia, H.L.; Huang, J.M.; Liang, Y.C.; Tan, H.; Geng, H.Z.; Guo, L.Y.; Yao, S.Z. Identification of biomarkers for lymph node metastasis in early-stage cervical cancer by tissue-based proteomics. Br. J. Cancer 2014, 110, 1748–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boichenko, A.P.; Govorukhina, N.; Klip, H.G.; Van Der Zee, A.G.J.; Güzel, C.; Luider, T.M.; Bischoff, R. A panel of regulated proteins in serum from patients with cervical intraepithelial neoplasia and cervical cancer. J. Proteome Res. 2014, 13, 4995–5007. [Google Scholar] [CrossRef] [PubMed]
- Dasari, S.; Pereira, L.; Reddy, A.P.; Michaels, J.E.A.; Lu, X.; Jacob, T.; Thomas, A.; Rodland, M.; Roberts, C.T.; Gravett, M.G.; et al. Comprehensive proteomic analysis of human cervical-vaginal fluid. J. Proteome Res. 2007, 6, 1258–1268. [Google Scholar] [CrossRef]
- Boylan, K.L.M.; Afiuni-Zadeh, S.; Geller, M.A.; Argenta, P.A.; Griffin, T.J.; Skubitz, A.P.N. Evaluation of the potential of Pap test fluid and cervical swabs to serve as clinical diagnostic biospecimens for the detection of ovarian cancer by mass spectrometry-based proteomics. Clin. Proteom. 2021, 18, 4. [Google Scholar] [CrossRef] [PubMed]
- Graves, P.R.; Haystead, T.A.J. Molecular Biologist’s Guide to Proteomics. Microbiol. Mol. Biol. Rev. 2002, 66, 39–63. [Google Scholar] [CrossRef] [Green Version]
- Chandramouli, K.; Qian, P.-Y. Proteomics: Challenges, Techniques and Possibilities to Overcome Biological Sample Complexity. Hum. Genom. Proteom. 2009, 1. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Eraslan, B.; Wieland, T.; Hallström, B.; Hopf, T.; Zolg, D.P.; Zecha, J.; Asplund, A.; Li, L.; Meng, C.; et al. A deep proteome and transcriptome abundance atlas of 29 healthy human tissues. Mol. Syst. Biol. 2019, 15, e8503. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.T.; Garneau-Tsodikova, S.; Gatto, G.J. Protein posttranslational modifications: The chemistry of proteome diversifications. Angew. Chem. Int. Ed. 2005, 44, 7342–7372. [Google Scholar] [CrossRef]
- Rao, V.S.; Srinivas, K.; Sujini, G.N.; Kumar, G.N.S. Protein-Protein Interaction Detection: Methods and Analysis. Int. J. Proteom. 2014, 2014, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Droit, A.; Poirier, G.G.; Hunter, J.M. Experimental and bioinformatic approaches for interrogating protein-protein interactions to determine protein function. J. Mol. Endocrinol. 2005, 34, 263–280. [Google Scholar] [CrossRef] [Green Version]
- Pascovici, D.; Wu, J.X.; McKay, M.J.; Joseph, C.; Noor, Z.; Kamath, K.; Wu, Y.; Ranganathan, S.; Gupta, V.; Mirzaei, M. Clinically relevant post-translational modification analyses—maturing workflows and bioinformatics tools. Int. J. Mol. Sci. 2019, 20, 16. [Google Scholar] [CrossRef] [Green Version]
- Jensen, O.N. Modification-specific proteomics: Characterization of post-translational modifications by mass spectrometry. Curr. Opin. Chem. Biol. 2004, 8, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Mishra, M.; Tiwari, S.; Gomes, A.V. Protein purification and analysis: Next generation western blotting techniques. Expert Rev. Proteom. 2017, 14, 1037–1053. [Google Scholar] [CrossRef] [PubMed]
- Klose, J. From 2-D electrophoresis to proteomics. Electrophoresis 2009, 30. [Google Scholar] [CrossRef] [PubMed]
- Palagi, P.M.; Hernandez, P.; Walther, D.; Appel, R.D. Proteome informatics I: Bioinformatics tools for processing experimental data. Proteomics 2006, 6, 5435–5444. [Google Scholar] [CrossRef]
- Deutsch, E.W.; Lam, H.; Aebersold, R. Data analysis and bioinformatics tools for tandem mass spectrometry in proteomics. Physiol. Genom. 2008, 33, 18–25. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Hou, J.; Tanner, J.J.; Cheng, J. Bioinformatics methods for mass spectrometry-based proteomics data analysis. Int. J. Mol. Sci. 2020, 21, 2873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, M.; Kelleher, N.L. Precision proteomics: The case for high resolution and high mass accuracy. Proc. Natl. Acad. Sci. USA 2008, 105, 18132–18138. [Google Scholar] [CrossRef] [Green Version]
- Gulcicek, E.E.; Colangelo, C.M.; McMurray, W.; Stone, K.; Williams, K.; Wu, T.; Zhao, H.; Spratt, H.; Kurosky, A.; Wu, B. Proteomics and the Analysis of Proteomic Data: An Overview of Current Protein-Profiling Technologies. Curr. Protoc. Bioinforma. 2005, 10. [Google Scholar] [CrossRef] [Green Version]
- Dupree, E.J.; Jayathirtha, M.; Yorkey, H.; Mihasan, M.; Petre, B.A.; Darie, C.C. A critical review of bottom-up proteomics: The good, the bad, and the future of this field. Proteomes 2020, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- Aslam, B.; Basit, M.; Nisar, M.A.; Khurshid, M.; Rasool, M.H. Proteomics: Technologies and their applications. J. Chromatogr. Sci. 2017, 55, 182–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perkins, R.C. Making the case for functional proteomics. In Methods in Molecular Biology; Humana Press Inc.: New York, NY, USA, 2019; Volume 1871, pp. 1–40. [Google Scholar]
- Shin, J.; Lee, W.; Lee, W. Structural proteomics by NMR spectroscopy. Expert Rev. Proteom. 2008, 5, 589–601. [Google Scholar] [CrossRef] [PubMed]
- Boersema, P.J.; Kahraman, A.; Picotti, P. Proteomics beyond large-scale protein expression analysis. Curr. Opin. Biotechnol. 2015, 34, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Banach, P.; Suchy, W.; Dereziński, P.; Matysiak, J.; Kokot, Z.J.; Nowak-Markwitz, E. Mass spectrometry as a tool for biomarkers searching in gynecological oncology. Biomed. Pharm. 2017, 92, 836–842. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, W.; Chen, J. Recent progress in mass spectrometry proteomics for biomedical research. Sci. China Life Sci. 2017, 60, 1093–1113. [Google Scholar] [CrossRef]
- Al-Wajeeh, A.S.; Salhimi, S.M.; Al-Mansoub, M.A.; Khalid, I.A.; Harvey, T.M.; Latiff, A.; Ismail, M.N. Comparative proteomic analysis of different stages of breast cancer tissues using ultra high performance liquid chromatography tandem mass spectrometer. PLoS ONE 2020, 15, e0227404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Chen, J.; Liu, H.; Chen, S.; Zhang, Y.; Li, P.; Thierry-Mieg, D.; Thierry-Mieg, J.; Mattes, W.; Ning, B.; et al. Comprehensive Identification and Characterization of Human Secretome Based on Integrative Proteomic and Transcriptomic Data. Front. Cell Dev. Biol. 2019, 7, 299. [Google Scholar] [CrossRef] [Green Version]
- Smithies, O.; Poulik, M.D. Two-dimensional electrophoresis of serum proteins. Nature 1956, 177, 1033. [Google Scholar] [CrossRef]
- O’Farrell, P.H. High resolution two dimensional electrophoresis of proteins. J. Biol. Chem. 1975, 250, 4007–4021. [Google Scholar] [CrossRef]
- Zong, C.; Young, G.W.; Wang, Y.; Lu, H.; Deng, N.; Drews, O.; Ping, P. Two-dimensional electrophoresis-based characterization of post-translational modifications of mammalian 20S proteasome complexes. Proteomics 2008, 8, 5025–5037. [Google Scholar] [CrossRef] [Green Version]
- Pietal, M.J.; Tuszynska, I.; Bujnicki, J.M. PROTMAP2D: Visualization, comparison and analysis of 2D maps of protein structure. Bioinformatics 2007, 23, 1429–1430. [Google Scholar] [CrossRef] [Green Version]
- López, J.L. Two-dimensional electrophoresis in proteome expression analysis. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2007, 849, 190–202. [Google Scholar] [CrossRef] [PubMed]
- Strohkamp, S.; Gemoll, T.; Habermann, J.K. Possibilities and limitations of 2DE-based analyses for identifying low-abundant tumor markers in human serum and plasma. Proteomics 2016, 16, 2519–2532. [Google Scholar] [CrossRef] [PubMed]
- Kendrick, N.; Darie, C.C.; Hoelter, M.; Powers, G.; Johansen, J. 2D SDS PAGE in Combination with Western Blotting and Mass Spectrometry Is a Robust Method for Protein Analysis with Many Applications. Adv. Exp. Med. Biol. 2019, 1140, 563–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meleady, P. Two-dimensional gel electrophoresis and 2D-DIGE. In Methods in Molecular Biology; Humana Press Inc.: New York, NY, USA, 2018; Volume 1664, pp. 3–14. [Google Scholar]
- Tannu, N.S.; Hemby, S.E. Two-dimensional fluorescence difference gel electrophoresis for comparative proteomics profiling. Nat. Protoc. 2006, 1, 1732–1742. [Google Scholar] [CrossRef] [PubMed]
- Moore, T.; Le, A.; Niemi, A.K.; Kwan, T.; Cusmano-Ozog, K.; Enns, G.M.; Cowan, T.M. A new LC-MS/MS method for the clinical determination of reduced and oxidized glutathione from whole blood. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2013, 929, 51–55. [Google Scholar] [CrossRef]
- Kang, L.; Weng, N.; Jian, W. LC–MS bioanalysis of intact proteins and peptides. Biomed. Chromatogr. 2020, 34, e4633. [Google Scholar] [CrossRef]
- Zhang, J.; Shou, W.; Ogura, T.; Li, S.; Weller, H. Optimization of microflow LC-MS/MS and its utility in quantitative discovery bioanalysis. Bioanalysis 2019, 11, 1117–1127. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.N.; Belanger, D.; Adams, R.P.; Sculley, D. Rapid Prediction of Electron-Ionization Mass Spectrometry Using Neural Networks. ACS Cent. Sci. 2019, 5, 700–708. [Google Scholar] [CrossRef]
- Longuespée, R.; Ly, A.; Casadonte, R.; Schwamborn, K.; Kazdal, D.; Zgorzelski, C.; Bollwein, C.; Kriegsmann, K.; Weichert, W.; Kriegsmann, J.; et al. Identification of MALDI Imaging Proteolytic Peptides Using LC-MS/MS-Based Biomarker Discovery Data: A Proof of Concept. Proteom. Clin. Appl. 2019, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whelan, L.C.; Power, K.A.R.; McDowell, D.T.; Kennedy, J.; Gallagher, W.M. Applications of SELDI-MS technology in oncology. J. Cell. Mol. Med. 2008, 12, 1535–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, S.; Mazumdar, S. Electrospray Ionization Mass Spectrometry: A Technique to Access the Information beyond the Molecular Weight of the Analyte. Int. J. Anal. Chem. 2012, 2012, 1–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seger, C.; Salzmann, L. After another decade: LC–MS/MS became routine in clinical diagnostics. Clin. Biochem. 2020, 82, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, S.; Umemura, H.; Nakayama, T. Current status of matrix-assisted laser desorption/ionization⇓time-of-flight mass spectrometry (MALDI-TOF MS) in clinical diagnostic microbiology. Molecules 2020, 25, 4775. [Google Scholar] [CrossRef] [PubMed]
- Swiatly, A.; Horala, A.; Hajduk, J.; Matysiak, J.; Nowak-Markwitz, E.; Kokot, Z.J. MALDI-TOF-MS analysis in discovery and identification of serum proteomic patterns of ovarian cancer. BMC Cancer 2017, 17, 1–9. [Google Scholar] [CrossRef]
- Liu, C. The application of SELDI-TOF-MS in clinical diagnosis of cancers. J. Biomed. Biotechnol. 2011, 2011. [Google Scholar] [CrossRef] [Green Version]
- Poon, T.C.W. Opportunities and limitations of SELDI-TOF-MS in biomedical research: Practical advices. Expert Rev. Proteom. 2007, 4, 51–65. [Google Scholar] [CrossRef]
- Ho, C.S.; Lam, C.W.K.; Chan, M.H.M.; Cheung, R.C.K.; Law, L.K.; Lit, L.C.W.; Ng, K.F.; Suen, M.W.M.; Tai, H.L. Electrospray ionisation mass spectrometry: Principles and clinical applications. Clin. Biochem. Rev. 2003, 24, 3–12. [Google Scholar]
- Leopold, J.; Popkova, Y.; Engel, K.M.; Schiller, J. Recent developments of useful MALDI matrices for the mass spectrometric characterization of lipids. Biomolecules 2018, 8, 173. [Google Scholar] [CrossRef] [Green Version]
- Dingle, T.C.; Butler-Wu, S.M. MALDI-TOF mass spectrometry for microorganism identification. Clin. Lab. Med. 2013, 33, 589–609. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.R.; McWhinney, B.C. Quadrupole Time-of-Flight Mass Spectrometry: A Paradigm Shift in Toxicology Screening Applications. Clin. Biochem. Rev. 2019, 40, 135–146. [Google Scholar] [CrossRef]
- Tsutsui, M.; Yokota, K.; Arima, A.; He, Y.; Kawai, T. Solid-State Nanopore Time-of-Flight Mass Spectrometer. ACS Sens. 2019, 4, 2974–2979. [Google Scholar] [CrossRef] [PubMed]
- Erne, R.; Bernhard, L.; Kawecki, M.; Baumgartner, M.R.; Kraemer, T. Using time-of-flight secondary ion mass spectrometry (ToF-SIMS) and matrix assisted laser desorption/ionization mass spectrometry (MALDI-MS) for investigations on single hair samples to solve the contamination: Versus incorporation issue of hair analysis in the case of cocaine and methadone. Analyst 2020, 145, 4906–4919. [Google Scholar] [CrossRef]
- Unsihuay, D.; Mesa Sanchez, D.; Laskin, J. Quantitative Mass Spectrometry Imaging of Biological Systems. Annu. Rev. Phys. Chem. 2020, 72, 307–329. [Google Scholar] [CrossRef] [PubMed]
- Chahrour, O.; Cobice, D.; Malone, J. Stable isotope labelling methods in mass spectrometry-based quantitative proteomics. J. Pharm. Biomed. Anal. 2015, 113, 2–20. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Zhao, B.; Weng, Y.; Shan, Y.; Li, X.; Hu, Y.; Liang, Z.; Yuan, H.; Zhang, L.; Zhang, Y. Site-Specific Quantification of Persulfidome by Combining an Isotope-Coded Affinity Tag with Strong Cation-Exchange-Based Fractionation. Anal. Chem. 2019, 91, 14860–14864. [Google Scholar] [CrossRef]
- Wang, X.; He, Y.; Ye, Y.; Zhao, X.; Deng, S.; He, G.; Zhu, H.; Xu, N.; Liang, S. SILAC-based quantitative MS approach for real-time recording protein-mediated cell-cell interactions. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.E.; Kim, E.K.; Song, M.J.; Kim, T.Y.; Jang, H.H.; Kang, D. Silac-based quantitative proteomic analysis of oxaliplatin-resistant pancreatic cancer cells. Cancers 2021, 13, 724. [Google Scholar] [CrossRef]
- Xu, Y.; Li, X.; Su, X. ITRAQ-based proteomics analysis of the therapeutic effects of combined anticancer bioactive peptides and oxaliplatin on gastric cancer cells. Oncol. Rep. 2020, 43, 201–217. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.C.Y.; Zhou, L.; Chan, E.C.Y. The isotope-coded affinity tag method for quantitative protein profile comparison and relative quantitation of cysteine redox modifications. Curr. Protoc. Protein Sci. 2015, 2015, 23.2.1–23.2.19. [Google Scholar] [CrossRef] [PubMed]
- Topf, U.; Suppanz, I.; Samluk, L.; Wrobel, L.; Böser, A.; Sakowska, P.; Knapp, B.; Pietrzyk, M.K.; Chacinska, A.; Warscheid, B. Quantitative proteomics identifies redox switches for global translation modulation by mitochondrially produced reactive oxygen species. Nat. Commun. 2018, 9, 1–17. [Google Scholar] [CrossRef]
- Merrill, A.E.; Coon, J.J. Quantifying proteomes and their post-translational modifications by stable isotope label-based mass spectrometry. Curr. Opin. Chem. Biol. 2013, 17, 779–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiley, C.D.; Liu, S.; Limbad, C.; Zawadzka, A.M.; Beck, J.; Demaria, M.; Artwood, R.; Alimirah, F.; Lopez-Dominguez, J.A.; Kuehnemann, C.; et al. SILAC Analysis Reveals Increased Secretion of Hemostasis-Related Factors by Senescent Cells. Cell Rep. 2019, 28, 3329–3337.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rauniyar, N.; Yates, J.R. Isobaric labeling-based relative quantification in shotgun proteomics. J. Proteome Res. 2014, 13, 5293–5309. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, J.A.; Weinstein, J.N. Biomarkers in cancer staging, prognosis and treatment selection. Nat. Rev. Cancer 2005, 5, 845–856. [Google Scholar] [CrossRef]
- Thun, M.J.; DeLancey, J.O.; Center, M.M.; Jemal, A.; Ward, E.M. The global burden of cancer: Priorities for prevention. Carcinogenesis 2009, 31, 100–110. [Google Scholar] [CrossRef] [Green Version]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Rusling, J.F.; Kumar, C.V.; Gutkind, J.S.; Patel, V. Measurement of biomarker proteins for point-of-care early detection and monitoring of cancer. Analyst 2010, 135, 2496–2511. [Google Scholar] [CrossRef] [Green Version]
- Montaner, J.; Ramiro, L.; Simats, A.; Tiedt, S.; Makris, K.; Jickling, G.C.; Debette, S.; Sanchez, J.C.; Bustamante, A. Multilevel omics for the discovery of biomarkers and therapeutic targets for stroke. Nat. Rev. Neurol. 2020, 16, 247–264. [Google Scholar] [CrossRef] [PubMed]
- Reymond, M.A.; Schlegel, W. Proteomics in Cancer. Adv. Clin. Chem. 2007, 44, 103–142. [Google Scholar]
- Huang, Z.; Ma, L.; Huang, C.; Li, Q.; Nice, E.C. Proteomic profiling of human plasma for cancer biomarker discovery. Proteomics 2017, 17, 1600240. [Google Scholar] [CrossRef] [PubMed]
- La Thangue, N.B.; Kerr, D.J. Predictive biomarkers: A paradigm shift towards personalized cancer medicine. Nat. Rev. Clin. Oncol. 2011, 8, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Ren, A.H.; Fiala, C.A.; Diamandis, E.P.; Kulasingam, V. Pitfalls in cancer biomarker discovery and validation with emphasis on circulating tumor DNA. Cancer Epidemiol. Biomark. Prev. 2020, 29, 2568–2574. [Google Scholar] [CrossRef] [Green Version]
- Sobsey, C.A.; Ibrahim, S.; Richard, V.R.; Gaspar, V.; Mitsa, G.; Lacasse, V.; Zahedi, R.P.; Batist, G.; Borchers, C.H. Targeted and Untargeted Proteomics Approaches in Biomarker Development. Proteomics 2020, 20, 1900029. [Google Scholar] [CrossRef]
- Zhou, B.X.; Li, Y. Significance of desmoglein-2 on cell malignant behaviors via mediating MAPK signaling in cervical cancer. Kaohsiung J. Med. Sci. 2020, 36, 336–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Chen, G.; Cai, H.; Wang, X.; Song, K.; Liu, L.; Qiu, T.; He, Y. Aberrantly enhanced melanoma-associated antigen (MAGE)-A3 expression facilitates cervical cancer cell proliferation and metastasis via actuating Wnt signaling pathway. Biomed. Pharm. 2020, 122, 109710. [Google Scholar] [CrossRef]
- Higareda-Almaraz, J.C.; del Rocío Enríquez-Gasca, M.; Hernández-Ortiz, M.; Resendis-Antonio, O.; Encarnación-Guevara, S. Proteomic patterns of cervical cancer cell lines, a network perspective. BMC Syst. Biol. 2011, 5, 96. [Google Scholar] [CrossRef] [Green Version]
- Leung, T.H.Y.; Tang, H.W.M.; Siu, M.K.Y.; Chan, D.W.; Chan, K.K.L.; Cheung, A.N.Y.; Ngan, H.Y.S. CD71+ population enriched by HPV-E6 protein promotes cancer aggressiveness and radioresistance in cervical cancer cells. Mol. Cancer Res. 2019, 17, 1867–1880. [Google Scholar] [CrossRef] [Green Version]
- Pappa, K.I.; Christou, P.; Xholi, A.; Mermelekas, G.; Kontostathi, G.; Lygirou, V.; Makridakis, M.; Zoidakis, J.; Anagnou, N.P. Membrane proteomics of cervical cancer cell lines reveal insights on the process of cervical carcinogenesis. Int. J. Oncol. 2018, 53, 2111–2122. [Google Scholar] [CrossRef] [Green Version]
- Xia, C.; Yang, F.; He, Z.; Cai, Y. iTRAQ-based quantitative proteomic analysis of the inhibition of cervical cancer cell invasion and migration by metformin. Biomed. Pharm. 2020, 123, 109762. [Google Scholar] [CrossRef] [PubMed]
- Zhan, F.; Zhong, Y.; Qin, Y.; Li, L.; Wu, W.; Yao, M. SND1 facilitates the invasion and migration of cervical cancer cells by Smurf1-mediated degradation of FOXA2. Exp. Cell Res. 2020, 388. [Google Scholar] [CrossRef]
- Zhang, B.; Chen, X.; Lin, Y.; Wang, S.; Bao, Q. Occludin protein expression in human cervical cancer and its association with patient’s clinical characteristics. J. Cancer Res. 2018, 14, 124–127. [Google Scholar] [CrossRef]
- Qi, Y.X.; Liu, K.; Yin, J.; Li, L. Evaluation of short- and long-term efficacy of chemoradiotherapy for advanced cervical cancer using HSP70 protein combined with multimodal MRI. J. Cell. Biochem. 2018, 119, 3017–3029. [Google Scholar] [CrossRef] [PubMed]
- Song, J.Y.; Bae, H.S.; Koo, D.H.; Lee, J.K.; Jung, H.H.; Lee, K.W.; Lee, N.W. Candidates for tumor markers of cervical cancer discovered by proteomic analysis. J. Korean Med. Sci. 2012, 27, 1479–1485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Y.; Yang, M.; She, S.; Min, H.; Xv, X.; Ran, X.; Wu, Y.; Wang, W.; Wang, L.; Yi, L.; et al. ITRAQ-based quantitative proteomic analysis of cervical cancer. Int. J. Oncol. 2015, 46, 1748–1758. [Google Scholar] [CrossRef]
- Xu, Q.; Chen, C.; Liu, B.; Lin, Y.; Zheng, P.; Zhou, D.; Xie, Y.; Lin, Y.; Guo, C.; Liu, J.; et al. Association of iRhom1 and iRhom2 expression with prognosis in patients with cervical cancer and possible signaling pathways. Oncol. Rep. 2020, 43, 41–54. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.W.; Cho, H.; Ylaya, K.; Kitano, H.; Chung, J.Y.; Hewitt, S.M.; Kim, J.H. Bcl-2-like protein 11 (BIM) expression is associated with favorable prognosis for patients with cervical cancer. Anticancer Res. 2017, 37, 4873–4879. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.J.; Li, H.L.; Guo, S.J.; Ma, H.; Liu, S.J.; Liu, D.; Xue, F.X. The increased PTK7 expression is a malignant factor in cervical cancer. Dis. Markers 2019, 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, S.-M.; Min, H.-J.; Ding, G.H.; Kwak, S.-Y.; Cho, Y.-L.; Nam, K.-H.; Park, C.H.; Kim, Y.-W.; Kim, C.-K.; Han, B.-D.; et al. Protein Expression Profile using Two-Dimensional Gel Analysis in Squamous Cervical Cancer Patients. Cancer Res. Treat. 2006, 38, 99. [Google Scholar] [CrossRef]
- Gu, Y.; Wu, S.L.; Meyer, J.L.; Hancock, W.S.; Burg, L.J.; Linder, J.; Hanlon, D.W.; Karger, B.L. Proteomic analysis of high-grade dysplastic cervical cells obtained from ThinPrep slides using laser capture microdissection and mass spectrometry. J. Proteome Res. 2007, 6, 4256–4268. [Google Scholar] [CrossRef]
- Zhu, X.; Lv, J.; Yu, L.; Zhu, X.; Wu, J.; Zou, S.; Jiang, S. Proteomic identification of differentially-expressed proteins in squamous cervical cancer. Gynecol. Oncol. 2009, 112, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; He, Y.; Wang, X.L.; Zhang, Y.X.; Wu, Y.M. Differentially expressed proteins among normal cervix, cervical intraepithelial neoplasia and cervical squamous cell carcinoma. Clin. Transl. Oncol. 2015, 17, 620–631. [Google Scholar] [CrossRef] [PubMed]
- Güzel, C.; Govorukhina, N.I.; Wisman, G.B.A.; Stingl, C.; Dekker, L.J.M.; Klip, H.G.; Hollema, H.; Guryev, V.; Horvatovich, P.L.; van der Zee, A.G.J.; et al. Proteomic alterations in early stage cervical cancer. Oncotarget 2018, 9, 18128–18147. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Acosta, P.; Molano, M.; Morales, N.; Acosta, J.; González-Prieto, C.; Mayorga, D.; Buitrago, L.; Gamboa, O.; Carlos Mejía, J.; Castro, J.; et al. hTERT Protein Expression in Cytoplasm and Nucleus and its Association with HPV Infection in Patients with Cervical Cancer. Cancer Genom. Proteom. 2020, 17, 615–625. [Google Scholar] [CrossRef]
- Anggraeni, T.D.; Rustamadji, P.; Aziz, M.F. Fas ligand (FasL) in association with Tumor-Infiltrating Lymphocytes (TILs) in early stage cervical cancer. Asian Pac. J. Cancer Prev. 2020, 21, 831–835. [Google Scholar] [CrossRef] [PubMed]
- Serafín-Higuera, I.; Garibay-Cerdenares, O.L.; Illades-Aguiar, B.; Flores-Alfaro, E.; Jiménez-López, M.A.; Sierra-Martínez, P.; del Carmen Alarcón-Romero, L. Differential proteins among normal cervix cells and cervical cancer cells with HPV-16 infection, through mass spectrometry-based Proteomics (2D-DIGE) in women from Southern México. Proteome Sci. 2016, 14, 10. [Google Scholar] [CrossRef] [Green Version]
- Mohd Isa, S.A.; Md Salleh, M.S.; Ismail, M.P.; Hairon, S.M. ADAM9 expression in uterine cervical cancer and its associated factors. Asian Pac. J. Cancer Prev. 2019, 20, 1081–1087. [Google Scholar] [CrossRef] [Green Version]
- Blancas, S.; Medina-Berlanga, R.; Ortíz-García, L.; Loredo-Ramírez, A.; Santos, L. Protein Expression Analysis in Uterine Cervical Cancer for Potential Targets in Treatment. Pathol. Oncol. Res. 2019, 25, 493–501. [Google Scholar] [CrossRef]
- Liu, L.; Xu, D.; Yang, S.; Li, X. Ebp1 protein expression in cervical cancer tissue and its significance. Genet. Mol. Res. 2015, 14, 5496–5500. [Google Scholar] [CrossRef] [PubMed]
- Van Raemdonck, G.A.A.; Tjalma, W.A.A.; Coen, E.P.; Depuydt, C.E.; Van Ostade, X.W.M. Identification of protein biomarkers for cervical cancer using human cervicovaginal fluid. PLoS ONE 2014, 9, e106488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, J.L.V.; Smith, C.R.; Diamandis, E.P. Proteomic analysis of human cervico-vaginal fluid. J. Proteome Res. 2007, 6, 2859–2865. [Google Scholar] [CrossRef]
- Zegels, G.; Van Raemdonck, G.A.A.; Coen, E.P.; Tjalma, W.A.A.; Van Ostade, X.W.M. Comprehensive proteomic analysis of human cervical-vaginal fluid using colposcopy samples. Proteome Sci. 2009, 7, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Qing, S.; Tulake, W.; Ru, M.; Li, X.; Yuemaier, R.; Lidifu, D.; Rouzibilali, A.; Hasimu, A.; Yang, Y.; Rouziahong, R.; et al. Proteomic identification of potential biomarkers for cervical squamous cell carcinoma and human papillomavirus infection. Tumor Biol. 2017, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Y.; Kim, S.C.; Kim, H.J.; Ju, W.; Kim, Y.H.; Kim, H.J. Use of protein-based biomarkers of exfoliated cervical cells for primary screening of cervical cancer. Arch. Pharm. Res. 2018, 41, 438–449. [Google Scholar] [CrossRef] [PubMed]
- Lokamani, I.; Looi, M.L.; Md Ali, S.A.; Mohd Dali, A.Z.H.; Ahmad Annuar, M.A.; Jamal, R. Gelsolin and ceruloplasmin as potential predictive biomarkers for cervical cancer by 2D-DIGE proteomics analysis. Pathol. Oncol. Res. 2014, 20, 119–129. [Google Scholar] [CrossRef]
- Qiu, F.; Chen, F.; Liu, D.; Xu, J.; He, J.; Xiao, J.; Cao, L.; Huang, X. LC-MS/MS-based screening of new protein biomarkers for cervical precancerous lesions and cervical cancer. Nan Fang Yi Ke Da Xue Xue Bao 2019, 39, 13–22. [Google Scholar] [CrossRef]
- Chokchaichamnankit, D.; Watcharatanyatip, K.; Subhasitanont, P.; Weeraphan, C.; Keeratichamroen, S.; Sritana, N.; Kantathavorn, N.; Ayudthaya, P.D.N.; Saharat, K.; Chantaraamporn, J.; et al. Urinary biomarkers for the diagnosis of cervical cancer by quantitative label-free mass spectrometry analysis. Oncol. Lett. 2019, 17, 5453–5468. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Chen, J.; Luan, T.; Chu, C.; Wu, W.; Zhu, Y.; Gu, Y. Proteomic analysis of human cervical adenocarcinoma mucus to identify potential protein biomarkers. PeerJ 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Hwang, Y.J.; Lee, S.P.; Kim, S.Y.; Choi, Y.H.; Kim, M.J.; Lee, C.H.; Lee, J.Y.; Kim, D.Y. Expression of heat shock protein 60 kDa is upregulated in cervical cancer. Yonsei Med. J. 2009, 50, 399–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, C.H.; Chung, J.Y.; Kang, J.H.; Paik, E.S.; Lee, Y.Y.; Park, W.; Byeon, S.J.; Chung, E.J.; Kim, B.G.; Hewitt, S.M.; et al. Chemoradiotherapy response prediction model by proteomic expressional profiling in patients with locally advanced cervical cancer. Gynecol. Oncol. 2020, 157, 437–443. [Google Scholar] [CrossRef]
- Guo, X.; Hao, Y.; Kamilijiang, M.; Hasimu, A.; Yuan, J.; Wu, G.; Reyimu, H.; Kadeer, N.; Abudula, A. Potential predictive plasma biomarkers for cervical cancer by 2D-DIGE proteomics and Ingenuity Pathway Analysis. Tumor Biol. 2015, 36, 1711–1720. [Google Scholar] [CrossRef]
- Berger, M.F.; Mardis, E.R. The emerging clinical relevance of genomics in cancer medicine. Nat. Rev. Clin. Oncol. 2018, 15, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Hiratani, I.; Takebayashi, S.-I.; Lu, J.; Gilbert, D.M. Replication timing and transcriptional control: Beyond cause and effect-part II. Curr. Opin. Genet. Dev. 2009, 19, 142–149. [Google Scholar] [CrossRef] [Green Version]
- Martisova, A.; Holcakova, J.; Izadi, N.; Sebuyoya, R.; Hrstka, R.; Bartosik, M. DNA methylation in solid tumors: Functions and methods of detection. Int. J. Mol. Sci. 2021, 22, 4247. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research, N.; Burk, R.D.; Chen, Z.; Saller, C.; Tarvin, K.; Carvalho, A.L.; Scapulatempo-Neto, C.; Silveira, H.C.; Fregnani, J.H.; Creighton, C.J.; et al. Integrated genomic and molecular characterization of cervical cancer. Nature 2017, 543, 378. Available online: https://www.nature.com/articles/nature21386#supplementary-information (accessed on 30 May 2021). [CrossRef]
- De Vuyst, H.; Franceschi, S.; Plummer, M.; Mugo, N.R.; Sakr, S.R.; Meijer, C.J.L.M.; Heideman, D.A.M.; Tenet, V.; Snijders, P.J.F.; Hesselink, A.T.; et al. Methylation levels of CADM1, MAL, and MIR124-2 in cervical scrapes for triage of HIV-infected, high-risk HPV-positive women in Kenya. J. Acquir. Immune Defic. Syndr. 2015, 70, 311–318. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Li, Y.; Du, P.; Zhang, X.; Li, X.; Cheng, G. Hypomethylation of the lncRNA SOX21-AS1 has clinical prognostic value in cervical cancer. Life Sci. 2019, 233. [Google Scholar] [CrossRef]
- Gong, J.M.; Shen, Y.; Shan, W.W.; He, Y.X. The association between MTHFR polymorphism and cervical cancer. Sci. Rep. 2018, 8, 7244. [Google Scholar] [CrossRef]
- Audirac-Chalifour, A.; Torres-Poveda, K.; Bahena-Román, M.; Téllez-Sosa, J.; Martínez-Barnetche, J.; Cortina-Ceballos, B.; López-Estrada, G.; Delgado-Romero, K.; Burguete-García, A.I.; Cantú, D.; et al. Cervical Microbiome and Cytokine Profile at Various Stages of Cervical Cancer: A Pilot Study. PLoS ONE 2016, 11, e0153274. [Google Scholar] [CrossRef] [PubMed]
- Usyk, M.; Zolnik, C.P.; Castle, P.E.; Porras, C.; Herrero, R.; Gradissimo, A.; Gonzalez, P.; Safaeian, M.; Schiffman, M.; Burk, R.D. Cervicovaginal microbiome and natural history of HPV in a longitudinal study. PLoS Pathog. 2020, 16, e1008376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, M.; Seo, S.S.; Kim, M.K.; Lee, D.O.; Lim, M.C. Compositional and functional differences between microbiota and cervical carcinogenesis as identified by shotgun metagenomic sequencing. Cancers 2019, 11, 309. [Google Scholar] [CrossRef] [Green Version]
- Oh, H.Y.; Seo, S.S.; Kong, J.S.; Lee, J.K.; Kim, M.K. Association between obesity and cervical microflora dominated by lactobacillus iners in Korean women. J. Clin. Microbiol. 2015, 53, 3304–3309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitra, A.; Grossman Biegert, G.W.; Delgado, A.Y.; Karpinets, T.V.; Solley, T.N.; Mezzari, M.P.; Yoshida-Court, K.; Petrosino, J.F.; Mikkelson, M.D.; Lin, L.; et al. Microbial Diversity and Composition Is Associated with Patient-Reported Toxicity during Chemoradiation Therapy for Cervical Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2020, 107, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Li, X.; Tian, R.; Gao, H.; Yang, Y.; Williams, B.R.G.; Gantier, M.P.; McMillan, N.A.J.; Xu, D.; Hu, Y.; Gao, Y. Identification of a histone family gene signature for predicting the prognosis of cervical cancer patients. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Liu, H.; Yang, Y.; Wang, X.; Liu, P.; Li, Y.; Meyers, C.; Banerjee, N.S.; Wang, H.K.; Cam, M.; et al. Genome-wide profiling of cervical RNA-binding proteins identifies human papillomavirus regulation of rnaRNASEH2A expression by viral e7 and e2f1. mBio 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brant, A.C.; Menezes, A.N.; Felix, S.P.; de Almeida, L.M.; Sammeth, M.; Moreira, M.A.M. Characterization of HPV integration, viral gene expression and E6E7 alternative transcripts by RNA-Seq: A descriptive study in invasive cervical cancer. Genomics 2019, 111, 1853–1861. [Google Scholar] [CrossRef]
- Hua, C.; Zhu, J.; Zhang, B.; Sun, S.; Song, Y.; van der Veen, S.; Cheng, H. Digital RNA Sequencing of Human Epidermal Keratinocytes Carrying Human Papillomavirus Type 16 E7. Front. Genet. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Tokarz, J.; Adamski, J.; Rižner, T.L. Metabolomics for diagnosis and prognosis of uterine diseases? A systematic review. J. Pers. Med. 2020, 10, 294. [Google Scholar] [CrossRef]
- Khan, I.; Nam, M.; Kwon, M.; Seo, S.S.; Jung, S.; Han, J.S.; Hwang, G.S.; Kim, M.K. LC/MS-based polar metabolite profiling identified unique biomarker signatures for cervical cancer and cervical intraepithelial neoplasia using global and targeted metabolomics. Cancers 2019, 11, 511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abudula, A.; Rouzi, N.; Xu, L.; Yang, Y.; Hasimu, A. Tissue-based metabolomics reveals potential biomarkers for cervical carcinoma and HPV infection. Bosn. J. Basic Med. Sci. 2020, 20, 78–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paraskevaidi, M.; Cameron, S.J.S.; Whelan, E.; Bowden, S.; Tzafetas, M.; Mitra, A.; Semertzidou, A.; Athanasiou, A.; Bennett, P.R.; MacIntyre, D.A.; et al. Laser-assisted rapid evaporative ionisation mass spectrometry (LA-REIMS) as a metabolomics platform in cervical cancer screening. EBioMedicine 2020, 60. [Google Scholar] [CrossRef] [PubMed]
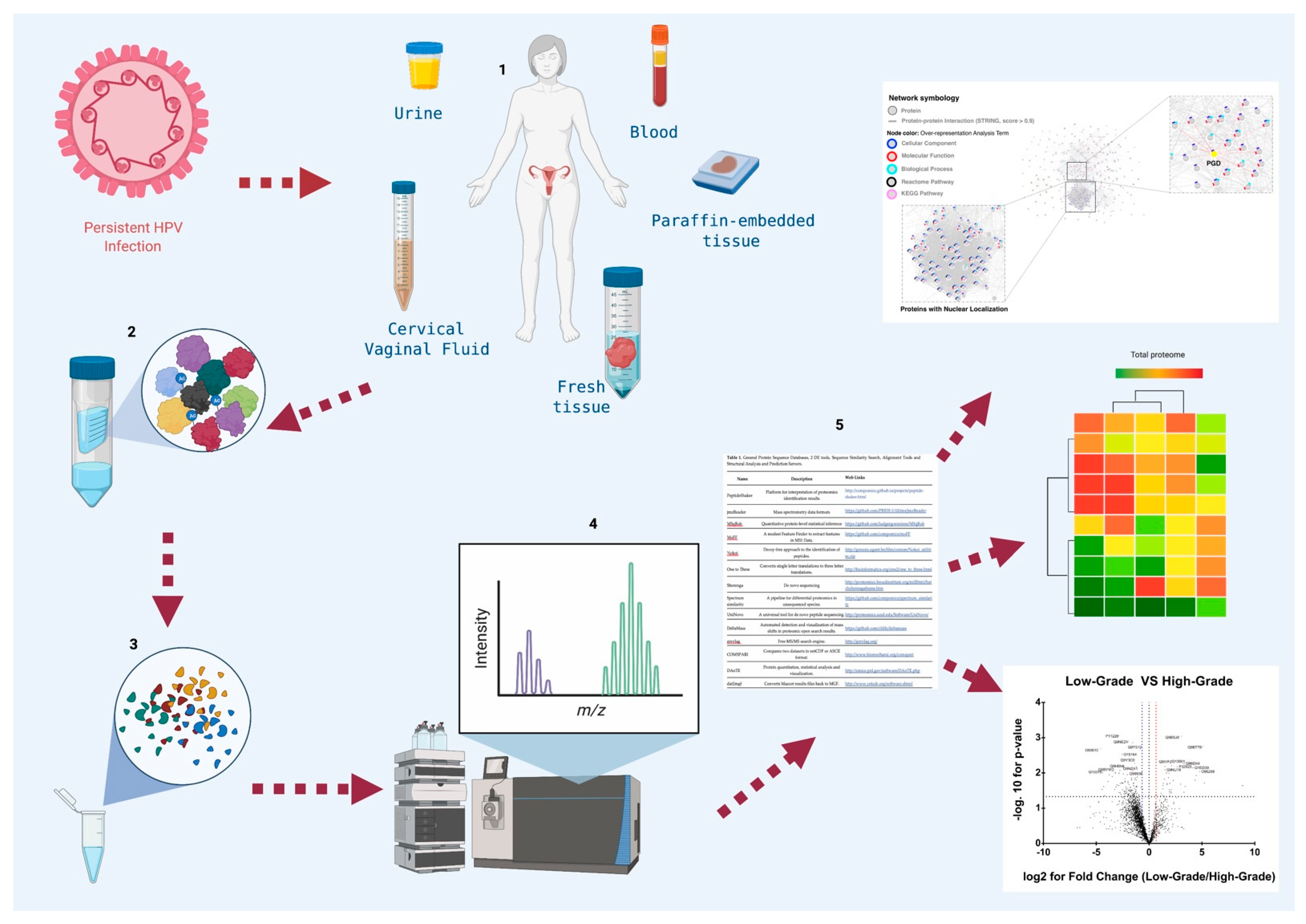

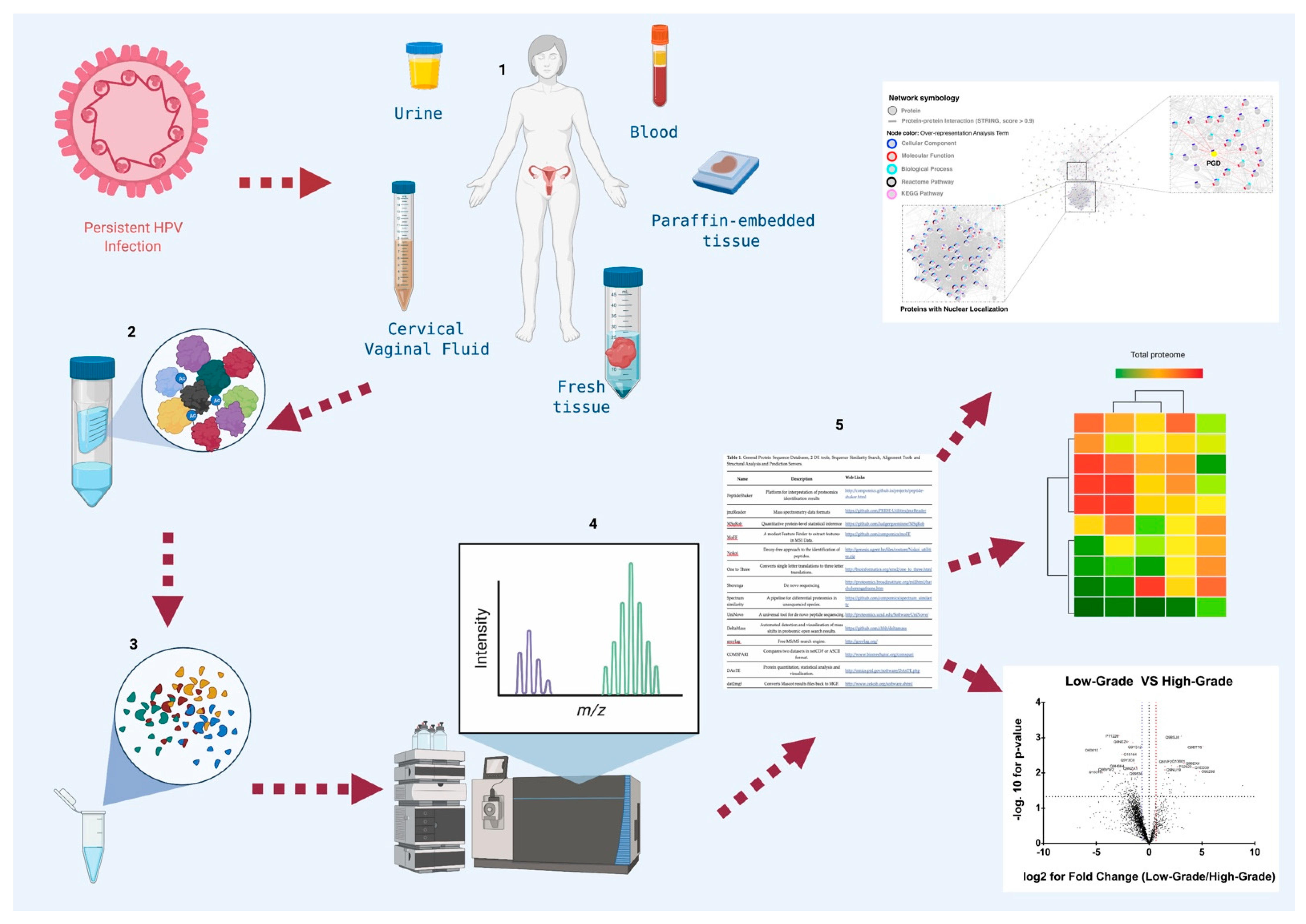{kind=link}
| Name | Description | Web Links |
|---|---|---|
| PeptideShaker | Platform for interpretation of proteomics identification results | http://compomics.github.io/projects/peptide-shaker.html |
| jmzReader | Mass spectrometry data formats | https://github.com/PRIDE-Utilities/jmzReader |
| MSqRob | Quantitative protein-level statistical inference | https://github.com/ludgergoeminne/MSqRob |
| MoFF | A modest Feature Finder to extract features in MS1 Data. | https://github.com/compomics/moFF |
| Nokoi | Decoy-free approach to the identification of peptides. | http://genesis.ugent.be/files/costore/Nokoi_utilities.zip |
| One to Three | Converts single-letter translations to three-letter translations. | http://bioinformatics.org/sms2/one_to_three.html |
| Sherenga | De novo sequencing | http://proteomics.broadinstitute.org/millhtml/batchsherengaframe.htm |
| Spectrum similarity | A pipeline for differential proteomics in unsequenced species. | https://github.com/compomics/spectrum_similarity |
| UniNovo | A universal tool for de novo peptide sequencing. | http://proteomics.ucsd.edu/Software/UniNovo/ |
| DeltaMass | Automated detection and visualization of mass shifts in proteomic open search results. | https://github.com/chhh/deltamass |
| greylag | Free MS/MS search engine. | http://greylag.org/ |
| COMSPARI | Compares two datasets in netCDF or ASCII format. | http://www.biomechanic.org/comspari |
| DAnTE | Protein quantitation, statistical analysis and visualization. | http://omics.pnl.gov/software/DAnTE.php |
| dat2mgf | Converts Mascot results files back to MGF. | http://www.ce4csb.org/software.shtml |
| DataAnalysis2TPP | Converts MGF from Bruker DataAnalysis to TPP-friendly format for use with XPRESS and ASAPRatio | http://www.ms-utils.org/DataAnalysis2TPP.html |
| MS-Spectre | Quantitiave analysis of multiple LC-MS(/MS) analyses in mzXML. | http://sourceforge.net/projects/ms-spectre |
| msaccess | Creates a pseudo-2D-gel representation. | http://proteowizard.sourceforge.net/tools/msaccess.html |
| mspire_mspire-sequest | MS data processing in Ruby, including mzML reader/writer/converter, ‘‘in-silico’’ digestion, isotopic pattern calculation etc. | http://mspire.rubyforge.org |
| Multi-Q | Tool for multiplexed iTRAQ-based quantitation. | http://ms.iis.sinica.edu.tw/Multi-Q/ |
| mzBruker | Converts analysis.baf files from Bruker into mzXML files. This software requires the CDAL library from Bruker | http://tools.proteomecenter.org/wiki/index.php?title=Software:mzBruker |
| MzJava | Library for the analysis of mass spectrometry data from large scale proteomic and glycomics experiments | http://mzjava.expasy.org/ |
| PROTICdb | Proteomic database to store, track, query and compare proteome data. | http://pappso.inra.fr/bioinfo/proticdb/ |
| ProtMAX | Fast and robust software tool for analyzing large shotgun proteomics mass spectrometry data sets. | http://www.univie.ac.at/mosys/software.html |
| MassWolf | Converts MassLynx format to mzXML | http://tools.proteomecenter.org/MassWolf.php |
| mres2x | A tool to process MASCOT results. | https://sourceforge.net/projects/protms/files/mres2x/ |
| mzXML2 | Converts mzXML and mzML files to SEQUEST dta, MASCOT mgf, and Micromass pkl files. | http://tools.proteomecenter.org/wiki/index.php?title=Software:MzXML2Search |
| nontarget | R function for compound, adducts and ion series detection using isotopic distributions. | http://cran.r-project.org/web/packages/nontarget/index.html |
| PAPPSO | Southwest Paris proteomic analysis platform. | http://pappso.inra.fr/bioinfo/ |
| PEAKS De Novo | Integrated ‘‘de novo’’ peptide sequencing, PTM finder, and homology search (demo available). | http://www.bioinfor.com/peaks/features/denovo.html |
| QuPE | Web application to support the analysis and integration of even complex mass spectrometry-based proteomics experiments | https://qupe.cebitec.uni-bielefeld.de |
| Skyline | Builds SRM/MRM methods and analyzes resulting data. | https://brendanx-uw1.gs.washington.edu/labkey/project/home/software/Skyline/begin.view |
| unfinnigan | Reading Thermo raw files without MsFileReader. | https://code.google.com/p/unfinnigan/ |
| ThermoRawFileParser | Open-source, crossplatform tool that converts Thermo RAW files into open file formats such as MGF and to the HUPO-PSI standard file format mzML | https://github.com/compomics/ThermoRawFileParser |
| BatchServer | Web application for batch effect evaluation, visualization and correction. | https://lifeinfo.shinyapps.io/batchserver/ |
| psims | Prototype work for a unified API for writing PSIMS standardized XML documents, currently just mzML and MzIdentML. | https://github.com/mobiusklein/psims |
| Compomics sigpep | Predicting peptide signatures for targeted proteomics. | https://github.com/compomics/compomics-sigpep |
| DACSIM | De novo peptide sequencing based on a divide-and-conquer algorithm and peptide tandem spectrum simulation | https://pubs.acs.org/doi/abs/10.1021/ac0491206 |
| Dinosaur | Peptide feature detector for LC-MS data | https://github.com/fickludd/dinosaur/ |
| Isoform Resolver | A peptide-centric algorithm for protein inference. | https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3167374/ |
| Jtraml | Java implementation of the PSI-MS Transitions Markup Language (TraML) specification. | https://github.com/compomics/jtraml |
| Protein Disorder | List is a list of Protein Disorder Predictors | http://www.disprot.org/predictors.php |
| Protein | Monthly review written by the Swiss-Prot team of the Swiss Institute of Bioinformatics. Spotlight articles describe a specific protein or family of proteins on an informal tone. | https://web.expasy.org/spotlight/ |
| Proteins API | Provide sequence feature annotations from UniProtKB, variation data from UniProtKB and mapped from LSS. | http://www.ebi.ac.uk/proteins/api |
| Proteinspector | Analysis of mass spectrometry proteomics quality control metrics. | https://bitbucket.org/proteinspector/qc_analysis/ |
| raw2mzDB | An extension of the ProteoWizard framework enabling the support of the mzDB format. | https://github.com/mzdb/pwiz-mzdb |
| SeqMS | De novo sequencing by tandem mass spectrometry | https://www.ncbi.nlm.nih.gov/pubmed/10870956/ |
| msInspect | A software platform for rapidly creating computational tools for mass spectrometry-based proteomics. | https://github.com/dhmay/msInspect |
| Peptizer | Automating manual validation of MS/MS search results. | https://github.com/compomics/peptizer |
| NIBR 2D- | World-wide gel-based proteomics database. | http://www.expasy.org/world-2dpage/ |
| OMSSA Parser | Java based parser for Open Mass Spectrometry Search Algorithm; omx files. | http://compomics.github.io/projects/omssa-parser.html |
| Proteins | Sample | Assay/Technique | OE/SE | Conclusion | Reference |
|---|---|---|---|---|---|
| CERVICAL CANCER CELL LINE | |||||
| DSG2 | HeLa, C33A | WB, qPCR | SE | Possible therapeutic target | [101] |
| MAGE-A3 | HeLA, SiHa, C33A, End1/E6E7 | WB | OE | Possible therapeutic target, prognostic | [102] |
| 14- 3-3ζ | HeLa, CaLo, SiHa, CasKi, ViBo, C-33A | 2D, MALDI-TOF-MS | OE | 14- 3-3ζ belong to “central core of CC” | [103] |
| CD71+, HPV-E6 | C33A, C4-1, CaSki | FC, Microarrays WB | OE | Possible therapeutic target | [104] |
| TMX2, FAM120A, CLPTM1, CKAP5, NCSTN | HeLa, SiHa, C33A | LC-MS/MS | OE | Possible therapeutic target | [105] |
| TGFβ-1, CCPG1, LGMN, SLC38A2, TRIM26, MTR, ATP6AP1, CIRBP, PTP4A1, CYR61, IGFBP7 | HeLa, SiHa | WB iTRAQ-MS | SE:TGFβ1,CCPG1LGMN, SLC38A2, TRIM26, MTR, ATP6AP1, CIRBP, PTP4A1 OE:CYR61, IGFBP7 | Possible therapeutic target | [106] |
| FRESH TISSUE | |||||
| SND1 | CC and control tissue | qRT-PCR WB | OE | Possible therapeutic target, prognostic | [107] |
| Occludin | CC and control tissue | IHC | OE | Diagnostic | [108] |
| HSP70 | CC and control tissue | IHC | OE | Possible therapeutic target, diagnostic | [109] |
| AIF-1, ALP-2, B-FABP, NCK-1, ICA69, PRSS1, CDK4. | CC and control tissue | ESI-MALDI-TOF-MS RT-PCR WB, IHC | OE | Diagnostic | [110] |
| G6PD | CC and control tissue | iTRAQ NanoLC-MS/MS qRT-PCR. WB Microrray | OE | Possible therapeutic target, | [111] |
| iRhom1 e iRhom2. | CC and control tissue | IHC, WB | OE | Possible therapeutic target, diagnostic, prognostic | [112] |
| BIM | CC and control tissue | IHC | OE | Possible therapeutic target, diagnostic, prognostic | [113] |
| PTK7 | CC and control tissue | IHC qRT-PCR. | OE. | Possible therapeutic target, diagnostic | [114] |
| 35 proteins | CC and control tissue | MALDI-TOF-MS | OE: 17 proteins SE: 18 proteins | Diagnostic | [115] |
| 200 proteins | CC and control tissue | LC-MS | OE | Diagnostic, prognostic | [116] |
| Tyk2, S100A9, ZNF217 | CC and control tissue | 2D-DIGE MALDI-TOF MS | OE | Possible therapeutic target, diagnostic | [117] |
| FABP5, HspB1, MnSOD | CC and control tissue | 2D-DIGE MALDI-TOF/TOF MS | OE | Diagnostic, prognostic | [25] |
| S100A9, eEF1A1, PKM2 | CC and control tissue | 2-D DIGE MALDI-TOF/TOF MS WB, IHC | OE: S100A9, PKM2 SE:eEF1A1 | Possible therapeutic target, diagnostic | [118] |
| MCM4, MT-ND4, RDH12 | CC and control tissue | Nano LC- MS/MS | OE | Diagnostic | [119] |
| FORMALIN-FIXED, PARAFFIN-EMBEDDED (FFPE) TISSUE | |||||
| hTERT | FFPE | IHC | OE | Possible therapeutic target, diagnostic | [120] |
| FasL y TIL | FFPE | IHC | OE: FasL SE: TIL | Possible therapeutic target, diagnostic | [121] |
| Mimecan, Aortic Smooth Muscle Actin, Lumican, Keratin, Type II Cytoskeleton 5, Peroxyredoxin-1, Sigma 14-3-3 | FFPE | IHC, 2-D, MALDI-TOF-MS | OE:Mimecan, Aortic Smooth Muscle Actine, Lumican SE: Keratin, Cytoskeleton 5, Peroxyredoxin-1, Sigma 14-3-3 | Possible therapeutic target, diagnostic | [122] |
| ADAM9 | FFPE | IHC | OE | Possible therapeutic target, diagnostic, prognostic | [123] |
| SEL1, Notch3, SOCS3 | FFPE | IHC | OE | Diagnostic, prognostic | [124] |
| Ebp1 | FFPE | IHC | OE | Diagnostic | [125] |
| CERVICAL VAGINAL FLUID (CVF) | |||||
| alpha-actinin-4 | CVF | MALDI-TOF-MS, ELISA | OE | Prognostic | [126] |
| haptoglobin, defensins, lactoferrin, azurocidin dermcidin, KLKs 6, 7, 10, 11, 12, 13 | CVF | 2D- MALDI-TOF/TOF-MS ELISA | OE | Diagnostic, prognostic | [127] |
| beta-defensin-2, cathelicidin | CVF | (RP)-LC MALDI-TOF/TOF-MS | Present | Prognostic. | [128] |
| ASAH1, PCBP2, DDX5, hnRNPA1, MCM4, MCM5, CYC, ENO1, TYPH | CVF | iTRAQ-MS | OE | Diagnostic, prognostic | [129] |
| SLeA, p53, HPV16 L1 | Swab vaginal | ELISA, WB | OE: p53, HPV16 L1 SE: SLeA | Diagnostic, prognostic | [130] |
| SERUM AND PLASMA | |||||
| HPT, A1AT, TRFE, FETUA, A1AG1, AACT, KNG1, VTDB | Serum | iTRAQ labelling. LC-MS/MS. Nanochip LC qTOF MS/MSCLS | OE | Diagnostic | [26] |
| Gelsoline y Ceruloplasmine | Serum | 2D, MS, ELISA, IHC | OE: Gelsoline SE: Ceruloplasmine | Prognostic | [131] |
| F9, CFI, AFM, HPR, ORM2 | Plasma | LC-MS/MS | OE | Diagnostic, prognostic | [132] |
| URINE | |||||
| MMRN1, LRG1, S100A8, SERPINB3, CD44 | Urine | LC-MS/MS WB | OE: MMRN1, LRG1 SE: S100A8, SERPINB3, CD44 | Diagnostic | [133] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Rodríguez, F.; Limones-González, J.E.; Mendoza-Almanza, B.; Esparza-Ibarra, E.L.; Gallegos-Flores, P.I.; Ayala-Luján, J.L.; Godina-González, S.; Salinas, E.; Mendoza-Almanza, G. Understanding Cervical Cancer through Proteomics. Cells 2021, 10, 1854. https://doi.org/10.3390/cells10081854
Martínez-Rodríguez F, Limones-González JE, Mendoza-Almanza B, Esparza-Ibarra EL, Gallegos-Flores PI, Ayala-Luján JL, Godina-González S, Salinas E, Mendoza-Almanza G. Understanding Cervical Cancer through Proteomics. Cells. 2021; 10(8):1854. https://doi.org/10.3390/cells10081854
Chicago/Turabian StyleMartínez-Rodríguez, Fátima, Jared E. Limones-González, Brenda Mendoza-Almanza, Edgar L. Esparza-Ibarra, Perla I. Gallegos-Flores, Jorge L. Ayala-Luján, Susana Godina-González, Eva Salinas, and Gretel Mendoza-Almanza. 2021. "Understanding Cervical Cancer through Proteomics" Cells 10, no. 8: 1854. https://doi.org/10.3390/cells10081854
APA StyleMartínez-Rodríguez, F., Limones-González, J. E., Mendoza-Almanza, B., Esparza-Ibarra, E. L., Gallegos-Flores, P. I., Ayala-Luján, J. L., Godina-González, S., Salinas, E., & Mendoza-Almanza, G. (2021). Understanding Cervical Cancer through Proteomics. Cells, 10(8), 1854. https://doi.org/10.3390/cells10081854