
CD36 Signal Transduction in Metabolic Diseases: Novel Insights and Therapeutic Targeting


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Comment on CD36-Assisted Fatty Acid Uptake
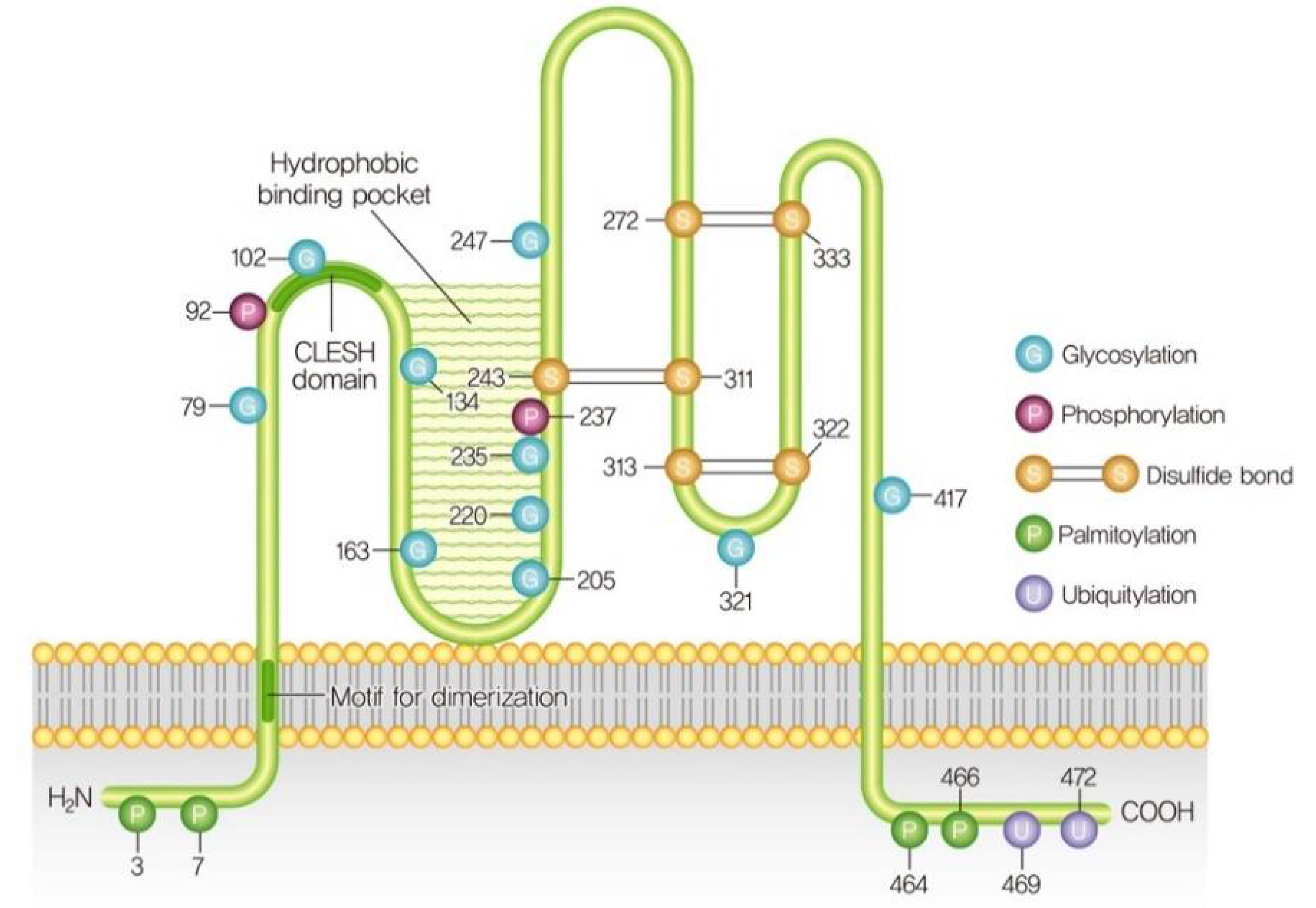
3. Structure and Post-Translational Modifications of CD36
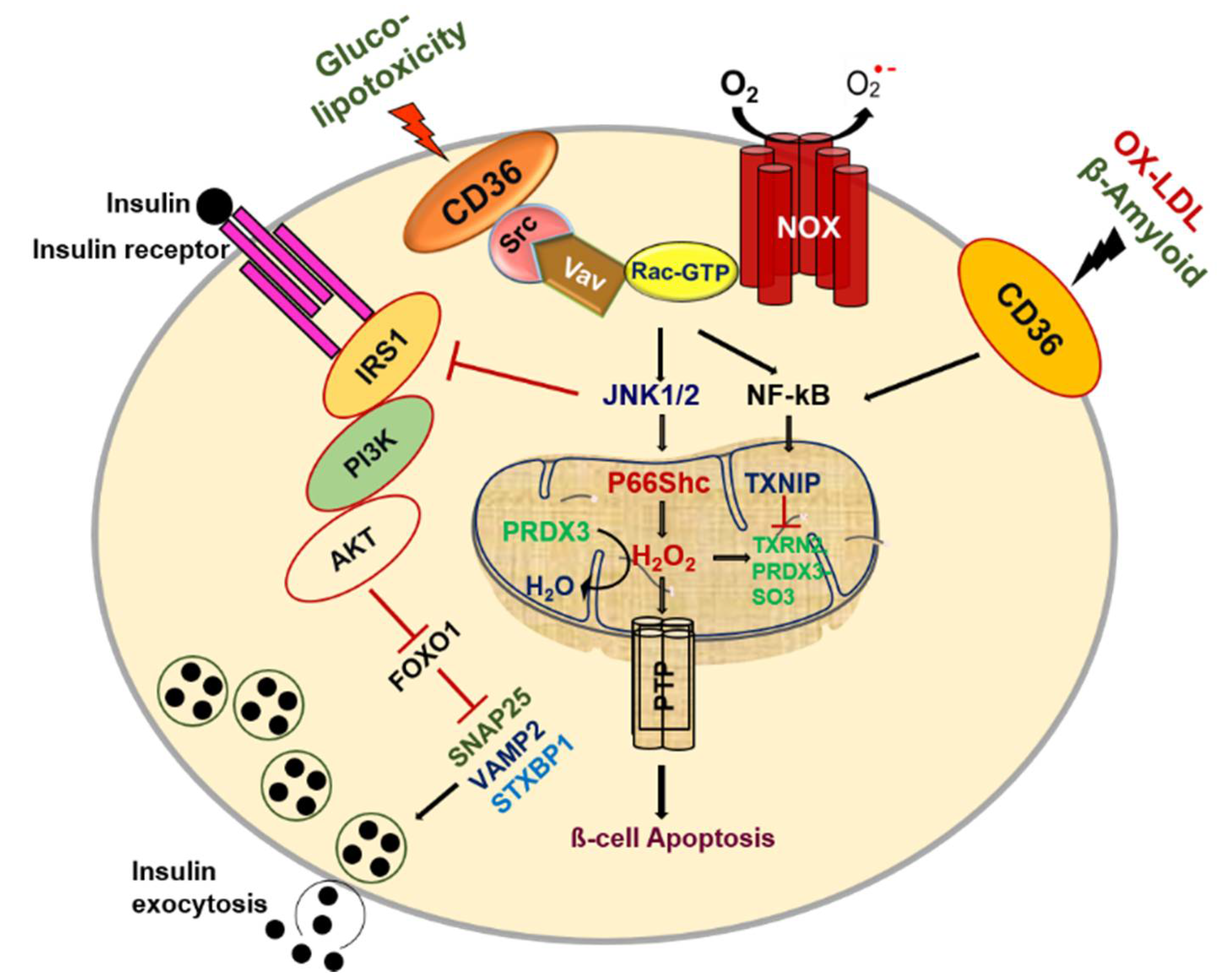
4. Role of CD36 in Pancreatic β-Cell Pathophysiology
4.1. Glucotoxicity
4.2. Lipotoxicity
4.3. OX-LDL and Amyloid Deposition
5. Role of CD36 in Peripheral Insulin Resistance and Metainflammation
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Harmon, C.M.; Abumrad, N.A. Binding of sulfosuccinimidyl fatty acids to adipocyte membrane proteins: Isolation and amino-terminal sequence of an 88-kD protein implicated in transport of long-chain fatty acids. J. Membr. Biol. 1993, 133, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Fukuwatari, T.; Kawada, T.; Tsuruta, M.; Hiraoka, T.; Iwanaga, T.; Sugimoto, E.; Fushiki, T. Expression of the putative membrane fatty acid transporter (FAT) in taste buds of the circumvallate papillae in rats. FEBS Lett. 1997, 414, 461–464. [Google Scholar] [PubMed]
- Van Nieuwenhoven, F.A.; Verstijnen, C.P.; Abumrad, N.A.; Willemsen, P.H.; Van Eys, G.J.; Van der Vusse, G.J.; Glatz, J.F. Putative membrane fatty acid translocase and cytoplasmic fatty acid-binding protein are co-expressed in rat heart and skeletal muscles. Biochem. Biophys. Res. Commun. 1995, 207, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Savill, J.; Hogg, N.; Ren, Y.; Haslett, C. Thrombospondin cooperates with CD36 and the vitronectin receptor in macrophage recognition of neutrophils undergoing apoptosis. J. Clin. Investig. 1992, 90, 1513–1522. [Google Scholar] [CrossRef]
- Ghosh, A.; Li, W.; Febbraio, M.; Espinola, R.G.; McCrae, K.R.; Cockrell, E.; Silverstein, R.L. Platelet CD36 mediates interactions with endothelial cell-derived microparticles and contributes to thrombosis in mice. J. Clin. Investig. 2008, 118, 1934–1943. [Google Scholar] [CrossRef]
- Podrez, E.A.; Byzova, T.V.; Febbraio, M.; Salomon, R.G.; Ma, Y.; Valiyaveettil, M.; Poliakov, E.; Sun, M.; Finton, P.J.; Curtis, B.R.; et al. Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nat. Med. 2007, 13, 1086–1095. [Google Scholar] [CrossRef] [PubMed]
- Koonen, D.P.; Jensen, M.K.; Handberg, A. Soluble CD36- a marker of the (pathophysiological) role of CD36 in the metabolic syndrome? Arch. Physiol. Biochem. 2011, 117, 57–63. [Google Scholar] [CrossRef]
- Wang, Y.; Koch, M.; di Giuseppe, R.; Evans, K.; Borggrefe, J.; Nöthlings, U.; Handberg, A.; Jensen, M.K.; Lieb, W. Associations of plasma CD36 and body fat distribution. J. Clin. Endocrinol. Metab. 2019, 104, 4016–4023. [Google Scholar] [CrossRef]
- Kennedy, D.J.; Kashyap, S.R. Pathogenic role of scavenger receptor CD36 in the metabolic syndrome and diabetes. Metab. Syndr. Relat. Disord. 2011, 9, 239–245. [Google Scholar] [CrossRef]
- Yang, J.; Park, K.W.; Cho, S. Inhibition of the CD36 receptor reduces visceral fat accumulation and improves insulin resistance in obese mice carrying the BDNF-Val66Metvariant. J. Biol. Chem. 2018, 293, 13338–13348. [Google Scholar] [CrossRef]
- Liu, M.; Tso, P.; Woods, S.C. Receptor CD36 links a risk-associated allele to obesity and metabolic disorders. J. Biol. Chem. 2018, 293, 13349–13350. [Google Scholar] [CrossRef] [PubMed]
- Abumrad, N.A.; el-Maghrabi, M.R.; Amri, E.Z.; Lopez, E.; Grimaldi, P.A. Cloning of a rat adipocyte membrane protein implicated in binding or transport of long-chain fatty acids that is induced during preadipocyte differentiation. Homology with human CD36. J. Biol. Chem. 1993, 268, 17665–17668. [Google Scholar] [CrossRef]
- Schwenk, R.W.; Dirkx, E.; Coumans, W.A.; Bonen, A.; Klip, A.; Glatz, J.F.; Luiken, J.J. Requirement for distinct vesicle-associated membrane proteins in insulin- and AMP-activated protein kinase (AMPK)-induced translocation of GLUT4 and CD36 in cultured cardiomyocytes. Diabetologia 2010, 53, 2209–2219. [Google Scholar] [CrossRef]
- Glatz, J.F.; Luiken, J.J.; Bonen, A. Membrane fatty acid transporters as regulators of lipid metabolism: Implications for metabolic disease. Physiol. Rev. 2010, 90, 367–417. [Google Scholar] [CrossRef]
- van Oort, M.M.; van Doorn, J.M.; Bonen, A.; Glatz, J.F.; van der Horst, D.J.; Rodenburg, K.W.; Luiken, J.J. Insulin-induced translocation of CD36 to the plasma membrane is reversible and shows similarity to that of GLUT4. Biochim. Biophys. Acta 2008, 1781, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Du, K.; Murakami, S.; Sun, Y.; Kilpatrick, C.L.; Luscher, B. DHHC7 Palmitoylates Glucose Transporter 4 (Glut4) and Regulates Glut4 Membrane Translocation. J. Biol. Chem. 2017, 292, 2979–2991. [Google Scholar] [CrossRef]
- Simons, K.; Toomre, D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000, 1, 31–39. [Google Scholar] [CrossRef]
- Pohl, J.; Ring, A.; Korkmaz, U.; Ehehalt, R.; Stremmel, W. FAT/CD36-mediated long-chain fatty acid uptake in adipocytes requires plasma membrane rafts. Mol. Biol. Cell. 2005, 16, 24–31. [Google Scholar] [CrossRef]
- Ehehalt, R.; Sparla, R.; Kulaksiz, H.; Herrmann, T.; Füllekrug, J.; Stremmel, W. Uptake of long chain fatty acids is regulated by dynamic interaction of FAT/CD36 with cholesterol/sphingolipid enriched microdomains (lipid rafts). BMC Cell Biol. 2008, 9, 45. [Google Scholar] [CrossRef]
- Ring, A.; Le Lay, S.; Pohl, J.; Verkade, P.; Stremmel, W. Caveolin-1 is required for fatty acid translocase (FAT/CD36) localization and function at the plasma membrane of mouse embryonic fibroblasts. Biochim. Biophys. Acta. 2006, 1761, 416–423. [Google Scholar] [CrossRef]
- Pohl, J.; Ring, A.; Ehehalt, R.; Schulze-Bergkamen, H.; Schad, A.; Verkade, P.; Stremmel, W. Long-chain fatty acid uptake into adipocytes depends on lipid raft function. Biochemistry 2004, 43, 4179–4187. [Google Scholar] [CrossRef]
- Campbell, S.E.; Tandon, N.N.; Woldegiorgis, G.; Luiken, J.J.; Glatz, J.F.; Bonen, A. A novel function for fatty acid translocase (FAT)/CD36: Involvement in long chain fatty acid transfer into the mitochondria. J. Biol. Chem. 2004, 279, 36235–36241. [Google Scholar] [CrossRef] [PubMed]
- Holloway, G.P.; Luiken, J.J.; Glatz, J.F.; Spriet, L.L.; Bonen, A. Contribution of FAT/CD36 to the regulation of skeletal muscle fatty acid oxidation: An overview. Acta Physiol. 2008, 194, 293–309. [Google Scholar] [CrossRef]
- Holloway, G.P.; Jain, S.S.; Bezaire, V.; Han, X.X.; Glatz, J.F.; Luiken, J.J.; Harper, M.E.; Bonen, A. FAT/CD36-null mice reveal that mitochondrial FAT/CD36 is required to upregulate mitochondrial fatty acid oxidation in contracting muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R960–R967. [Google Scholar] [CrossRef]
- Samovski, D.; Sun, J.; Pietka, T.; Gross, R.W.; Eckel, R.H.; Su, X.; Stahl, P.D.; Abumrad, N.A. Regulation of AMPK activation by CD36 links fatty acid uptake to β-oxidation. Diabetes 2015, 64, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Pepino, M.Y.; Kuda, O.; Samovski, D.; Abumrad, N.A. Structure-function of CD36 and importance of fatty acid signal transduction in fat metabolism. Annu. Rev. Nutr. 2014, 34, 281–303. [Google Scholar] [CrossRef] [PubMed]
- Abumrad, N.A.; Goldberg, I.J. CD36 actions in the heart: Lipids, calcium, inflammation, repair and more? Biochim. Biophys. Acta 2016, 1861, 1442–1449. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.S.; Karunakaran, U.; Suma, E.; Chung, S.M.; Won, K.C. The role of CD36 in type 2 diabetes nellitus: β-cell dysfunction and beyond. Diabetes Metab. J. 2020, 44, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Glatz, J.F.; Luiken, J.J. From fat to FAT (CD36/SR-B2): Understanding the regulation of cellular fatty acid uptake. Biochimie 2017, 136, 21–26. [Google Scholar] [CrossRef]
- Glatz, J.F.C.; Luiken, J.J.F.P. Time for a détente in the war on the mechanism of cellular fatty acid uptake. J. Lipid. Res. 2020, 61, 1300–1303. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Abumrad, N.A. Cellular fatty acid uptake: A pathway under construction. Trends Endocrinol. Metab. 2009, 20, 72–77. [Google Scholar] [CrossRef] [PubMed]
- PrabhuDas, M.R.; Baldwin, C.L.; Bollyky, P.L.; Bowdish, D.M.E.; Drickamer, K.; Febbraio, M.; Herz, J.; Kobzik, L.; Krieger, M.; Loike, J.; et al. A Consensus Definitive Classification of Scavenger Receptors and Their Roles in Health and Disease. J. Immunol. 2017, 198, 3775–3789. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Ruiz, E.; Armesilla, A.L.; Sanchez-Madrid, F.; Vega, M.A. Gene encoding the collagen type I and thrombospondin receptor CD36 is located on chromosome 7q11.2. Genomics 1993, 17, 759–761. [Google Scholar] [CrossRef]
- Armesilla, A.L.; Vega, M.A. Structural organization of the gene for human CD36 glycoprotein. J. Biol. Chem. 1994, 269, 18985–18991. [Google Scholar] [CrossRef]
- Ohgami, N.; Nagai, R.; Ikemoto, M.; Arai, H.; Kuniyasu, A.; Horiuchi, S.; Nakayama, H. Cd36, a member of the class b scavenger receptor family, as a receptor for advanced glycation end products. J. Biol. Chem. 2001, 276, 3195–3202. [Google Scholar] [CrossRef]
- Puente Navazo, M.D.; Daviet, L.; Ninio, E.; McGregor, J.L. Identification on human CD36 of a domain (155–183) implicated in binding oxidized low-density lipoproteins (Ox-LDL). Arterioscler. Thromb. Vasc. Biol. 1996, 16, 1033–1039. [Google Scholar] [CrossRef]
- Neculai, D.; Schwake, M.; Ravichandran, M.; Zunke, F.; Collins, R.F.; Peters, J.; Neculai, M.; Plumb, J.; Loppnau, P.; Pizarro, J.C.; et al. Structure of LIMP-2 provides functional insights with implications for SR-BI and CD36. Nature 2013, 504, 172–176. [Google Scholar] [CrossRef]
- Hoosdally, S.J.; Andress, E.J.; Wooding, C.; Martin, C.A.; Linton, K.J. The Human Scavenger Receptor CD36: Glycosylation status and its role in trafficking and function. J. Biol. Chem. 2009, 284, 16277–16288. [Google Scholar] [CrossRef]
- Laczy, B.; Fulop, N.; Onay-Besikci, A.; Des Rosiers, C.; Chatham, J.C. Acute regulation of cardiac metabolism by the hexosamine biosynthesis pathway and protein O-GlcNAcylation. PLoS ONE 2011, 6, e18417. [Google Scholar] [CrossRef] [PubMed]
- Guinez, C.; Morelle, W.; Michalski, J.C.; Lefebvre, T. O-GlcNAc glycosylation: A signal for the nuclear transport of cytosolic proteins? Int. J. Biochem. Cell Biol. 2005, 37, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Wells, L.; Vosseller, K.; Hart, G.W. Glycosylation of nucleocytoplasmic proteins: Signal transduction and O-GlcNAc. Science 2001, 291, 2376–2378. [Google Scholar] [CrossRef]
- Jiang, M.; Wu, N.; Xu, B.; Chu, Y.; Li, X.; Su, S.; Chen, D.; Li, W.; Shi, Y.; Gao, X.; et al. Fatty acid-induced CD36 expression via O-GlcNAcylation drives gastric cancer metastasis. Theranostics 2019, 9, 5359–5373. [Google Scholar] [CrossRef] [PubMed]
- Nabeebaccus, A.A.; Zoccarato, A.; Hafstad, A.D.; Santos, C.X.; Aasum, E.; Brewer, A.C.; Zhang, M.; Beretta, M.; Yin, X.; West, J.A.; et al. Nox4 reprograms cardiac substrate metabolism via protein O-GlcNAcylation to enhance stress adaptation. JCI Insight 2017, 2. [Google Scholar] [CrossRef]
- Aicart-Ramos, C.; Valero, R.A.; Rodriguez-Crespo, I. Protein palmitoylation and subcellular trafficking. Biochim. Biophys. Acta 2011, 1808, 2981–2994. [Google Scholar] [CrossRef] [PubMed]
- Tao, N.; Wagner, S.J.; Lublin, D.M. CD36 is palmitoylated on both N- and C-terminal cytoplasmic tails. J. Biol. Chem. 1996, 271, 22315–22320. [Google Scholar] [CrossRef] [PubMed]
- Thorne, R.F.; Ralston, K.J.; de Bock, C.E.; Mhaidat, N.M.; Zhang, X.D.; Boyd, A.W.; Burns, G.F. Palmitoylation of CD36/FAT regulates the rate of its post-transcriptional processing in the endoplasmic reticulum. Biochim. Biophys. Acta 2010, 1803, 1298–1307. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.; Su, X.; El-Maghrabi, R.; Stahl, P.D.; Abumrad, N.A. Opposite regulation of CD36 ubiquitination by fatty acids and insulin: Effects on fatty acid uptake. J. Biol. Chem. 2008, 283, 13578–13585. [Google Scholar] [CrossRef]
- Xia, X.; Hu, T.; He, J.; Xu, Q.; Yu, C.; Liu, X.; Shao, Z.; Liao, Y.; Huang, H.; Liu, N. USP10 deletion inhibits macrophage-derived foam cell formation and cellular-oxidized low density lipoprotein uptake by promoting the degradation of CD36. Aging 2020, 12, 22892–22905. [Google Scholar] [CrossRef]
- Kim, K.Y.; Stevens, M.V.; Akter, M.H.; Rusk, S.E.; Huang, R.J.; Cohen, A.; Noguchi, A.; Springer, D.; Bocharov, A.V.; Eggerman, T.L.; et al. Parkin is a lipid-responsive regulator of fat uptake in mice and mutant human cells. J. Clin. Investig. 2011, 121, 3701–3712. [Google Scholar] [CrossRef] [PubMed]
- Asch, A.S.; Liu, I.; Briccetti, F.M.; Barnwell, J.W.; Kwakye-Berko, F.; Dokun, A.; Goldberger, J.; Pernambuco, M. Analysis of CD36 binding domains: Ligand specificity controlled by dephosphorylation of an ectodomain. Science 1993, 262, 1436–1440. [Google Scholar] [CrossRef]
- Hatmi, M.; Gavaret, J.M.; Elalamy, I.; Vargaftig, B.B.; Jacquemin, C. Evidence for cAMP-dependent platelet ectoprotein kinase activity that phosphorylates platelet glycoprotein IV (CD36). J. Biol. Chem. 1996, 271, 24776–24780. [Google Scholar] [CrossRef]
- Lundby, A.; Lage, K.; Weinert, B.T.; Bekker-Jensen, D.B.; Secher, A.; Skovgaard, T.; Kelstrup, C.D.; Dmytriyev, A.; Choudhary, C.; Lundby, C.; et al. Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Rep. 2012, 2, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Kuda, O.; Pietka, T.A.; Demianova, Z.; Kudova, E.; Cvacka, J.; Kopecky, J.; Abumrad, N.A. Sulfo-N-succinimidyl oleate (SSO) inhibits fatty acid uptake and signaling for intracellular calcium via binding CD36 lysine 164: SSO also inhibits oxidized low density lipoprotein uptake by macrophages. J. Biol. Chem. 2013, 288, 15547–15555. [Google Scholar] [CrossRef]
- Noushmehr, H.; D’Amico, E.; Farilla, L.; Hui, H.; Wawrowsky, K.A.; Mlynarski, W.; Doria, A.; Abumrad, N.A.; Perfetti, R. Fatty acid translocase (FAT/CD36) is localized on insulin-containing granules in human pancreatic beta-cells and mediates fatty acid effects on insulin secretion. Diabetes 2005, 54, 472–481. [Google Scholar] [CrossRef]
- Schwenk, R.W.; Holloway, G.P.; Luiken, J.J.; Bonen, A.; Glatz, J.F. Fatty acid transport across the cell membrane: Regulation by fatty acid transporters. Prostaglandins Leukot. Essent. Fatty Acids 2010, 82, 149–154. [Google Scholar] [CrossRef]
- Maedler, K.; Oberholzer, J.; Bucher, P.; Spinas, G.A.; Donath, M.Y. Monounsaturated fatty acids prevent the deleterious effects of palmitate and high glucose on human pancreatic beta-cell turnover and function. Diabetes 2003, 52, 726–733. [Google Scholar] [CrossRef] [PubMed]
- Maedler, K.; Spinas, G.A.; Dyntar, D.; Moritz, W.; Kaiser, N.; Donath, M.Y. Distinct effects of saturated and monounsaturated fatty acids on beta-cell turnover and function. Diabetes 2001, 50, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Wallin, T.; Ma, Z.; Ogata, H.; Jorgensen, I.H.; Iezzi, M.; Wang, H.; Wollheim, C.B.; Bjorklund, A. Facilitation of fatty acid uptake by CD36 in insulin-producing cells reduces fatty-acid-induced insulin secretion and glucose regulation of fatty acid oxidation. Biochim. Biophys. Acta 2010, 1801, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.W.; Moon, J.S.; Seo, Y.J.; Park, S.Y.; Kim, J.Y.; Yoon, J.S.; Lee, I.K.; Lee, H.W.; Won, K.C. Inhibition of fatty acid translocase cluster determinant 36 (CD36), stimulated by hyperglycemia, prevents glucotoxicity in INS-1 cells. Biochem. Biophys. Res. Commun. 2012, 420, 462–466. [Google Scholar] [CrossRef] [PubMed]
- Elumalai, S.; Karunakaran, U.; Lee, I.K.; Moon, J.S.; Won, K.C. Rac1-NADPH oxidase signaling promotes CD36 activation under glucotoxic conditions in pancreatic beta cells. Redox Biol. 2017, 11, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Asahara, S.; Shibutani, Y.; Teruyama, K.; Inoue, H.Y.; Kawada, Y.; Etoh, H.; Matsuda, T.; Kimura-Koyanagi, M.; Hashimoto, N.; Sakahara, M.; et al. Ras-related C3 botulinum toxin substrate 1 (RAC1) regulates glucose-stimulated insulin secretion via modulation of F-actin. Diabetologia 2013, 56, 1088–1097. [Google Scholar] [CrossRef]
- Li, J.; Luo, R.; Kowluru, A.; Li, G. Novel regulation by Rac1 of glucose- and forskolin-induced insulin secretion in INS-1 beta-cells. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E818–E827. [Google Scholar] [CrossRef] [PubMed]
- Thurmond, D.C.; Gonelle-Gispert, C.; Furukawa, M.; Halban, P.A.; Pessin, J.E. Glucose-stimulated insulin secretion is coupled to the interaction of actin with the t-SNARE (target membrane soluble N-ethylmaleimide-sensitive factor attachment protein receptor protein) complex. Mol. Endocrinol. 2003, 17, 732–742. [Google Scholar] [CrossRef]
- Tomas, A.; Yermen, B.; Min, L.; Pessin, J.E.; Halban, P.A. Regulation of pancreatic beta-cell insulin secretion by actin cytoskeleton remodelling: Role of gelsolin and cooperation with the MAPK signalling pathway. J. Cell Sci. 2006, 119, 2156–2167. [Google Scholar] [CrossRef]
- Band, A.M.; Ali, H.; Vartiainen, M.K.; Welti, S.; Lappalainen, P.; Olkkonen, V.M.; Kuismanen, E. Endogenous plasma membrane t-SNARE syntaxin 4 is present in rab11 positive endosomal membranes and associates with cortical actin cytoskeleton. FEBS Lett. 2002, 531, 513–519. [Google Scholar] [CrossRef]
- Jewell, J.L.; Luo, W.; Oh, E.; Wang, Z.; Thurmond, D.C. Filamentous actin regulates insulin exocytosis through direct interaction with Syntaxin 4. J. Biol. Chem. 2008, 283, 10716–10726. [Google Scholar] [CrossRef]
- Rondas, D.; Tomas, A.; Soto-Ribeiro, M.; Wehrle-Haller, B.; Halban, P.A. Novel mechanistic link between focal adhesion remodeling and glucose-stimulated insulin secretion. J. Biol. Chem. 2012, 287, 2423–2436. [Google Scholar] [CrossRef] [PubMed]
- Nagamatsu, S.; Nakamichi, Y.; Yamamura, C.; Matsushima, S.; Watanabe, T.; Ozawa, S.; Furukawa, H.; Ishida, H. Decreased expression of t-SNAR.E.; syntaxin 1, and SNAP-25 in pancreatic beta-cells is involved in impaired insulin secretion from diabetic GK rat islets: Restoration of decreased t-SNARE proteins improves impaired insulin secretion. Diabetes 1999, 48, 2367–2373. [Google Scholar] [CrossRef] [PubMed]
- Thurmond, D.C.; Gaisano, H.Y. Recent Insights into Beta-cell Exocytosis in Type 2 Diabetes. J. Mol. Biol. 2020, 432, 1310–1325. [Google Scholar] [CrossRef] [PubMed]
- Torrejon-Escribano, B.; Escoriza, J.; Montanya, E.; Blasi, J. Glucose-dependent changes in SNARE protein levels in pancreatic beta-cells. Endocrinology 2011, 152, 1290–1299. [Google Scholar] [CrossRef] [PubMed]
- Gaisano, H.Y. Recent new insights into the role of SNARE and associated proteins in insulin granule exocytosis. Diabetes Obes. Metab. 2017, 19 (Suppl. 1), 115–123. [Google Scholar] [CrossRef]
- Nagao, M.; Esguerra, J.L.S.; Asai, A.; Ofori, J.K.; Edlund, A.; Wendt, A.; Sugihara, H.; Wollheim, C.B.; Oikawa, S.; Eliasson, L. Potential Protection Against Type 2 Diabetes in Obesity Through Lower CD36 Expression and Improved Exocytosis in beta-Cells. Diabetes 2020, 69, 1193–1205. [Google Scholar] [CrossRef]
- Cnop, M.; Welsh, N.; Jonas, J.C.; Jorns, A.; Lenzen, S.; Eizirik, D.L. Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: Many differences, few similarities. Diabetes 2005, 54 (Suppl. 2), S97–S107. [Google Scholar] [CrossRef]
- Ihara, Y.; Toyokuni, S.; Uchida, K.; Odaka, H.; Tanaka, T.; Ikeda, H.; Hiai, H.; Seino, Y.; Yamada, Y. Hyperglycemia causes oxidative stress in pancreatic beta-cells of GK rats, a model of type 2 diabetes. Diabetes 1999, 48, 927–932. [Google Scholar] [CrossRef]
- Guichard, C.; Moreau, R.; Pessayre, D.; Epperson, T.K.; Krause, K.H. NOX family NADPH oxidases in liver and in pancreatic islets: A role in the metabolic syndrome and diabetes? Biochem. Soc. Trans. 2008, 36, 920–929. [Google Scholar] [CrossRef] [PubMed]
- Gharib, M.; Tao, H.; Fungwe, T.V.; Hajri, T. Cluster Differentiating 36 (CD36) Deficiency Attenuates Obesity-Associated Oxidative Stress in the Heart. PLoS ONE 2016, 11, e0155611. [Google Scholar] [CrossRef] [PubMed]
- Veluthakal, R.; Kumar, B.; Mohammad, G.; Kowluru, A.; Kowluru, R.A. Tiam1-Rac1 Axis Promotes Activation of p38 MAP Kinase in the Development of Diabetic Retinopathy: Evidence for a Requisite Role for Protein Palmitoylation. Cell Physiol. Biochem. 2015, 36, 208–220. [Google Scholar] [CrossRef]
- Li, W.; Febbraio, M.; Reddy, S.P.; Yu, D.Y.; Yamamoto, M.; Silverstein, R.L. CD36 participates in a signaling pathway that regulates ROS formation in murine VSMCs. J. Clin. Investig. 2010, 120, 3996–4006. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, B.; Volpert, O.V.; Crawford, S.E.; Febbraio, M.; Silverstein, R.L.; Bouck, N. Signals leading to apoptosis-dependent inhibition of neovascularization by thrombospondin-1. Nat. Med. 2000, 6, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; El Khoury, J.; Medeiros, L.A.; Terada, K.; Geula, C.; Luster, A.D.; Freeman, M.W. A CD36-initiated signaling cascade mediates inflammatory effects of beta-amyloid. J. Biol. Chem. 2002, 277, 47373–47379. [Google Scholar] [CrossRef] [PubMed]
- Rahaman, S.O.; Lennon, D.J.; Febbraio, M.; Podrez, E.A.; Hazen, S.L.; Silverstein, R.L. A CD36-dependent signaling cascade is necessary for macrophage foam cell formation. Cell Metab. 2006, 4, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, P.; Bugliani, M.; Lupi, R.; Marselli, L.; Masini, M.; Boggi, U.; Filipponi, F.; Weir, G.C.; Eizirik, D.L.; Cnop, M. The endoplasmic reticulum in pancreatic beta cells of type 2 diabetes patients. Diabetologia 2007, 50, 2486–2494. [Google Scholar] [CrossRef] [PubMed]
- Nakatani, Y.; Kaneto, H.; Kawamori, D.; Yoshiuchi, K.; Hatazaki, M.; Matsuoka, T.A.; Ozawa, K.; Ogawa, S.; Hori, M.; Yamasaki, Y.; et al. Involvement of endoplasmic reticulum stress in insulin resistance and diabetes. J. Biol. Chem. 2005, 280, 847–851. [Google Scholar] [CrossRef]
- Moon, J.S.; Karunakaran, U.; Elumalai, S.; Lee, I.K.; Lee, H.W.; Kim, Y.W.; Won, K.C. Metformin prevents glucotoxicity by alleviating oxidative and ER stress-induced CD36 expression in pancreatic beta cells. J. Diabetes Complicat. 2017, 31, 21–30. [Google Scholar] [CrossRef]
- Karunakaran, U.; Kim, H.J.; Kim, J.Y.; Lee, I.K. Guards and culprits in the endoplasmic reticulum: Glucolipotoxicity and beta-cell failure in type II diabetes. Exp. Diabetes Res. 2012, 2012, 639762. [Google Scholar] [CrossRef]
- Lytrivi, M.; Castell, A.L.; Poitout, V.; Cnop, M. Recent Insights Into Mechanisms of beta-Cell Lipo- and Glucolipotoxicity in Type 2 Diabetes. J. Mol. Biol. 2020, 432, 1514–1534. [Google Scholar] [CrossRef] [PubMed]
- Poitout, V.; Robertson, R.P. Glucolipotoxicity: Fuel excess and beta-cell dysfunction. Endocr. Rev. 2008, 29, 351–366. [Google Scholar] [CrossRef]
- Boslem, E.; Meikle, P.J.; Biden, T.J. Roles of ceramide and sphingolipids in pancreatic beta-cell function and dysfunction. Islets 2012, 4, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Maestre, I.; Jordan, J.; Calvo, S.; Reig, J.A.; Cena, V.; Soria, B.; Prentki, M.; Roche, E. Mitochondrial dysfunction is involved in apoptosis induced by serum withdrawal and fatty acids in the beta-cell line INS-1. Endocrinology 2003, 144, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Shimabukuro, M.; Ohneda, M.; Lee, Y.; Unger, R.H. Role of nitric oxide in obesity-induced beta cell disease. J. Clin. Investig. 1997, 100, 290–295. [Google Scholar] [CrossRef]
- Shimabukuro, M.; Zhou, Y.T.; Levi, M.; Unger, R.H. Fatty acid-induced beta cell apoptosis: A link between obesity and diabetes. Proc. Natl. Acad. Sci. USA 1998, 95, 2498–2502. [Google Scholar] [CrossRef] [PubMed]
- Bindokas, V.P.; Kuznetsov, A.; Sreenan, S.; Polonsky, K.S.; Roe, M.W.; Philipson, L.H. Visualizing superoxide production in normal and diabetic rat islets of Langerhans. J. Biol. Chem. 2003, 278, 9796–9801. [Google Scholar] [CrossRef] [PubMed]
- Lupi, R.; Dotta, F.; Marselli, L.; Del Guerra, S.; Masini, M.; Santangelo, C.; Patane, G.; Boggi, U.; Piro, S.; Anello, M.; et al. Prolonged exposure to free fatty acids has cytostatic and pro-apoptotic effects on human pancreatic islets: Evidence that beta-cell death is caspase mediated, partially dependent on ceramide pathway, and Bcl-2 regulated. Diabetes 2002, 51, 1437–1442. [Google Scholar] [CrossRef]
- Haus, J.M.; Kashyap, S.R.; Kasumov, T.; Zhang, R.; Kelly, K.R.; Defronzo, R.A.; Kirwan, J.P. Plasma ceramides are elevated in obese subjects with type 2 diabetes and correlate with the severity of insulin resistance. Diabetes 2009, 58, 337–343. [Google Scholar] [CrossRef]
- Samad, F.; Hester, K.D.; Yang, G.; Hannun, Y.A.; Bielawski, J. Altered adipose and plasma sphingolipid metabolism in obesity: A potential mechanism for cardiovascular and metabolic risk. Diabetes 2006, 55, 2579–2587. [Google Scholar] [CrossRef]
- Wigger, L.; Cruciani-Guglielmacci, C.; Nicolas, A.; Denom, J.; Fernandez, N.; Fumeron, F.; Marques-Vidal, P.; Ktorza, A.; Kramer, W.; Schulte, A.; et al. Plasma Dihydroceramides Are Diabetes Susceptibility Biomarker Candidates in Mice and Humans. Cell Rep. 2017, 18, 2269–2279. [Google Scholar] [CrossRef]
- Bustelo, X.R. Vav family exchange factors: An integrated regulatory and functional view. Small GTPases 2014, 5, 9. [Google Scholar] [CrossRef]
- Kominato, R.; Fujimoto, S.; Mukai, E.; Nakamura, Y.; Nabe, K.; Shimodahira, M.; Nishi, Y.; Funakoshi, S.; Seino, Y.; Inagaki, N. Src activation generates reactive oxygen species and impairs metabolism-secretion coupling in diabetic Goto-Kakizaki and ouabain-treated rat pancreatic islets. Diabetologia 2008, 51, 1226–1235. [Google Scholar] [CrossRef]
- Karunakaran, U.; Elumalai, S.; Moon, J.S.; Won, K.C. CD36 dependent redoxosomes promotes ceramide-mediated pancreatic beta-cell failure via p66Shc activation. Free Radic. Biol. Med. 2019, 134, 505–515. [Google Scholar] [CrossRef]
- Holzer, R.G.; Park, E.J.; Li, N.; Tran, H.; Chen, M.; Choi, C.; Solinas, G.; Karin, M. Saturated fatty acids induce c-Src clustering within membrane subdomains, leading to JNK activation. Cell 2011, 147, 173–184. [Google Scholar] [CrossRef]
- Hirosumi, J.; Tuncman, G.; Chang, L.; Gorgun, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336. [Google Scholar] [CrossRef]
- Kant, S.; Standen, C.L.; Morel, C.; Jung, D.Y.; Kim, J.K.; Swat, W.; Flavell, R.A.; Davis, R.J. A Protein Scaffold Coordinates SRC-Mediated JNK Activation in Response to Metabolic Stress. Cell Rep. 2017, 20, 2775–2783. [Google Scholar] [CrossRef]
- Hua, W.; Huang, H.Z.; Tan, L.T.; Wan, J.M.; Gui, H.B.; Zhao, L.; Ruan, X.Z.; Chen, X.M.; Du, X.G. CD36 Mediated Fatty Acid-Induced Podocyte Apoptosis via Oxidative Stress. PLoS ONE 2015, 10, e0127507. [Google Scholar] [CrossRef]
- Xu, S.; Nam, S.M.; Kim, J.H.; Das, R.; Choi, S.K.; Nguyen, T.T.; Quan, X.; Choi, S.J.; Chung, C.H.; Lee, E.Y.; et al. Palmitate induces ER calcium depletion and apoptosis in mouse podocytes subsequent to mitochondrial oxidative stress. Cell Death Dis. 2015, 6, e1976. [Google Scholar] [CrossRef]
- Natalicchio, A.; Tortosa, F.; Labarbuta, R.; Biondi, G.; Marrano, N.; Carchia, E.; Leonardini, A.; Cignarelli, A.; Bugliani, M.; Marchetti, P.; et al. The p66(Shc) redox adaptor protein is induced by saturated fatty acids and mediates lipotoxicity-induced apoptosis in pancreatic beta cells. Diabetologia 2015, 58, 1260–1271. [Google Scholar] [CrossRef]
- Khalid, S.; Drasche, A.; Thurner, M.; Hermann, M.; Ashraf, M.I.; Fresser, F.; Baier, G.; Kremser, L.; Lindner, H.; Troppmair, J. cJun N-terminal kinase (JNK) phosphorylation of serine 36 is critical for p66Shc activation. Sci. Rep. 2016, 6, 20930. [Google Scholar] [CrossRef]
- Yang, M.; Li, W.; Harberg, C.; Chen, W.; Yue, H.; Ferreira, R.B.; Wynia-Smith, S.L.; Carroll, K.S.; Zielonka, J.; Flaumenhaft, R.; et al. Cysteine sulfenylation by CD36 signaling promotes arterial thrombosis in dyslipidemia. Blood Adv. 2020, 4, 4494–4507. [Google Scholar] [CrossRef]
- Kang, S.W.; Chae, H.Z.; Seo, M.S.; Kim, K.; Baines, I.C.; Rhee, S.G. Mammalian peroxiredoxin isoforms can reduce hydrogen peroxide generated in response to growth factors and tumor necrosis factor-alpha. J. Biol. Chem. 1998, 273, 6297–6302. [Google Scholar] [CrossRef]
- Lim, Y.S.; Cha, M.K.; Kim, H.K.; Uhm, T.B.; Park, J.W.; Kim, K.; Kim, I.H. Removals of hydrogen peroxide and hydroxyl radical by thiol-specific antioxidant protein as a possible role in vivo. Biochem. Biophys. Res. Commun. 1993, 192, 273–280. [Google Scholar] [CrossRef]
- Cox, A.G.; Pullar, J.M.; Hughes, G.; Ledgerwood, E.C.; Hampton, M.B. Oxidation of mitochondrial peroxiredoxin 3 during the initiation of receptor-mediated apoptosis. Free Radic. Biol. Med. 2008, 44, 1001–1009. [Google Scholar] [CrossRef]
- Karunakaran, U.; Moon, J.S.; Lee, H.W.; Won, K.C. CD36 initiated signaling mediates ceramide-induced TXNIP expression in pancreatic beta-cells. Biochim. Biophys. Acta 2015, 1852, 2414–2422. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Park, K.S.; Wiederkehr, A.; Kirkpatrick, C.; Mattenberger, Y.; Martinou, J.C.; Marchetti, P.; Demaurex, N.; Wollheim, C.B. Selective actions of mitochondrial fission/fusion genes on metabolism-secretion coupling in insulin-releasing cells. J. Biol. Chem. 2008, 283, 33347–33356. [Google Scholar] [CrossRef]
- Wasilewski, M.; Scorrano, L. The changing shape of mitochondrial apoptosis. Trends Endocrinol. Metab. 2009, 20, 287–294. [Google Scholar] [CrossRef]
- Koshkin, V.; Dai, F.F.; Robson-Doucette, C.A.; Chan, C.B.; Wheeler, M.B. Limited mitochondrial permeabilization is an early manifestation of palmitate-induced lipotoxicity in pancreatic beta-cells. J. Biol. Chem. 2008, 283, 7936–7948. [Google Scholar] [CrossRef] [PubMed]
- De Stefani, D.; Bononi, A.; Romagnoli, A.; Messina, A.; De Pinto, V.; Pinton, P.; Rizzuto, R. VDAC1 selectively transfers apoptotic Ca2+ signals to mitochondria. Cell Death Differ. 2012, 19, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Marinello, M.; Bononi, A.; Bonora, M.; Giorgi, C.; Rimessi, A.; Pinton, P. Selective modulation of subtype III IP(3)R by Akt regulates ER Ca(2)(+) release and apoptosis. Cell Death Dis. 2012, 3, e304. [Google Scholar] [CrossRef]
- Plaisance, V.; Brajkovic, S.; Tenenbaum, M.; Favre, D.; Ezanno, H.; Bonnefond, A.; Bonner, C.; Gmyr, V.; Kerr-Conte, J.; Gauthier, B.R.; et al. Endoplasmic Reticulum Stress Links Oxidative Stress to Impaired Pancreatic Beta-Cell Function Caused by Human Oxidized LDL. PLoS ONE 2016, 11, e0163046. [Google Scholar] [CrossRef]
- Abderrahmani, A.; Niederhauser, G.; Favre, D.; Abdelli, S.; Ferdaoussi, M.; Yang, J.Y.; Regazzi, R.; Widmann, C.; Waeber, G. Human high-density lipoprotein particles prevent activation of the JNK pathway induced by human oxidised low-density lipoprotein particles in pancreatic beta cells. Diabetologia 2007, 50, 1304–1314. [Google Scholar] [CrossRef]
- Cnop, M.; Hannaert, J.C.; Grupping, A.Y.; Pipeleers, D.G. Low density lipoprotein can cause death of islet beta-cells by its cellular uptake and oxidative modification. Endocrinology 2002, 143, 3449–3453. [Google Scholar] [CrossRef]
- Grupping, A.Y.; Cnop, M.; Van Schravendijk, C.F.; Hannaert, J.C.; Van Berkel, T.J.; Pipeleers, D.G. Low density lipoprotein binding and uptake by human and rat islet beta cells. Endocrinology 1997, 138, 4064–4068. [Google Scholar] [CrossRef]
- Shi, Y.; Cosentino, F.; Camici, G.G.; Akhmedov, A.; Vanhoutte, P.M.; Tanner, F.C.; Luscher, T.F. Oxidized low-density lipoprotein activates p66Shc via lectin-like oxidized low-density lipoprotein receptor-1, protein kinase C-beta, and c-Jun N-terminal kinase kinase in human endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2090–2097. [Google Scholar] [CrossRef]
- Ma, Z.; Ketelhuth, D.F.J.; Wirstrom, T.; Ohki, T.; Forteza, M.J.; Wang, H.; Grill, V.; Wollheim, C.B.; Bjorklund, A. Increased uptake of oxLDL does not exert lipotoxic effects in insulin-secreting cells. J. Mol. Endocrinol. 2019, 62, 159–168. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, M.; Huang, W.; Chen, W.; Zhao, Y.; Schulte, M.L.; Volberding, P.; Gerbec, Z.; Zimmermann, M.T.; Zeighami, A.; et al. Mitochondrial Metabolic Reprogramming by CD36 Signaling Drives Macrophage Inflammatory Responses. Circ. Res. 2019, 125, 1087–1102. [Google Scholar] [CrossRef]
- Zhao, M.; Pan, W.; Shi, R.Z.; Bai, Y.P.; You, B.Y.; Zhang, K.; Fu, Q.M.; Schuchman, E.H.; He, X.X.; Zhang, G.G. Acid Sphingomyelinase Mediates Oxidized-LDL Induced Apoptosis in Macrophage via Endoplasmic Reticulum Stress. J. Atheroscler. Thromb. 2016, 23, 1111–1125. [Google Scholar] [CrossRef] [PubMed]
- Benáková, Š.; Holendová, B.; Plecitá-Hlavatá, L. Redox Homeostasis in Pancreatic β-Cells: From Development to Failure. Antioxidants 2021, 10, 526. [Google Scholar] [CrossRef] [PubMed]
- Ježek, P.; Holendová, B.; Jabůrek, M.; Tauber, J.; Dlasková, A.; Plecitá-Hlavatá, L. The Pancreatic β-Cell: The Perfect Redox System. Antioxidants 2021, 10, 197. [Google Scholar] [CrossRef]
- Lenzen, S.; Drinkgern, J.; Tiedge, M. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radic. Biol. Med. 1996, 20, 463–466. [Google Scholar] [CrossRef]
- Kaneto, H.; Kajimoto, Y.; Miyagawa, J.; Matsuoka, T.; Fujitani, Y.; Umayahara, Y.; Hanafusa, T.; Matsuzawa, Y.; Yamasaki, Y.; Hori, M. Beneficial effects of antioxidants in diabetes: Possible protection of pancreatic beta-cells against glucose toxicity. Diabetes 1999, 48, 2398–2406. [Google Scholar] [CrossRef] [PubMed]
- Podszun, M.C.; Alawad, A.S.; Lingala, S.; Morris, N.; Huang, W.A.; Yang, S.; Schoenfeld, M.; Rolt, A.; Ouwerkerk, R.; Valdez, K.; et al. Vitamin E treatment in NAFLD patients demonstrates that oxidative stress drives steatosis through upregulation of de-novo lipogenesis. Redox Biol. 2020, 37, 101710. [Google Scholar] [CrossRef]
- Ziegler, M.; Wallert, M.; Lorkowski, S.; Peter, K. Cardiovascular and Metabolic Protection by Vitamin E: A Matter of Treatment Strategy? Antioxidants 2020, 9, 935. [Google Scholar] [CrossRef] [PubMed]
- Ungurianu, A.; Zanfirescu, A.; Nițulescu, G.; Margină, D. Vitamin E beyond Its Antioxidant Label. Antioxidants 2021, 10, 634. [Google Scholar] [CrossRef] [PubMed]
- Di Vincenzo, A.; Tana, C.; El Hadi, H.; Pagano, C.; Vettor, R.; Rossato, M. Antioxidant, Anti-Inflammatory, and Metabolic Properties of Tocopherols and Tocotrienols: Clinical Implications for Vitamin E Supplementation in Diabetic Kidney Disease. Int. J. Mol. Sci. 2019, 20, 5101. [Google Scholar] [CrossRef] [PubMed]
- Ozer, N.K.; Negis, Y.; Aytan, N.; Villacorta, L.; Ricciarelli, R.; Zingg, J.M.; Azzi, A. Vitamin E inhibits CD36 scavenger receptor expression in hypercholesterolemic rabbits. Atherosclerosis 2006, 184, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Ricciarelli, R.; Zingg, J.M.; Azzi, A. Vitamin E reduces the uptake of oxidized LDL by inhibiting CD36 scavenger receptor expression in cultured aortic smooth muscle cells. Circulation 2000, 102, 82–87. [Google Scholar] [CrossRef]
- Munteanu, A.; Taddei, M.; Tamburini, I.; Bergamini, E.; Azzi, A.; Zingg, J.M. Antagonistic effects of oxidized low density lipoprotein and alpha-tocopherol on CD36 scavenger receptor expression in monocytes: Involvement of protein kinase B and peroxisome proliferator-activated receptor-gamma. J. Biol. Chem. 2006, 281, 6489–6497. [Google Scholar] [CrossRef] [PubMed]
- Zingg, J.M.; Azzi, A.; Meydani, M. α-Tocopheryl Phosphate Induces VEGF Expression via CD36/PI3Kγ in THP-1 Monocytes. J. Cell Biochem. 2017, 118, 1855–1867. [Google Scholar] [CrossRef]
- Brissova, M.; Shostak, A.; Shiota, M.; Wiebe, P.O.; Poffenberger, G.; Kantz, J.; Chen, Z.; Carr, C.; Jerome, W.G.; Chen, J.; et al. Pancreatic islet production of vascular endothelial growth factor—A is essential for islet vascularization, revascularization, and function. Diabetes 2006, 55, 2974–2985. [Google Scholar] [CrossRef]
- Chia, L.L.; Jantan, I.; Chua, K.H. Tocotrienols Stimulate Insulin Secretion of Rat Pancreatic Isolated Islets in a Dynamic Culture. Curr. Pharm. Biotechnol. 2017, 18, 560–568. [Google Scholar] [CrossRef]
- Hani, H.; Allaudin, Z.N.; Mohd-Lila, M.A.; Sarsaifi, K.; Rasouli, M.; Tam, Y.J.; Tengku-Ibrahim, T.A.; Othman, A.M. Improvement of isolated caprine islet survival and functionality in vitro by enhancing of PDX1 gene expression. Xenotransplantation 2017, 24, e12302. [Google Scholar] [CrossRef]
- Abedini, A.; Schmidt, A.M. Mechanisms of islet amyloidosis toxicity in type 2 diabetes. FEBS Lett. 2013, 587, 1119–1127. [Google Scholar] [CrossRef]
- Bishoyi, A.K.; Roham, P.H.; Rachineni, K.; Save, S.; Hazari, M.A.; Sharma, S.; Kumar, A. Human islet amyloid polypeptide (hIAPP)—A curse in type II diabetes mellitus: Insights from structure and toxicity studies. Biol. Chem. 2021, 402, 133–153. [Google Scholar] [CrossRef]
- Subramanian, S.L.; Hull, R.L.; Zraika, S.; Aston-Mourney, K.; Udayasankar, J.; Kahn, S.E. cJUN N-terminal kinase (JNK) activation mediates islet amyloid-induced beta cell apoptosis in cultured human islet amyloid polypeptide transgenic mouse islets. Diabetologia 2012, 55, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liu, J.; Dragunow, M.; Cooper, G.J. Fibrillogenic amylin evokes islet beta-cell apoptosis through linked activation of a caspase cascade and JNK1. J. Biol. Chem. 2003, 278, 52810–52819. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.R.; Stuart, L.M.; Wilkinson, K.; van Gils, J.M.; Deng, J.; Halle, A.; Rayner, K.J.; Boyer, L.; Zhong, R.; Frazier, W.A.; et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 2010, 11, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Bonen, A.; Jain, S.S.; Snook, L.A.; Han, X.X.; Yoshida, Y.; Buddo, K.H.; Lally, J.S.; Pask, E.D.; Paglialunga, S.; Beaudoin, M.S.; et al. Extremely rapid increase in fatty acid transport and intramyocellular lipid accumulation but markedly delayed insulin resistance after high fat feeding in rats. Diabetologia 2015, 58, 2381–2391. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Del Prato, S. Insulin resistance and diabetes mellitus. J. Diabetes Complicat. 1996, 10, 243–245. [Google Scholar] [CrossRef]
- Turner, N.; Kowalski, G.M.; Leslie, S.J.; Risis, S.; Yang, C.; Lee-Young, R.S.; Babb, J.R.; Meikle, P.J.; Lancaster, G.I.; Henstridge, D.C.; et al. Distinct patterns of tissue-specific lipid accumulation during the induction of insulin resistance in mice by high-fat feeding. Diabetologia 2013, 56, 1638–1648. [Google Scholar] [CrossRef]
- Gustafson, B.; Hedjazifar, S.; Gogg, S.; Hammarstedt, A.; Smith, U. Insulin resistance and impaired adipogenesis. Trends Endocrinol. Metab. 2015, 26, 193–200. [Google Scholar] [CrossRef]
- Lessard, J.; Laforest, S.; Pelletier, M.; Leboeuf, M.; Blackburn, L.; Tchernof, A. Low abdominal subcutaneous preadipocyte adipogenesis is associated with visceral obesity, visceral adipocyte hypertrophy, and a dysmetabolic state. Adipocyte 2014, 3, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Osborn, O.; Olefsky, J.M. The cellular and signaling networks linking the immune system and metabolism in disease. Nat. Med. 2012, 18, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, B.; Sultana, R.; Greene, M.W. Adipose tissue and insulin resistance in obese. Biomed. Pharmacother. 2021, 137, 111315. [Google Scholar] [CrossRef]
- Cai, L.; Wang, Z.; Ji, A.; Meyer, J.M.; van der Westhuyzen, D.R. Scavenger receptor CD36 expression contributes to adipose tissue inflammation and cell death in diet-induced obesity. PLoS ONE 2012, 7, e36785. [Google Scholar] [CrossRef] [PubMed]
- Jiao, P.; Ma, J.; Feng, B.; Zhang, H.; Diehl, J.A.; Chin, Y.E.; Yan, W.; Xu, H. FFA-induced adipocyte inflammation and insulin resistance: Involvement of ER stress and IKKbeta pathways. Obesity 2011, 19, 483–491. [Google Scholar] [CrossRef]
- Kawasaki, N.; Asada, R.; Saito, A.; Kanemoto, S.; Imaizumi, K. Obesity-induced endoplasmic reticulum stress causes chronic inflammation in adipose tissue. Sci. Rep. 2012, 2, 799. [Google Scholar] [CrossRef]
- Vandanmagsar, B.; Youm, Y.H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef]
- Roden, M. Mechanisms of Disease: Hepatic steatosis in type 2 diabetes—Pathogenesis and clinical relevance. Nat. Clin. Pract. Endocrinol. Metab. 2006, 2, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Dostert, C.; Petrilli, V.; Van Bruggen, R.; Steele, C.; Mossman, B.T.; Tschopp, J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 2008, 320, 674–677. [Google Scholar] [CrossRef]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef]
- Oslowski, C.M.; Hara, T.; O’Sullivan-Murphy, B.; Kanekura, K.; Lu, S.; Hara, M.; Ishigaki, S.; Zhu, L.J.; Hayashi, E.; Hui, S.T.; et al. Thioredoxin-interacting protein mediates ER stress-induced beta cell death through initiation of the inflammasome. Cell Metab. 2012, 16, 265–273. [Google Scholar] [CrossRef]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.; Brickey, W.J.; Ting, J.P. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef]
- Sheedy, F.J.; Grebe, A.; Rayner, K.J.; Kalantari, P.; Ramkhelawon, B.; Carpenter, S.B.; Becker, C.E.; Ediriweera, H.N.; Mullick, A.E.; Golenbock, D.T.; et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat. Immunol. 2013, 14, 812–820. [Google Scholar] [CrossRef]
- Luo, X.; Li, Y.; Yang, P.; Chen, Y.; Wei, L.; Yu, T.; Xia, J.; Ruan, X.Z.; Zhao, L.; Chen, Y. Obesity induces preadipocyte CD36 expression promoting inflammation via the disruption of lysosomal calcium homeostasis and lysosome function. EBioMedicine 2020, 56, 102797. [Google Scholar] [CrossRef] [PubMed]
- Mizunoe, Y.; Sudo, Y.; Okita, N.; Hiraoka, H.; Mikami, K.; Narahara, T.; Negishi, A.; Yoshida, M.; Higashibata, R.; Watanabe, S.; et al. Involvement of lysosomal dysfunction in autophagosome accumulation and early pathologies in adipose tissue of obese mice. Autophagy 2017, 13, 642–653. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.; Schilling, J.D. Lysosomes integrate metabolic-inflammatory cross-talk in primary macrophage inflammasome activation. J. Biol. Chem. 2014, 289, 9158–9171. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hao, J.W.; Wang, X.; Guo, H.; Sun, H.H.; Lai, X.Y.; Liu, L.Y.; Zhu, M.; Wang, H.Y.; Li, Y.F.; et al. DHHC4 and DHHC5 Facilitate Fatty Acid Uptake by Palmitoylating and Targeting CD36 to the Plasma Membrane. Cell Rep. 2019, 26, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhang, C.; Luo, X.; Wang, P.; Zhou, W.; Zhong, S.; Xie, Y.; Jiang, Y.; Yang, P.; Tang, R.; et al. CD36 palmitoylation disrupts free fatty acid metabolism and promotes tissue inflammation in non-alcoholic steatohepatitis. J. Hepatol. 2018, 69, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Sheedfar, F.; Sung, M.M.; Aparicio-Vergara, M.; Kloosterhuis, N.J.; Miquilena-Colina, M.E.; Vargas-Castrillon, J.; Febbraio, M.; Jacobs, R.L.; de Bruin, A.; Vinciguerra, M.; et al. Increased hepatic CD36 expression with age is associated with enhanced susceptibility to nonalcoholic fatty liver disease. Aging 2014, 6, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Chong, M.; Yin, T.; Chen, R.; Xiang, H.; Yuan, L.; Ding, Y.; Pan, C.C.; Tang, Z.; Alexander, P.B.; Li, Q.J.; et al. CD36 initiates the secretory phenotype during the establishment of cellular senescence. EMBO Rep. 2018, 19. [Google Scholar] [CrossRef]
- Febbraio, M.; Hajjar, D.P.; Silverstein, R.L. CD36: A class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J. Clin. Investig. 2001, 108, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Rao, X.; Zhao, S.; Braunstein, Z.; Mao, H.; Razavi, M.; Duan, L.; Wei, Y.; Toomey, A.C.; Rajagopalan, S.; Zhong, J. Oxidized LDL upregulates macrophage DPP4 expression via TLR4/TRIF/CD36 pathways. EBioMedicine 2019, 41, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Seimon, T.A.; Nadolski, M.J.; Liao, X.; Magallon, J.; Nguyen, M.; Feric, N.T.; Koschinsky, M.L.; Harkewicz, R.; Witztum, J.L.; Tsimikas, S.; et al. Atherogenic lipids and lipoproteins trigger CD36-TLR2-dependent apoptosis in macrophages undergoing endoplasmic reticulum stress. Cell Metab. 2010, 12, 467–482. [Google Scholar] [CrossRef]
- Glatz, J.F.C.; Luiken, J.; Nabben, M. CD36 (SR-B2) as a Target to Treat Lipid Overload-Induced Cardiac Dysfunction. J. Lipid. Atheroscler. 2020, 9, 66–78. [Google Scholar] [CrossRef] [PubMed]
- Shu, H.; Peng, Y.; Hang, W.; Nie, J.; Zhou, N.; Wang, D.W. The role of CD36 in cardiovascular disease. Cardiovasc. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Silverstein, R.L. CD36 signaling in vascular redox stress. Free Radic. Biol. Med. 2019, 136, 159–171. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karunakaran, U.; Elumalai, S.; Moon, J.-S.; Won, K.-C. CD36 Signal Transduction in Metabolic Diseases: Novel Insights and Therapeutic Targeting. Cells 2021, 10, 1833. https://doi.org/10.3390/cells10071833
Karunakaran U, Elumalai S, Moon J-S, Won K-C. CD36 Signal Transduction in Metabolic Diseases: Novel Insights and Therapeutic Targeting. Cells. 2021; 10(7):1833. https://doi.org/10.3390/cells10071833
Chicago/Turabian StyleKarunakaran, Udayakumar, Suma Elumalai, Jun-Sung Moon, and Kyu-Chang Won. 2021. "CD36 Signal Transduction in Metabolic Diseases: Novel Insights and Therapeutic Targeting" Cells 10, no. 7: 1833. https://doi.org/10.3390/cells10071833
APA StyleKarunakaran, U., Elumalai, S., Moon, J.-S., & Won, K.-C. (2021). CD36 Signal Transduction in Metabolic Diseases: Novel Insights and Therapeutic Targeting. Cells, 10(7), 1833. https://doi.org/10.3390/cells10071833