
Diet-Induced Models of Non-Alcoholic Fatty Liver Disease: Food for Thought on Sugar, Fat, and Cholesterol


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. NAFLD, NASH and MAFLD
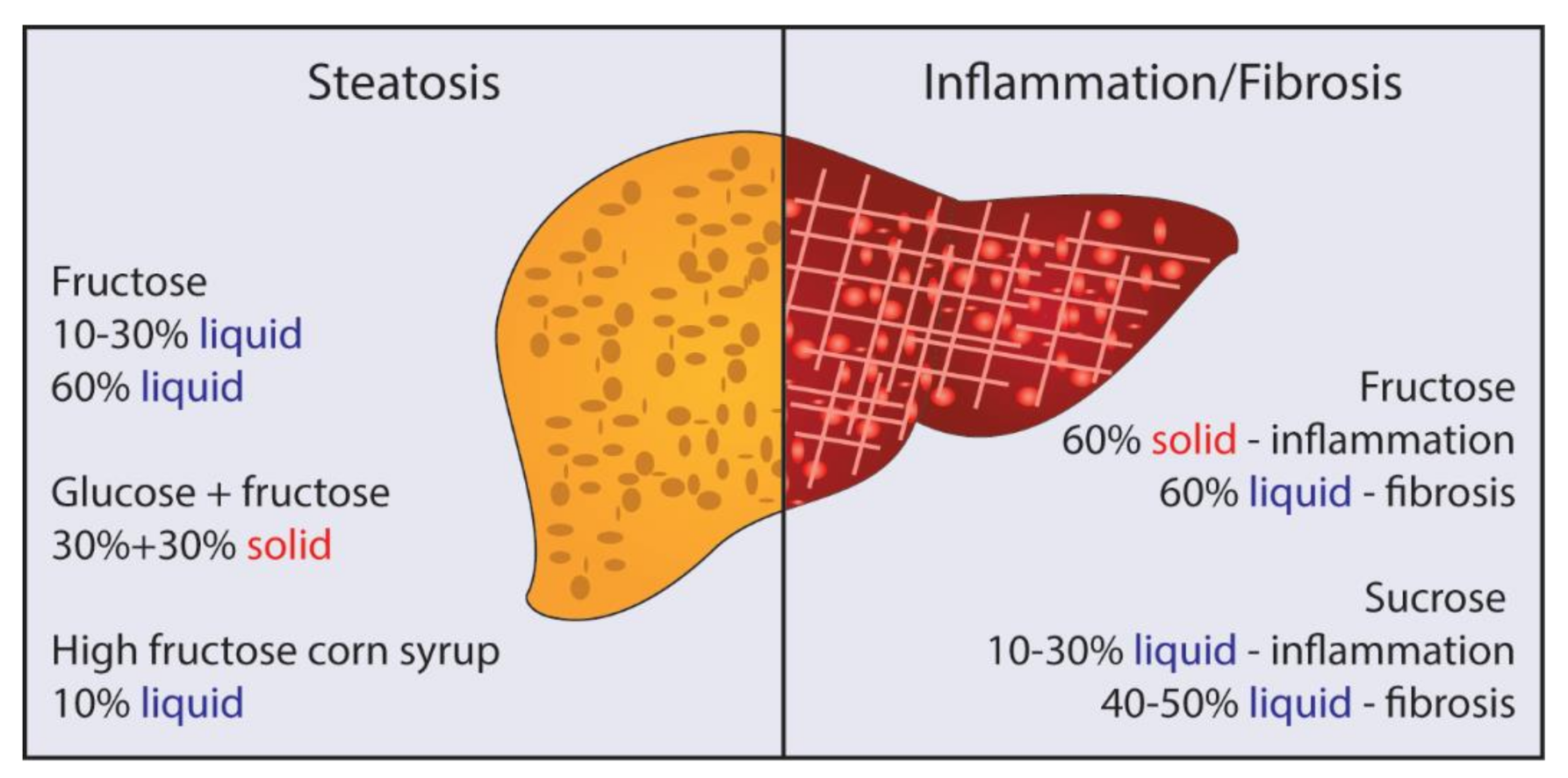
3. Fructose, Glucose, and Sucrose: Is There Really a Difference?
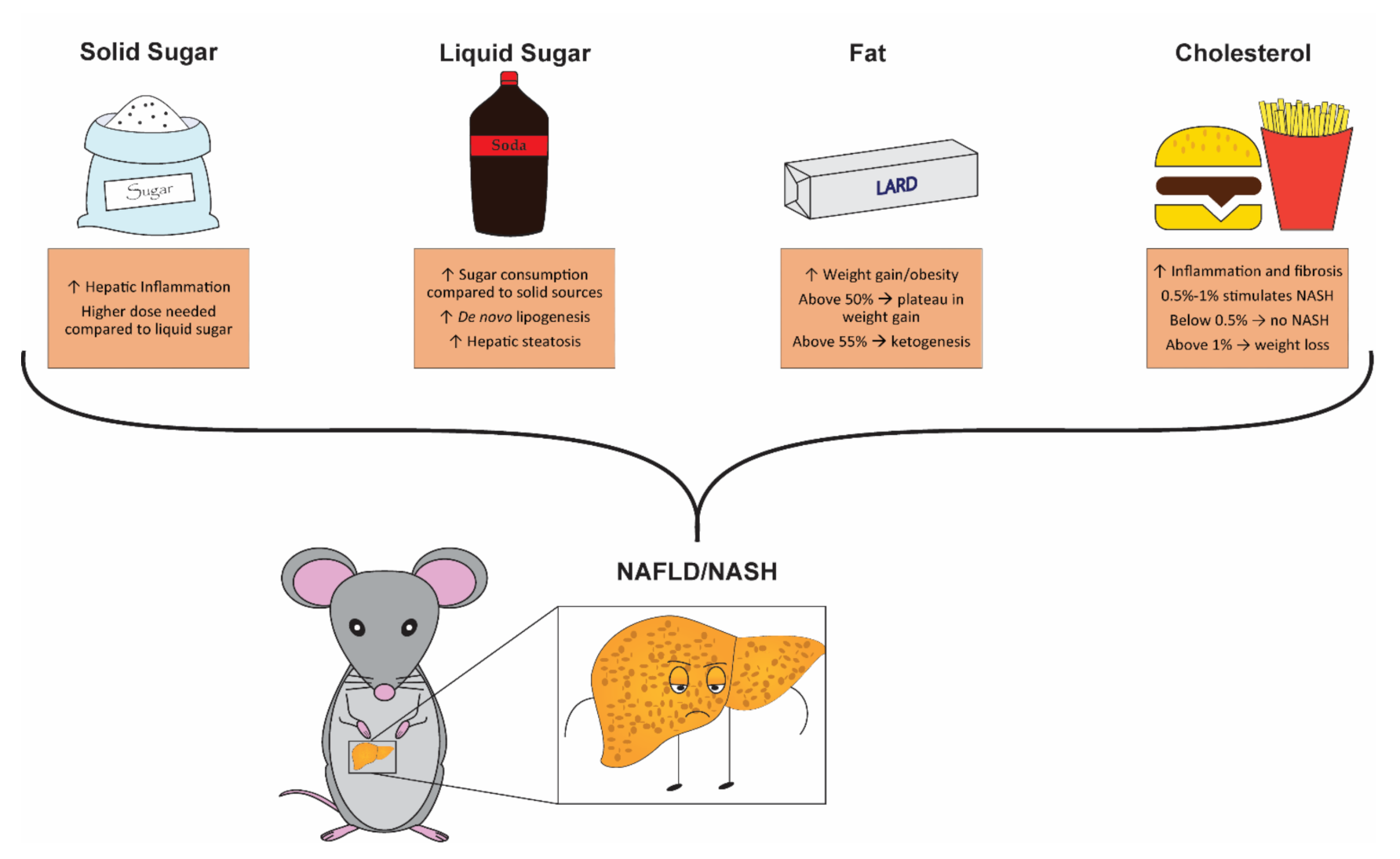
4. Solid vs. Liquid Sugar: Which Is Better for Promoting NAFLD/NASH?
5. Is High Sugar Enough?
6. The Great Fat Debate
7. Cholesterol: A Question of Quantity?
8. Is Trans Fat the New Cholesterol?
9. Matched Control Diets: What Should I Be Using for “Normal” Chow?
10. Is Mouse Strain Important?
11. How Long Do I Need to Feed Mice to Promote NASH?
12. Late-Stage Endpoints
13. So… What Model Should I Use to Study NAFLD/NASH in Rodents?
14. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mitra, S.; De, A.; Chowdhury, A. Epidemiology of non-alcoholic and alcoholic fatty liver diseases. Transl. Gastroenterol. Hepatol. 2020, 5, 16. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Van Herck, M.A.; Vonghia, L.; Francque, S.M. Animal Models of Nonalcoholic Fatty Liver Disease-A Starter’s Guide. Nutrients 2017, 9, 1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Goldin, R.D. Mouse models in non-alcoholic fatty liver disease and steatohepatitis research. Int J. Exp. Pathol 2006, 87, 1–16. [Google Scholar] [CrossRef]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Animal Models of Fibrosis in Nonalcoholic Steatohepatitis: Do They Reflect Human Disease? Adv. Nutr 2020, 11, 1696–1711. [Google Scholar] [CrossRef]
- Eslam, M.; Sanyal, A.J.; George, J.; International Consensus, P. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1. [Google Scholar] [CrossRef]
- Chan, A.W.; Wong, G.L.; Chan, H.Y.; Tong, J.H.; Yu, Y.H.; Choi, P.C.; Chan, H.L.; To, K.F.; Wong, V.W. Concurrent fatty liver increases risk of hepatocellular carcinoma among patients with chronic hepatitis B. J. Gastroenterol. Hepatol. 2017, 32, 667–676. [Google Scholar] [CrossRef]
- Jensen, T.; Abdelmalek, M.F.; Sullivan, S.; Nadeau, K.J.; Green, M.; Roncal, C.; Nakagawa, T.; Kuwabara, M.; Sato, Y.; Kang, D.H.; et al. Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 1063–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Softic, S.; Gupta, M.K.; Wang, G.X.; Fujisaka, S.; O’Neill, B.T.; Rao, T.N.; Willoughby, J.; Harbison, C.; Fitzgerald, K.; Ilkayeva, O.; et al. Divergent effects of glucose and fructose on hepatic lipogenesis and insulin signaling. J. Clin. Investig. 2017, 127, 4059–4074. [Google Scholar] [CrossRef] [Green Version]
- Basaranoglu, M.; Basaranoglu, G.; Bugianesi, E. Carbohydrate intake and nonalcoholic fatty liver disease: Fructose as a weapon of mass destruction. Hepatobiliary Surg. Nutr. 2015, 4, 109–116. [Google Scholar] [CrossRef]
- Sanchez-Lozada, L.G.; Mu, W.; Roncal, C.; Sautin, Y.Y.; Abdelmalek, M.; Reungjui, S.; Le, M.; Nakagawa, T.; Lan, H.Y.; Yu, X.; et al. Comparison of free fructose and glucose to sucrose in the ability to cause fatty liver. Eur. J. Nutr. 2010, 49, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, J.S.; Mietus-Snyder, M.; Valente, A.; Schwarz, J.M.; Lustig, R.H. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 251–264. [Google Scholar] [CrossRef] [PubMed]
- DiNicolantonio, J.J.; Subramonian, A.M.; O’Keefe, J.H. Added fructose as a principal driver of non-alcoholic fatty liver disease: A public health crisis. Open Heart 2017, 4, e000631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linden, A.G.; Li, S.; Choi, H.Y.; Fang, F.; Fukasawa, M.; Uyeda, K.; Hammer, R.E.; Horton, J.D.; Engelking, L.J.; Liang, G. Interplay between ChREBP and SREBP-1c coordinates postprandial glycolysis and lipogenesis in livers of mice. J. Lipid Res. 2018, 59, 475–487. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Fernandez, S.; Garces-Rimon, M.; Vera, G.; Astier, J.; Landrier, J.F.; Miguel, M. High Fat/High Glucose Diet Induces Metabolic Syndrome in an Experimental Rat Model. Nutrients 2018, 10, 1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronnevik, A.K.; Gudbrandsen, O.A. Substitution of high-dose sucrose with fructose in high-fat diets resulted in higher plasma concentrations of aspartic acid, cystine, glutamic acid, ornithine and phenylalanine, and higher urine concentrations of arginine and citrulline. Nutr. Res. 2020, 79, 100–110. [Google Scholar] [CrossRef]
- Sadowska, J.; Bruszkowska, M. Comparing the effects of sucrose and high-fructose corn syrup on lipid metabolism and the risk of cardiovascular disease in male rats. Acta Sci. Pol. Technol. Aliment. 2017, 16, 231–240. [Google Scholar] [CrossRef] [Green Version]
- Mock, K.; Lateef, S.; Benedito, V.A.; Tou, J.C. High-fructose corn syrup-55 consumption alters hepatic lipid metabolism and promotes triglyceride accumulation. J. Nutr. Biochem. 2017, 39, 32–39. [Google Scholar] [CrossRef]
- Andres-Hernando, A.; Orlicky, D.J.; Kuwabara, M.; Ishimoto, T.; Nakagawa, T.; Johnson, R.J.; Lanaspa, M.A. Deletion of Fructokinase in the Liver or in the Intestine Reveals Differential Effects on Sugar-Induced Metabolic Dysfunction. Cell Metab. 2020, 32, 117–127.e3. [Google Scholar] [CrossRef]
- Liu, X.J.; Duan, N.N.; Liu, C.; Niu, C.; Liu, X.P.; Wu, J. Characterization of a murine nonalcoholic steatohepatitis model induced by high fat high calorie diet plus fructose and glucose in drinking water. Lab. Investig. 2018, 98, 1184–1199. [Google Scholar] [CrossRef] [PubMed]
- Rosinger, A.; Herrick, K.; Gahche, J.; Park, S. Sugar-sweetened Beverage Consumption Among U.S. Adults, 2011–2014. NCHS Data Brief 2017, 270, 1–8. [Google Scholar]
- Chen, H.; Wang, J.; Li, Z.; Lam, C.W.K.; Xiao, Y.; Wu, Q.; Zhang, W. Consumption of Sugar-Sweetened Beverages Has a Dose-Dependent Effect on the Risk of Non-Alcoholic Fatty Liver Disease: An Updated Systematic Review and Dose-Response Meta-Analysis. Int. J. Environ. Res. Public Health 2019, 16, 2192. [Google Scholar] [CrossRef] [Green Version]
- Zelber-Sagi, S.; Nitzan-Kaluski, D.; Goldsmith, R.; Webb, M.; Blendis, L.; Halpern, Z.; Oren, R. Long term nutritional intake and the risk for non-alcoholic fatty liver disease (NAFLD): A population based study. J. Hepatol. 2007, 47, 711–717. [Google Scholar] [CrossRef]
- Toop, C.R.; Gentili, S. Fructose Beverage Consumption Induces a Metabolic Syndrome Phenotype in the Rat: A Systematic Review and Meta-Analysis. Nutrients 2016, 8, 577. [Google Scholar] [CrossRef] [Green Version]
- De Souza, L.; Barros, W.M.; De Souza, R.M.; Delanogare, E.; Machado, A.E.; Braga, S.P.; Rosa, G.K.; Nardi, G.M.; Rafacho, A.; Speretta, G.F.F.; et al. Impact of different fructose concentrations on metabolic and behavioral parameters of male and female mice. Physiol. Behav. 2021, 228, 113187. [Google Scholar] [CrossRef] [PubMed]
- Togo, J.; Hu, S.; Li, M.; Niu, C.; Speakman, J.R. Impact of dietary sucrose on adiposity and glucose homeostasis in C57BL/6J mice depends on mode of ingestion: Liquid or solid. Mol. Metab. 2019, 27, 22–32. [Google Scholar] [CrossRef]
- Mastrocola, R.; Ferrocino, I.; Liberto, E.; Chiazza, F.; Cento, A.S.; Collotta, D.; Querio, G.; Nigro, D.; Bitonto, V.; Cutrin, J.C.; et al. Fructose liquid and solid formulations differently affect gut integrity, microbiota composition and related liver toxicity: A comparative in vivo study. J. Nutr. Biochem. 2018, 55, 185–199. [Google Scholar] [CrossRef]
- Sundborn, G.; Thornley, S.; Merriman, T.R.; Lang, B.; King, C.; Lanaspa, M.A.; Johnson, R.J. Are Liquid Sugars Different from Solid Sugar in Their Ability to Cause Metabolic Syndrome? Obesity 2019, 27, 879–887. [Google Scholar] [CrossRef]
- Welsh, J.A.; Sharma, A.J.; Grellinger, L.; Vos, M.B. Consumption of added sugars is decreasing in the United States. Am. J. Clin. Nutr. 2011, 94, 726–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vos, M.B.; Kimmons, J.E.; Gillespie, C.; Welsh, J.; Blanck, H.M. Dietary fructose consumption among US children and adults: The Third National Health and Nutrition Examination Survey. Medscape J. Med. 2008, 10, 160. [Google Scholar]
- Sadowska, J.; Rygielska, M. The effect of high fructose corn syrup on the plasma insulin and leptin concentration, body weight gain and fat accumulation in rat. Adv. Clin. Exp. Med. 2019, 28, 879–884. [Google Scholar] [CrossRef]
- Baiges-Gaya, G.; Fernandez-Arroyo, S.; Luciano-Mateo, F.; Cabre, N.; Rodriguez-Tomas, E.; Hernandez-Aguilera, A.; Castane, H.; Romeu, M.; Nogues, M.R.; Camps, J.; et al. Hepatic metabolic adaptation and adipose tissue expansion are altered in mice with steatohepatitis induced by high-fat high sucrose diet. J. Nutr. Biochem. 2020, 89, 108559. [Google Scholar] [CrossRef]
- Malik, A.N.; Simoes, I.C.M.; Rosa, H.S.; Khan, S.; Karkucinska-Wieckowska, A.; Wieckowski, M.R. A Diet Induced Maladaptive Increase in Hepatic Mitochondrial DNA Precedes OXPHOS Defects and May Contribute to Non-Alcoholic Fatty Liver Disease. Cells 2019, 8, 1222. [Google Scholar] [CrossRef] [Green Version]
- Acosta-Cota, S.J.; Aguilar-Medina, E.M.; Ramos-Payan, R.; Ruiz-Quinonez, A.K.; Romero-Quintana, J.G.; Montes-Avila, J.; Rendon-Maldonado, J.G.; Sanchez-Lopez, A.; Centurion, D.; Osuna-Martinez, U. Histopathological and biochemical changes in the development of nonalcoholic fatty liver disease induced by high-sucrose diet at different times. Can. J. Physiol. Pharmacol. 2019, 97, 23–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simoes, I.C.M.; Karkucinska-Wieckowska, A.; Janikiewicz, J.; Szymanska, S.; Pronicki, M.; Dobrzyn, P.; Dabrowski, M.; Dobrzyn, A.; Oliveira, P.J.; Zischka, H.; et al. Western Diet Causes Obesity-Induced Nonalcoholic Fatty Liver Disease Development by Differentially Compromising the Autophagic Response. Antioxidants 2020, 9, 995. [Google Scholar] [CrossRef]
- Luo, Y.; Burrington, C.M.; Graff, E.C.; Zhang, J.; Judd, R.L.; Suksaranjit, P.; Kaewpoowat, Q.; Davenport, S.K.; O’Neill, A.M.; Greene, M.W. Metabolic phenotype and adipose and liver features in a high-fat Western diet-induced mouse model of obesity-linked NAFLD. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E418–E439. [Google Scholar] [CrossRef] [Green Version]
- Ito, M.; Suzuki, J.; Tsujioka, S.; Sasaki, M.; Gomori, A.; Shirakura, T.; Hirose, H.; Ito, M.; Ishihara, A.; Iwaasa, H.; et al. Longitudinal analysis of murine steatohepatitis model induced by chronic exposure to high-fat diet. Hepatol. Res. 2007, 37, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Wang, L.; Yang, D.; Li, L.; Togo, J.; Wu, Y.; Liu, Q.; Li, B.; Li, M.; Wang, G.; et al. Dietary Fat, but Not Protein or Carbohydrate, Regulates Energy Intake and Causes Adiposity in Mice. Cell Metab 2018, 28, 415–431.e4. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, K.C.; Strohacker, K.; McFarlin, B.K. Considerations to maximize fat mass gain in a mouse model of diet-induced weight gain. Lab. Anim. 2013, 47, 266–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Recena Aydos, L.; Aparecida do Amaral, L.; Serafim de Souza, R.; Jacobowski, A.C.; Freitas Dos Santos, E.; Rodrigues Macedo, M.L. Nonalcoholic Fatty Liver Disease Induced by High-Fat Diet in C57bl/6 Models. Nutrients 2019, 11, 3067. [Google Scholar] [CrossRef] [Green Version]
- Henkel, J.; Alfine, E.; Sain, J.; Johrens, K.; Weber, D.; Castro, J.P.; Konig, J.; Stuhlmann, C.; Vahrenbrink, M.; Jonas, W.; et al. Soybean Oil-Derived Poly-Unsaturated Fatty Acids Enhance Liver Damage in NAFLD Induced by Dietary Cholesterol. Nutrients 2018, 10, 1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henkel, J.; Coleman, C.D.; Schraplau, A.; Jhrens, K.; Weber, D.; Castro, J.P.; Hugo, M.; Schulz, T.J.; Kramer, S.; Schurmann, A.; et al. Induction of steatohepatitis (NASH) with insulin resistance in wildtype B6 mice by a western-type diet containing soybean oil and cholesterol. Mol. Med. 2017, 23, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Bough, K.J.; Eagles, D.A. A ketogenic diet increases the resistance to pentylenetetrazole-induced seizures in the rat. Epilepsia 1999, 40, 138–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, A.R.; Pissios, P.; Otu, H.; Roberson, R.; Xue, B.; Asakura, K.; Furukawa, N.; Marino, F.E.; Liu, F.F.; Kahn, B.B.; et al. A high-fat, ketogenic diet induces a unique metabolic state in mice. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E1724–E1739. [Google Scholar] [CrossRef]
- Sikder, K.; Shukla, S.K.; Patel, N.; Singh, H.; Rafiq, K. High Fat Diet Upregulates Fatty Acid Oxidation and Ketogenesis via Intervention of PPAR-gamma. Cell. Physiol. Biochem. 2018, 48, 1317–1331. [Google Scholar] [CrossRef]
- Matsuzawa, N.; Takamura, T.; Kurita, S.; Misu, H.; Ota, T.; Ando, H.; Yokoyama, M.; Honda, M.; Zen, Y.; Nakanuma, Y.; et al. Lipid-induced oxidative stress causes steatohepatitis in mice fed an atherogenic diet. Hepatology 2007, 46, 1392–1403. [Google Scholar] [CrossRef]
- Buettner, R.; Ascher, M.; Gabele, E.; Hellerbrand, C.; Kob, R.; Bertsch, T.; Bollheimer, L.C. Olive oil attenuates the cholesterol-induced development of nonalcoholic steatohepatitis despite increased insulin resistance in a rodent model. Horm. Metab. Res. 2013, 45, 795–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Blazquez, R.; Alcala, M.; Fernandez-Alfonso, M.S.; Villa-Valverde, P.; Viana, M.; Gil-Ortega, M.; Somoza, B. Relevance of control diet choice in metabolic studies: Impact in glucose homeostasis and vascular function. Sci. Rep. 2020, 10, 2902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ioannou, G.N.; Subramanian, S.; Chait, A.; Haigh, W.G.; Yeh, M.M.; Farrell, G.C.; Lee, S.P.; Savard, C. Cholesterol crystallization within hepatocyte lipid droplets and its role in murine NASH. J. Lipid Res. 2017, 58, 1067–1079. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.H.; Choi, D.; Kim, J.Y.; Lee, J.H.; Koo, S.H. Fast food diet-induced non-alcoholic fatty liver disease exerts early protective effect against acetaminophen intoxication in mice. BMC Gastroenterol. 2017, 17, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, J.Q.; Teoh, N.; Xu, L.; Pok, S.; Li, X.; Chu, E.S.H.; Chiu, J.; Dong, L.; Arfianti, E.; Haigh, W.G.; et al. Dietary cholesterol promotes steatohepatitis related hepatocellular carcinoma through dysregulated metabolism and calcium signaling. Nat. Commun. 2018, 9, 4490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puschel, G.P.; Henkel, J. Dietary cholesterol does not break your heart but kills your liver. Porto Biomed. J. 2018, 3, e12. [Google Scholar] [CrossRef]
- Savari, F.; Mard, S.A.; Badavi, M.; Rezaie, A.; Gharib-Naseri, M.K. A new method to induce nonalcoholic steatohepatitis (NASH) in mice. BMC Gastroenterol. 2019, 19, 125. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Leveille, M.; Courty, E.; Gunes, A.; B, N.N.; Estall, J.L. Differences in metabolic and liver pathobiology induced by two dietary mouse models of nonalcoholic fatty liver disease. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E863–E876. [Google Scholar] [CrossRef]
- Krishnan, A.; Abdullah, T.S.; Mounajjed, T.; Hartono, S.; McConico, A.; White, T.; LeBrasseur, N.; Lanza, I.; Nair, S.; Gores, G.; et al. A longitudinal study of whole body, tissue, and cellular physiology in a mouse model of fibrosing NASH with high fidelity to the human condition. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G666–G680. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, G.; Nonomura, T.; Sasaki, M.; Ishida, Y.; Arai, S.; Miyazaki, T. AIM-deficient mouse fed a high-trans fat, high-cholesterol diet: A new animal model for nonalcoholic fatty liver disease. Exp. Anim. 2019, 68, 147–158. [Google Scholar] [CrossRef]
- Soliman, G.A. Dietary Cholesterol and the Lack of Evidence in Cardiovascular Disease. Nutrients 2018, 10, 780. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; McClure, S.T.; Appel, L.J. Dietary Cholesterol Intake and Sources among U.S. Adults: Results from National Health and Nutrition Examination Surveys (NHANES), 2001–2014. Nutrients 2018, 10, 771. [Google Scholar] [CrossRef] [Green Version]
- Ding, X.; Zhang, W.; Li, S.; Yang, H. The role of cholesterol metabolism in cancer. Am. J. Cancer Res. 2019, 9, 219–227. [Google Scholar] [PubMed]
- Wang, X.; Tanaka, N.; Hu, X.; Kimura, T.; Lu, Y.; Jia, F.; Sato, Y.; Nakayama, J.; Moriya, K.; Koike, K.; et al. A high-cholesterol diet promotes steatohepatitis and liver tumorigenesis in HCV core gene transgenic mice. Arch. Toxicol 2019, 93, 1713–1725. [Google Scholar] [CrossRef]
- De Pergola, G.; Silvestris, F. Obesity as a major risk factor for cancer. J. Obes. 2013, 2013, 291546. [Google Scholar] [CrossRef] [Green Version]
- Esposito, K.; Chiodini, P.; Colao, A.; Lenzi, A.; Giugliano, D. Metabolic syndrome and risk of cancer: A systematic review and meta-analysis. Diabetes Care 2012, 35, 2402–2411. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.Q.; Carey, M.C. Measurement of intestinal cholesterol absorption by plasma and fecal dual-isotope ratio, mass balance, and lymph fistula methods in the mouse: An analysis of direct versus indirect methodologies. J. Lipid Res. 2003, 44, 1042–1059. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, M.; Davis, D.L.; Vick, B.R.; Russell, D.W. Genetic analysis of intestinal cholesterol absorption in inbred mice. J. Lipid Res. 2001, 42, 1801–1811. [Google Scholar] [CrossRef]
- Sontag, T.J.; Chellan, B.; Getz, G.S.; Reardon, C.A. Differing rates of cholesterol absorption among inbred mouse strains yield differing levels of HDL-cholesterol. J. Lipid Res. 2013, 54, 2515–2524. [Google Scholar] [CrossRef] [Green Version]
- Carson, J.A.S.; Lichtenstein, A.H.; Anderson, C.A.M.; Appel, L.J.; Kris-Etherton, P.M.; Meyer, K.A.; Petersen, K.; Polonsky, T.; Van Horn, L.; On behalf of the American Heart Association Nutrition Committee of the Council on Lifestyle and Cardiometabolic Health. Dietary Cholesterol and Cardiovascular Risk: A Science Advisory From the American Heart Association. Circulation 2020, 141, e39–e53. [Google Scholar] [CrossRef] [Green Version]
- Ascherio, A.; Willett, W.C. Health effects of trans fatty acids. Am. J. Clin. Nutr. 1997, 66, 1006S–1010S. [Google Scholar] [CrossRef] [PubMed]
- Koppe, S.W.; Elias, M.; Moseley, R.H.; Green, R.M. Trans fat feeding results in higher serum alanine aminotransferase and increased insulin resistance compared with a standard murine high-fat diet. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297, G378–G384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado, R.M.; Stefano, J.T.; Oliveira, C.P.; Mello, E.S.; Ferreira, F.D.; Nunes, V.S.; de Lima, V.M.; Quintao, E.C.; Catanozi, S.; Nakandakare, E.R.; et al. Intake of trans fatty acids causes nonalcoholic steatohepatitis and reduces adipose tissue fat content. J. Nutr. 2010, 140, 1127–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obara, N.; Fukushima, K.; Ueno, Y.; Wakui, Y.; Kimura, O.; Tamai, K.; Kakazu, E.; Inoue, J.; Kondo, Y.; Ogawa, N.; et al. Possible involvement and the mechanisms of excess trans-fatty acid consumption in severe NAFLD in mice. J. Hepatol. 2010, 53, 326–334. [Google Scholar] [CrossRef]
- Drescher, H.K.; Weiskirchen, R.; Fulop, A.; Hopf, C.; de San Roman, E.G.; Huesgen, P.F.; de Bruin, A.; Bongiovanni, L.; Christ, A.; Tolba, R.; et al. The Influence of Different Fat Sources on Steatohepatitis and Fibrosis Development in the Western Diet Mouse Model of Non-alcoholic Steatohepatitis (NASH). Front. Physiol. 2019, 10, 770. [Google Scholar] [CrossRef] [PubMed]
- Boland, M.L.; Oro, D.; Tolbol, K.S.; Thrane, S.T.; Nielsen, J.C.; Cohen, T.S.; Tabor, D.E.; Fernandes, F.; Tovchigrechko, A.; Veidal, S.S.; et al. Towards a standard diet-induced and biopsy-confirmed mouse model of non-alcoholic steatohepatitis: Impact of dietary fat source. World J. Gastroenterol. 2019, 25, 4904–4920. [Google Scholar] [CrossRef] [PubMed]
- Fengler, V.H.; Macheiner, T.; Kessler, S.M.; Czepukojc, B.; Gemperlein, K.; Muller, R.; Kiemer, A.K.; Magnes, C.; Haybaeck, J.; Lackner, C.; et al. Susceptibility of Different Mouse Wild Type Strains to Develop Diet-Induced NAFLD/AFLD-Associated Liver Disease. PLoS ONE 2016, 11, e0155163. [Google Scholar] [CrossRef]
- Simon, M.M.; Greenaway, S.; White, J.K.; Fuchs, H.; Gailus-Durner, V.; Wells, S.; Sorg, T.; Wong, K.; Bedu, E.; Cartwright, E.J.; et al. A comparative phenotypic and genomic analysis of C57BL/6J and C57BL/6N mouse strains. Genome Biol. 2013, 14, R82. [Google Scholar] [CrossRef]
- Toye, A.A.; Lippiat, J.D.; Proks, P.; Shimomura, K.; Bentley, L.; Hugill, A.; Mijat, V.; Goldsworthy, M.; Moir, L.; Haynes, A.; et al. A genetic and physiological study of impaired glucose homeostasis control in C57BL/6J mice. Diabetologia 2005, 48, 675–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fergusson, G.; Ethier, M.; Guevremont, M.; Chretien, C.; Attane, C.; Joly, E.; Fioramonti, X.; Prentki, M.; Poitout, V.; Alquier, T. Defective insulin secretory response to intravenous glucose in C57Bl/6J compared to C57Bl/6N mice. Mol. Metab. 2014, 3, 848–854. [Google Scholar] [CrossRef]
- Kawashita, E.; Ishihara, K.; Nomoto, M.; Taniguchi, M.; Akiba, S. A comparative analysis of hepatic pathological phenotypes in C57BL/6J and C57BL/6N mouse strains in non-alcoholic steatohepatitis models. Sci. Rep. 2019, 9, 204. [Google Scholar] [CrossRef] [Green Version]
- Oldford, C.; Kuksal, N.; Gill, R.; Young, A.; Mailloux, R.J. Estimation of the hydrogen peroxide producing capacities of liver and cardiac mitochondria isolated from C57BL/6N and C57BL/6J mice. Free Radic. Biol. Med. 2019, 135, 15–27. [Google Scholar] [CrossRef]
- Asgharpour, A.; Cazanave, S.C.; Pacana, T.; Seneshaw, M.; Vincent, R.; Banini, B.A.; Kumar, D.P.; Daita, K.; Min, H.K.; Mirshahi, F.; et al. A diet-induced animal model of non-alcoholic fatty liver disease and hepatocellular cancer. J. Hepatol. 2016, 65, 579–588. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Allen, A.M.; Wang, Z.; Prokop, L.J.; Murad, M.H.; Loomba, R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: A systematic review and meta-analysis of paired-biopsy studies. Clin. Gastroenterol. Hepatol. 2015, 13, 643–654.e9. [Google Scholar] [CrossRef] [Green Version]
- Harris, S.E.; Poolman, T.M.; Arvaniti, A.; Cox, R.D.; Gathercole, L.L.; Tomlinson, J.W. The American lifestyle-induced obesity syndrome diet in male and female rodents recapitulates the clinical and transcriptomic features of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 319, G345–G360. [Google Scholar] [CrossRef]
- Fouret, G.; Gaillet, S.; Lecomte, J.; Bonafos, B.; Djohan, F.; Barea, B.; Badia, E.; Coudray, C.; Feillet-Coudray, C. 20-Week follow-up of hepatic steatosis installation and liver mitochondrial structure and activity and their interrelation in rats fed a high-fat-high-fructose diet. Br. J. Nutr. 2018, 119, 368–380. [Google Scholar] [CrossRef] [Green Version]
- Casagrande, B.P.; Gomes, M.F.P.; Moura, E.O.C.; Santos, A.C.C.; Kubota, M.C.; Ribeiro, D.A.; Pisani, L.P.; Medeiros, A.; Estadella, D. Age-dependent hepatic alterations induced by a high-fat high-fructose diet. Inflamm. Res. 2019, 68, 359–368. [Google Scholar] [CrossRef]
- Brown, Z.J.; Heinrich, B.; Greten, T.F. Mouse models of hepatocellular carcinoma: An overview and highlights for immunotherapy research. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 536–554. [Google Scholar] [CrossRef]
- Zhang, X.; Coker, O.O.; Chu, E.S.; Fu, K.; Lau, H.C.H.; Wang, Y.X.; Chan, A.W.H.; Wei, H.; Yang, X.; Sung, J.J.Y.; et al. Dietary cholesterol drives fatty liver-associated liver cancer by modulating gut microbiota and metabolites. Gut 2021, 70, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Straniero, S.; Laskar, A.; Savva, C.; Hardfeldt, J.; Angelin, B.; Rudling, M. Of mice and men: Murine bile acids explain species differences in the regulation of bile acid and cholesterol metabolism. J. Lipid Res. 2020, 61, 480–491. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S.M.; Li, H.; Zhu, X.; Shah, A.S.; Lu, L.J.; Davidson, W.S. A comparison of the mouse and human lipoproteome: Suitability of the mouse model for studies of human lipoproteins. J. Proteome Res. 2015, 14, 2686–2695. [Google Scholar] [CrossRef] [Green Version]
- Krych, L.; Hansen, C.H.; Hansen, A.K.; van den Berg, F.W.; Nielsen, D.S. Quantitatively different, yet qualitatively alike: A meta-analysis of the mouse core gut microbiome with a view towards the human gut microbiome. PLoS ONE 2013, 8, e62578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coskun, T.; Bina, H.A.; Schneider, M.A.; Dunbar, J.D.; Hu, C.C.; Chen, Y.; Moller, D.E.; Kharitonenkov, A. Fibroblast growth factor 21 corrects obesity in mice. Endocrinology 2008, 149, 6018–6027. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Saxena, N.K.; Lin, S.; Gupta, N.A.; Anania, F.A. Exendin-4, a glucagon-like protein-1 (GLP-1) receptor agonist, reverses hepatic steatosis in ob/ob mice. Hepatology 2006, 43, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Gilroy, C.A.; Capozzi, M.E.; Varanko, A.K.; Tong, J.; D’Alessio, D.A.; Campbell, J.E.; Chilkoti, A. Sustained release of a GLP-1 and FGF21 dual agonist from an injectable depot protects mice from obesity and hyperglycemia. Sci. Adv. 2020, 6, eaaz9890. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eng, J.M.; Estall, J.L. Diet-Induced Models of Non-Alcoholic Fatty Liver Disease: Food for Thought on Sugar, Fat, and Cholesterol. Cells 2021, 10, 1805. https://doi.org/10.3390/cells10071805
Eng JM, Estall JL. Diet-Induced Models of Non-Alcoholic Fatty Liver Disease: Food for Thought on Sugar, Fat, and Cholesterol. Cells. 2021; 10(7):1805. https://doi.org/10.3390/cells10071805
Chicago/Turabian StyleEng, James M., and Jennifer L. Estall. 2021. "Diet-Induced Models of Non-Alcoholic Fatty Liver Disease: Food for Thought on Sugar, Fat, and Cholesterol" Cells 10, no. 7: 1805. https://doi.org/10.3390/cells10071805
APA StyleEng, J. M., & Estall, J. L. (2021). Diet-Induced Models of Non-Alcoholic Fatty Liver Disease: Food for Thought on Sugar, Fat, and Cholesterol. Cells, 10(7), 1805. https://doi.org/10.3390/cells10071805