
Vasculogenic Mimicry in Breast Cancer: Clinical Relevance and Drivers

 ,
, 
 , and
, and 
Abstract
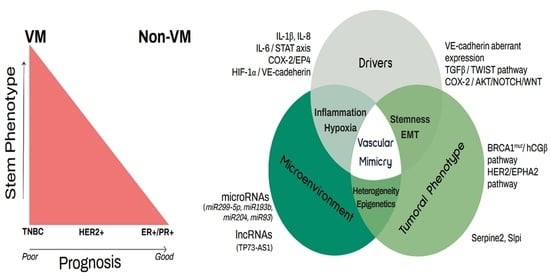
1. Introduction
What Is Vascular Mimicry?
2. First Highlights of VM in Breast Cancer
3. Clinical Relevance of VM in Breast Cancer and Association with Clinicopathological Parameters
4. Relationship between VM and Tumor Phenotype
5. Drivers of Vascular Mimicry in Breast Cancer
6. VM Regulation by Noncoding RNAs in Breast Cancer
7. Targeting Microenvironment to Overcome VM in Breast Cancer
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DDAH1 | Dimethylarginine dimethylaminohydrolase 1 |
| ECM | Extracellular matrix |
| ECs | Endothelial cells |
| EMT | Epithelial-to-mesenchymal transition |
| hCG | Human chorionic gonadotropin |
| HOTAIR | HOX transcript antisense RNA |
| IL | Interleukin |
| LATS2 | Large tumor suppressor, homology 2 |
| LH | Luteinizing hormone |
| LHCGR | Luteinizing hormone/chorionic gonadotropin receptor |
| lncRNAs | Long noncoding RNAs |
| LRIG1 | Leucine rich repeats and immunoglobulin like domains 1 |
| miRNAs | MicroRNAs |
| ncRNAs | Noncoding RNAs |
| PAS | Periodic acid-Schiff |
| RGS3 | G protein signaling 3 |
| RR | Relative risk |
| S1PR1 | Sphingosine-1 phosphate receptor 1 |
| STAT3 | Signal transducer and activator of transcription 3 |
| TNBC | Triple-negative breast cancer |
| TKR | Tyrosine kinase receptor |
| TP73-AS1 | P73 antisense RNA 1T |
| UTR | Untranslated region |
| VM | Vasculogenic mimicry |
References
- Kolte, D.; McClung, J.A.; Aronow, W.S. Chapter 6—Vasculogenesis and Angiogenesis. In Translational Research in Coronary Artery Disease; Aronow, W.S., McClung, J.A., Eds.; Academic Press: Boston, MA, USA, 2016; pp. 49–65. [Google Scholar]
- Patan, S.; Munn, L.L.; Jain, R.K. Intussusceptive Microvascular Growth in a Human Colon Adenocarcinoma Xenograft: A Novel Mechanism of Tumor Angiogenesis. Microvasc. Res. 1996, 51, 260–272. [Google Scholar] [CrossRef]
- Rodríguez-Núñez, I.; Romero, F.; González, M.; Campos, R.R. Biología del Desarrollo Vascular: Mecanismos en Condiciones Físiológicas y Estrés Flujo. Int. J. Morphol. 2015, 33, 1348–1354. [Google Scholar] [CrossRef]
- Kuczynski, E.A.; Vermeulen, P.B.; Pezzella, F.; Kerbel, R.S.; Reynolds, A.R. Vessel co-option in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 469–493. [Google Scholar] [CrossRef]
- Bergers, G.; Benjamin, L.E. Tumorigenesis and the angiogenic switch. Nat. Reviews Cancer 2003, 3, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Folberg, R.; Hendrix, M.J.; Maniotis, A.J. Vasculogenic mimicry and tumor angiogenesis. Am. J. Pathol. 2000, 156, 361–381. [Google Scholar] [CrossRef]
- Maniotis, A.J.; Folberg, R.; Hess, A.; Seftor, E.A.; Gardner, L.M.; Pe’er, J.; Trent, J.M.; Meltzer, P.S.; Hendrix, M.J. Vascular channel formation by human melanoma cells in vivo and in vitro: Vasculogenic mimicry. Am. J. Pathol. 1999, 155, 739–752. [Google Scholar] [CrossRef]
- Folberg, R.; Arbieva, Z.; Moses, J.; Hayee, A.; Sandal, T.; Kadkol, S.; Lin, A.Y.; Valyi-Nagy, K.; Setty, S.; Leach, L.; et al. Tumor cell plasticity in uveal melanoma: Microenvironment directed dampening of the invasive and metastatic genotype and phenotype accompanies the generation of vasculogenic mimicry patterns. Am. J. Pathol. 2006, 169, 1376–1389. [Google Scholar] [CrossRef] [PubMed]
- Basu, G.D.; Liang, W.S.; Stephan, D.A.; Wegener, L.T.; Conley, C.R.; Pockaj, B.A.; Mukherjee, P. A novel role for cyclooxygenase-2 in regulating vascular channel formation by human breast cancer cells. Breast Cancer Res. 2006, 8, 69. [Google Scholar] [CrossRef]
- Chiao, M.T.; Yang, Y.C.; Cheng, W.Y.; Shen, C.C.; Ko, J.L. CD133+ glioblastoma stem-like cells induce vascular mimicry in vivo. Curr. Neurovascular Res. 2011, 8, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Bellido, D.; Serrano-Saenz, S.; Fernández-Cortés, M.; Oliver, F.J. Vasculogenic mimicry signaling revisited: Focus on non-vascular VE-cadherin. Mol. Cancer 2017, 16, 65. [Google Scholar] [CrossRef]
- Andonegui-Elguera, M.A.; Alfaro-Mora, Y.; Caceres-Gutierrez, R.; Caro-Sanchez, C.H.S.; Herrera, L.A.; Diaz-Chavez, J. An Overview of Vasculogenic Mimicry in Breast Cancer. Front. Oncol. 2020, 10, 220. [Google Scholar] [CrossRef]
- Breier, G.; Grosser, M.; Rezaei, M. Endothelial cadherins in cancer. Cell Tissue Res. 2014, 355, 523–527. [Google Scholar] [CrossRef]
- Hendrix, M.J.; Seftor, E.A.; Hess, A.R.; Seftor, R.E. Vasculogenic mimicry and tumour-cell plasticity: Lessons from melanoma. Nat. Rev. Cancer 2003, 3, 411–421. [Google Scholar] [CrossRef]
- van der Schaft, D.W.; Seftor, R.E.; Seftor, E.A.; Hess, A.R.; Gruman, L.M.; Kirschmann, D.A.; Yokoyama, Y.; Griffioen, A.W.; Hendrix, M.J. Effects of angiogenesis inhibitors on vascular network formation by human endothelial and melanoma cells. J. Natl. Cancer Inst. 2004, 96, 1473–1477. [Google Scholar] [CrossRef]
- Mitra, D.; Bhattacharyya, S.; Alam, N.; Sen, S.; Mitra, S.; Mandal, S.; Vignesh, S.; Majumder, B.; Murmu, N. Phosphorylation of EphA2 receptor and vasculogenic mimicry is an indicator of poor prognosis in invasive carcinoma of the breast. Breas Cancer Res Treat. 2020, 179, 359–370. [Google Scholar] [CrossRef]
- Zhang, D.; Sun, B.; Zhao, X.; Ma, Y.; Ji, R.; Gu, Q.; Dong, X.; Li, J.; Liu, F.; Jia, X.; et al. Twist1 expression induced by sunitinib accelerates tumor cell vasculogenic mimicry by increasing the population of CD133+ cells in triple-negative breast cancer. Mol. Cancer 2014, 13, 207. [Google Scholar] [CrossRef]
- Brugnoli, F.; Grassilli, S.; Al-Qassab, Y.; Capitani, S.; Bertagnolo, V. CD133 in Breast Cancer Cells: More than a Stem Cell Marker. J. Oncol. 2019, 2019, 7512632. [Google Scholar] [CrossRef]
- Liu, T.J.; Sun, B.C.; Zhao, X.L.; Zhao, X.M.; Sun, T.; Gu, Q.; Yao, Z.; Dong, X.Y.; Zhao, N.; Liu, N. CD133+ cells with cancer stem cell characteristics associates with vasculogenic mimicry in triple-negative breast cancer. Oncogene 2013, 32, 544–553. [Google Scholar] [CrossRef]
- Rossi, E.; Poirault-Chassac, S.; Bieche, I.; Chocron, R.; Schnitzler, A.; Lokajczyk, A.; Bourdoncle, P.; Dizier, B.; Bacha, N.C.; Gendron, N.; et al. Human Endothelial Colony Forming Cells Express Intracellular CD133 that Modulates their Vasculogenic Properties. Stem Cell Rev Rep. 2019, 15, 590–600. [Google Scholar] [CrossRef]
- Ribatti, D.; Pezzella, F. Overview on the Different Patterns of Tumor Vascularization. Cells 2021, 10, 639. [Google Scholar] [CrossRef]
- Folberg, R.; Maniotis, A.J. Vasculogenic mimicry. Acta Pathol. Microbiol. Et Immunol. Scand. 2004, 112, 508–525. [Google Scholar] [CrossRef]
- Garcia-Quiroz, J.; Garcia-Becerra, R.; Santos-Cuevas, C.; Ramirez-Nava, G.J.; Morales-Guadarrama, G.; Cardenas-Ochoa, N.; Segovia-Mendoza, M.; Prado-Garcia, H.; Ordaz-Rosado, D.; Avila, E.; et al. Synergistic Antitumorigenic Activity of Calcitriol with Curcumin or Resveratrol is Mediated by Angiogenesis Inhibition in Triple Negative Breast Cancer Xenografts. Cancers 2019, 11, 1739. [Google Scholar] [CrossRef]
- Shirakawa, K.; Tsuda, H.; Heike, Y.; Kato, K.; Asada, R.; Inomata, M.; Sasaki, H.; Kasumi, F.; Yoshimoto, M.; Iwanaga, T.; et al. Absence of endothelial cells, central necrosis, and fibrosis are associated with aggressive inflammatory breast cancer. Cancer Res. 2001, 61, 445–451. [Google Scholar]
- Shirakawa, K.; Wakasugi, H.; Heike, Y.; Watanabe, I.; Yamada, S.; Saito, K.; Konishi, F. Vasculogenic mimicry and pseudo-comedo formation in breast cancer. Int. J. Cancer 2002, 99, 821–828. [Google Scholar] [CrossRef]
- Shirakawa, K.; Kobayashi, H.; Heike, Y.; Kawamoto, S.; Brechbiel, M.W.; Kasumi, F.; Iwanaga, T.; Konishi, F.; Terada, M.; Wakasugi, H. Hemodynamics in vasculogenic mimicry and angiogenesis of inflammatory breast cancer xenograft. Cancer Res. 2002, 62, 560–566. [Google Scholar]
- Silvestri, V.L.; Henriet, E.; Linville, R.M.; Wong, A.D.; Searson, P.C.; Ewald, A.J. A Tissue-Engineered 3D Microvessel Model Reveals the Dynamics of Mosaic Vessel Formation in Breast Cancer. Cancer Res. 2020, 80, 4288–4301. [Google Scholar] [CrossRef]
- Xu, Y.; Li, Q.; Li, X.Y.; Yang, Q.Y.; Xu, W.W.; Liu, G.L. Short-term anti-vascular endothelial growth factor treatment elicits vasculogenic mimicry formation of tumors to accelerate metastasis. J. Exp. Clin. Cancer Res. 2012, 31, 1–7. [Google Scholar] [CrossRef]
- Valencia-Cervantes, J.; Huerta-Yepez, S.; Aquino-Jarquín, G.; Rodríguez-Enríquez, S.; Martínez-Fong, D.; Arias-Montaño, J.A.; Dávila-Borja, V.M. Hypoxia increases chemoresistance in human medulloblastoma DAOY cells via hypoxia-inducible factor 1α-mediated downregulation of the CYP2B6, CYP3A4 and CYP3A5 enzymes and inhibition of cell proliferation. Oncol. Rep. 2019, 41, 178–190. [Google Scholar] [CrossRef]
- Weidner, N.; Semple, J.P.; Welch, W.R.; Folkman, J. Tumor angiogenesis and metastasis--correlation in invasive breast carcinoma. N. Engl. J. Med. 1991, 324, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Zhang, S.; Zhang, D.; Du, J.; Guo, H.; Zhao, X.; Zhang, W.; Hao, X. Vasculogenic mimicry is associated with high tumor grade, invasion and metastasis, and short survival in patients with hepatocellular carcinoma. Oncol. Rep. 2006, 16, 693–698. [Google Scholar] [CrossRef]
- Shen, Y.; Quan, J.; Wang, M.; Li, S.; Yang, J.; Lv, M.; Chen, Z.; Zhang, L.; Zhao, X.; Yang, J. Tumor vasculogenic mimicry formation as an unfavorable prognostic indicator in patients with breast cancer. Oncotarget 2017, 8, 56408–56416. [Google Scholar] [CrossRef]
- Jafarian, A.H.; Kooshkiforooshani, M.; Rasoliostadi, A.; Mohamadian Roshan, N. Vascular Mimicry Expression in Invasive Ductal Carcinoma; A New Technique for Prospect of Aggressiveness. Iran. J. Pathol. 2019, 14, 232–235. [Google Scholar] [CrossRef]
- Liu, T.; Sun, B.; Zhao, X.; Gu, Q.; Dong, X.; Yao, Z.; Zhao, N.; Chi, J.; Liu, N.; Sun, R.; et al. HER2/neu expression correlates with vasculogenic mimicry in invasive breast carcinoma. J. Cell. Mol. Med. 2013, 17, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.P.; Liao, Y.D.; Mai, D.M.; Xie, P.; Qiang, Y.Y.; Zheng, L.S.; Wang, M.Y.; Mei, Y.; Meng, D.F.; Xu, L.; et al. Tumor vasculogenic mimicry predicts poor prognosis in cancer patients: A meta-analysis. Angiogenesis 2016, 19, 191–200. [Google Scholar] [CrossRef]
- Quiros-Gonzalez, I.; Tomaszewski, M.R.; Aitken, S.J.; Ansel-Bollepalli, L.; McDuffus, L.A.; Gill, M.; Hacker, L.; Brunker, J.; Bohndiek, S.E. Optoacoustics delineates murine breast cancer models displaying angiogenesis and vascular mimicry. Br. J. Cancer 2018, 118, 1098–1106. [Google Scholar] [CrossRef]
- Luan, Y.Y.; Liu, Z.M.; Zhong, J.Y.; Yao, R.Y.; Yu, H.S. Effect of grape seed proanthocyanidins on tumor vasculogenic mimicry in human triple-negative breast cancer cells. Asian Pac. J. Cancer Prev. 2015, 16, 531–535. [Google Scholar] [CrossRef] [PubMed]
- Nisar, M.A.; Zheng, Q.; Saleem, M.Z.; Ahmmed, B.; Ramzan, M.N.; Ud Din, S.R.; Tahir, N.; Liu, S.; Yan, Q. IL-1β Promotes Vasculogenic Mimicry of Breast Cancer Cells Through p38/MAPK and PI3K/Akt Signaling Pathways. Front. Oncol. 2021, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Xing, P.; Dong, H.; Liu, Q.; Zhao, T.; Yao, F.; Xu, Y.; Chen, B.; Zheng, X.; Wu, Y.; Jin, F.; et al. ALDH1 Expression and Vasculogenic Mimicry Are Positively Associated with Poor Prognosis in Patients with Breast Cancer. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 49, 961–970. [Google Scholar] [CrossRef]
- Sorlie, T.; Tibshirani, R.; Parker, J.; Hastie, T.; Marron, J.S.; Nobel, A.; Deng, S.; Johnsen, H.; Pesich, R.; Geisler, S.; et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc. Natl. Acad. Sci. USA 2003, 100, 8418–8423. [Google Scholar] [CrossRef] [PubMed]
- Hess, A.R.; Seftor, E.A.; Gruman, L.M.; Kinch, M.S.; Seftor, R.E.; Hendrix, M.J. VE-cadherin regulates EphA2 in aggressive melanoma cells through a novel signaling pathway: Implications for vasculogenic mimicry. Cancer Biol. Ther. 2006, 5, 228–233. [Google Scholar] [CrossRef]
- Hess, A.R.; Seftor, E.A.; Gardner, L.M.; Carles-Kinch, K.; Schneider, G.B.; Seftor, R.E.; Kinch, M.S.; Hendrix, M.J. Molecular regulation of tumor cell vasculogenic mimicry by tyrosine phosphorylation: Role of epithelial cell kinase (Eck/EphA2). Cancer Res. 2001, 61, 3250–3255. [Google Scholar] [PubMed]
- Hendrix, M.J.; Seftor, E.A.; Meltzer, P.S.; Gardner, L.M.; Hess, A.R.; Kirschmann, D.A.; Schatteman, G.C.; Seftor, R.E. Expression and functional significance of VE-cadherin in aggressive human melanoma cells: Role in vasculogenic mimicry. Proc. Natl. Acad. Sci. USA 2001, 98, 8018–8023. [Google Scholar] [CrossRef] [PubMed]
- Seftor, R.E.; Seftor, E.A.; Koshikawa, N.; Meltzer, P.S.; Gardner, L.M.; Bilban, M.; Stetler-Stevenson, W.G.; Quaranta, V.; Hendrix, M.J. Cooperative interactions of laminin 5 gamma2 chain, matrix metalloproteinase-2, and membrane type-1-matrix/metalloproteinase are required for mimicry of embryonic vasculogenesis by aggressive melanoma. Cancer Res. 2001, 61, 6322–6327. [Google Scholar] [PubMed]
- Hori, A.; Shimoda, M.; Naoi, Y.; Kagara, N.; Tanei, T.; Miyake, T.; Shimazu, K.; Kim, S.J.; Noguchi, S. Vasculogenic mimicry is associated with trastuzumab resistance of HER2-positive breast cancer. Breast Cancer Res. 2019, 21, 88. [Google Scholar] [CrossRef]
- Kotiyal, S.; Bhattacharya, S. Epithelial Mesenchymal Transition and Vascular Mimicry in Breast Cancer Stem Cells. Crit. Rev. Eukaryot. Gene Expr. 2015, 25, 269–280. [Google Scholar] [CrossRef]
- Yang, J.; Lu, Y.; Lin, Y.Y.; Zheng, Z.Y.; Fang, J.H.; He, S.; Zhuang, S.M. Vascular mimicry formation is promoted by paracrine TGF-β and SDF1 of cancer-associated fibroblasts and inhibited by miR-101 in hepatocellular carcinoma. Cancer Lett. 2016, 383, 18–27. [Google Scholar] [CrossRef]
- Tang, N.N.; Zhu, H.; Zhang, H.J.; Zhang, W.F.; Jin, H.L.; Wang, L.; Wang, P.; He, G.J.; Hao, B.; Shi, R.H. HIF-1α induces VE-cadherin expression and modulates vasculogenic mimicry in esophageal carcinoma cells. World J. Gastroenterol. 2014, 20, 17894–17904. [Google Scholar] [CrossRef]
- Li, S.; Meng, W.; Guan, Z.; Guo, Y.; Han, X. The hypoxia-related signaling pathways of vasculogenic mimicry in tumor treatment. Biomed. Pharmacother. 2016, 80, 127–135. [Google Scholar] [CrossRef]
- Wei, X.; Chen, Y.; Jiang, X.; Peng, M.; Liu, Y.; Mo, Y.; Ren, D.; Hua, Y.; Yu, B.; Zhou, Y.; et al. Mechanisms of vasculogenic mimicry in hypoxic tumor microenvironments. Mol. Cancer 2021, 20, 7. [Google Scholar] [CrossRef]
- Li, S.; Zhang, Q.; Zhou, L.; Guan, Y.; Chen, S.; Zhang, Y.; Han, X. Inhibitory effects of compound DMBT on hypoxia-induced vasculogenic mimicry in human breast cancer. Biomed. Pharmacother. 2017, 96, 982–992. [Google Scholar] [CrossRef]
- Majumder, M.; Xin, X.; Liu, L.; Tutunea-Fatan, E.; Rodriguez-Torres, M.; Vincent, K.; Postovit, L.M.; Hess, D.; Lala, P.K. COX-2 Induces Breast Cancer Stem Cells via EP4/PI3K/AKT/NOTCH/WNT Axis. Stem Cells 2016, 34, 2290–2305. [Google Scholar] [CrossRef]
- Majumder, M.; Xin, X.; Liu, L.; Girish, G.V.; Lala, P.K. Prostaglandin E2 receptor EP4 as the common target on cancer cells and macrophages to abolish angiogenesis, lymphangiogenesis, metastasis, and stem-like cell functions. Cancer Sci. 2014, 105, 1142–1151. [Google Scholar] [CrossRef]
- Liu, S.; Ni, C.; Zhang, D.; Sun, H.; Dong, X.; Che, N.; Liang, X.; Chen, C.; Liu, F.; Bai, J.; et al. S1PR1 regulates the switch of two angiogenic modes by VE-cadherin phosphorylation in breast cancer. Cell Death Dis. 2019, 10, 200. [Google Scholar] [CrossRef] [PubMed]
- Wagenblast, E.; Soto, M.; Gutiérrez-Ángel, S.; Hartl, C.A.; Gable, A.L.; Maceli, A.R.; Erard, N.; Williams, A.M.; Kim, S.Y.; Dickopf, S.; et al. A model of breast cancer heterogeneity reveals vascular mimicry as a driver of metastasis. Nature 2015, 520, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Stanton, S.E.; Adams, S.; Disis, M.L. Variation in the Incidence and Magnitude of Tumor-Infiltrating Lymphocytes in Breast Cancer Subtypes: A Systematic Review. JAMA Oncol. 2016, 2, 1354–1360. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.H.; Zheng, Z.Y.; Liu, J.Y.; Xie, C.; Zhang, Z.J.; Zhuang, S.M. Regulatory Role of the MicroRNA-29b-IL-6 Signaling in the Formation of Vascular Mimicry. Mol. Therapy. Nucleic Acids 2017, 8, 90–100. [Google Scholar] [CrossRef]
- Aikins, A.R.; Kim, M.; Raymundo, B.; Kim, C.-W. Downregulation of transgelin blocks interleukin-8 utilization and suppresses vasculogenic mimicry in breast cancer cells. Exp. Biol Med. 2017, 242, 573–583. [Google Scholar] [CrossRef]
- Wright, M.H.; Calcagno, A.M.; Salcido, C.D.; Carlson, M.D.; Ambudkar, S.V.; Varticovski, L. Brca1 breast tumors contain distinct CD44+/CD24- and CD133+ cells with cancer stem cell characteristics. Breast Cancer Res. 2008, 10, 10. [Google Scholar] [CrossRef]
- Sengodan, S.K.; Nadhan, R.; Nair, R.S.; Hemalatha, S.K.; Somasundaram, V.; Sushama, R.R.; Rajan, A.; Latha, N.R.; Varghese, G.R.; Thankappan, R.K.; et al. BRCA1 regulation on β-hCG: A mechanism for tumorigenicity in BRCA1 defective breast cancer. Oncogenesis 2017, 6, 376. [Google Scholar] [CrossRef]
- Zygmunt, M.; Herr, F.; Keller-Schoenwetter, S.; Kunzi-Rapp, K.; Münstedt, K.; Rao, C.V.; Lang, U.; Preissner, K.T. Characterization of human chorionic gonadotropin as a novel angiogenic factor. J. Clin. Endocrinol. Metab. 2002, 87, 5290–5296. [Google Scholar] [CrossRef]
- Su, M.; Xu, X.; Wei, W.; Gao, S.; Wang, X.; Chen, C.; Zhang, Y. Involvement of human chorionic gonadotropin in regulating vasculogenic mimicry and hypoxia-inducible factor-1α expression in ovarian cancer cells. Cancer Cell Int. 2016, 16, 50. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yuri, T.; Kinoshita, Y.; Emoto, Y.; Yoshizawa, K.; Tsubura, A. Human chorionic gonadotropin suppresses human breast cancer cell growth directly via p53-mediated mitochondrial apoptotic pathway and indirectly via ovarian steroid secretion. Anticancer Res. 2014, 34, 1347–1354. [Google Scholar] [PubMed]
- Iezzi, M.; Quaglino, E.; Cappello, P.; Toto, V.; Sabatini, F.; Curcio, C.; Garotta, G.; Musiani, P.; Cavallo, F. HCG hastens both the development of mammary carcinoma and the metastatization of HCG/LH and ERBB-2 receptor-positive cells in mice. Int. J. Immunopathol. Pharmacol. 2011, 24, 621–630. [Google Scholar] [CrossRef]
- Laederich, M.B.; Funes-Duran, M.; Yen, L.; Ingalla, E.; Wu, X.; Carraway, K.L., 3rd; Sweeney, C. The leucine-rich repeat protein LRIG1 is a negative regulator of ErbB family receptor tyrosine kinases. J. Biol. Chem. 2004, 279, 47050–47056. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.K.; Shattuck, D.L.; Ingalla, E.Q.; Yen, L.; Borowsky, A.D.; Young, L.J.; Cardiff, R.D.; Carraway, K.L., 3rd; Sweeney, C. Suppression of the negative regulator LRIG1 contributes to ErbB2 overexpression in breast cancer. Cancer Res. 2008, 68, 8286–8294. [Google Scholar] [CrossRef]
- Krig, S.R.; Frietze, S.; Simion, C.; Miller, J.K.; Fry, W.H.; Rafidi, H.; Kotelawala, L.; Qi, L.; Griffith, O.L.; Gray, J.W.; et al. Lrig1 is an estrogen-regulated growth suppressor and correlates with longer relapse-free survival in ERα-positive breast cancer. Mol. Cancer Res. 2011, 9, 1406–1417. [Google Scholar] [CrossRef] [PubMed]
- Yokdang, N.; Hatakeyama, J.; Wald, J.H.; Simion, C.; Tellez, J.D.; Chang, D.Z.; Swamynathan, M.M.; Chen, M.; Murphy, W.J.; Carraway Iii, K.L.; et al. LRIG1 opposes epithelial-to-mesenchymal transition and inhibits invasion of basal-like breast cancer cells. Oncogene 2016, 35, 2932–2947. [Google Scholar] [CrossRef] [PubMed]
- van Schooneveld, E.; Wouters, M.C.; Van der Auwera, I.; Peeters, D.J.; Wildiers, H.; Van Dam, P.A.; Vergote, I.; Vermeulen, P.B.; Dirix, L.Y.; Van Laere, S.J. Expression profiling of cancerous and normal breast tissues identifies microRNAs that are differentially expressed in serum from patients with (metastatic) breast cancer and healthy volunteers. Breast Cancer Res. 2012, 14, R34. [Google Scholar] [CrossRef]
- Li, C.; Wang, A.; Chen, Y.; Liu, Y.; Zhang, H.; Zhou, J. MicroRNA-299-5p inhibits cell metastasis in breast cancer by directly targeting serine/threonine kinase 39. Oncol. Rep. 2020, 43, 1221–1233. [Google Scholar] [CrossRef]
- Shevde, L.A.; Metge, B.J.; Mitra, A.; Xi, Y.; Ju, J.; King, J.A.; Samant, R.S. Spheroid-forming subpopulation of breast cancer cells demonstrates vasculogenic mimicry via hsa-miR-299-5p regulated de novo expression of osteopontin. J. Cell. Mol. Med. 2010, 14, 1693–1706. [Google Scholar] [CrossRef]
- Yang, Z.; He, M.; Wang, K.; Sun, G.; Tang, L.; Xu, Z. Tumor suppressive microRNA-193b promotes breast cancer progression via targeting DNAJC13 and RAB22A. Int. J. Clin. Exp. Pathol. 2014, 7, 7563–7570. [Google Scholar]
- Hulin, J.A.; Tommasi, S.; Elliot, D.; Hu, D.G.; Lewis, B.C.; Mangoni, A.A. MiR-193b regulates breast cancer cell migration and vasculogenic mimicry by targeting dimethylarginine dimethylaminohydrolase 1. Sci. Rep. 2017, 7, 13996. [Google Scholar] [CrossRef]
- Zou, Q.; Zhou, E.; Xu, F.; Zhang, D.; Yi, W.; Yao, J. A TP73-AS1/miR-200a/ZEB1 regulating loop promotes breast cancer cell invasion and migration. J. Cell. Biochem. 2018, 119, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Xu, F.; Zhang, D.; Yi, W.; Chen, X.; Chen, G.; Zhou, E. TP73-AS1 promotes breast cancer cell proliferation through miR-200a-mediated TFAM inhibition. J. Cell. Biochem. 2018, 119, 680–690. [Google Scholar] [CrossRef]
- Tao, W.; Sun, W.; Zhu, H.; Zhang, J. Knockdown of long non-coding RNA TP73-AS1 suppresses triple negative breast cancer cell vasculogenic mimicry by targeting miR-490-3p/TWIST1 axis. Biochem. Biophys. Res. Commun. 2018, 504, 629–634. [Google Scholar] [CrossRef]
- Li, W.; Jin, X.; Zhang, Q.; Zhang, G.; Deng, X.; Ma, L. Decreased expression of miR-204 is associated with poor prognosis in patients with breast cancer. Int. J. Clin. Exp. Pathol. 2014, 7, 3287–3292. [Google Scholar]
- Salinas-Vera, Y.M.; Marchat, L.A.; García-Vázquez, R.; González de la Rosa, C.H.; Castañeda-Saucedo, E.; Tito, N.N.; Flores, C.P.; Pérez-Plasencia, C.; Cruz-Colin, J.L.; Carlos-Reyes, Á.; et al. Cooperative multi-targeting of signaling networks by angiomiR-204 inhibits vasculogenic mimicry in breast cancer cells. Cancer Lett. 2018, 432, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Lozano-Romero, A.; Astudillo-de la Vega, H.; Terrones-Gurrola, M.; Marchat, L.A.; Hernandez-Sotelo, D.; Salinas-Vera, Y.M.; Ramos-Payan, R.; Silva-Cazares, M.B.; Nunez-Olvera, S.I.; Hernandez-de la Cruz, O.N.; et al. HOX Transcript Antisense RNA HOTAIR Abrogates Vasculogenic Mimicry by Targeting the AngiomiR-204/FAK Axis in Triple Negative Breast Cancer Cells. Non-Coding RNA 2020, 6, 19. [Google Scholar] [CrossRef] [PubMed]
- Hong, Z.; Hong, C.; Ma, B.; Wang, Q.; Zhang, X.; Li, L.; Wang, C.; Chen, D. MicroRNA-126-3p inhibits the proliferation, migration, invasion, and angiogenesis of triple-negative breast cancer cells by targeting RGS3. Oncol. Rep. 2019, 42, 1569–1579. [Google Scholar] [CrossRef] [PubMed]
- Rossi, J.F.; Lu, Z.Y.; Jourdan, M.; Klein, B. Interleukin-6 as a therapeutic target. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 1248–1257. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Niu, X.L.; Qu, Y.; Wu, J.; Zhu, Y.Q.; Sun, W.J.; Li, L.Z. Autocrine production of interleukin-6 confers cisplatin and paclitaxel resistance in ovarian cancer cells. Cancer Lett. 2010, 295, 110–123. [Google Scholar] [CrossRef]
- Zhang, G.J.; Adachi, I. Serum interleukin-6 levels correlate to tumor progression and prognosis in metastatic breast carcinoma. Anticancer Res. 1999, 19, 1427–1432. [Google Scholar]
- Park, Y.; Kim, J. Regulation of IL-6 signaling by miR-125a and let-7e in endothelial cells controls vasculogenic mimicry formation of breast cancer cells. BMB Rep. 2019, 52, 214–219. [Google Scholar] [CrossRef]
- Lim, D.; Cho, J.G.; Yun, E.; Lee, A.; Ryu, H.Y.; Lee, Y.J.; Yoon, S.; Chang, W.; Lee, M.S.; Kwon, B.S.; et al. MicroRNA 34a-AXL Axis Regulates Vasculogenic Mimicry Formation in Breast Cancer Cells. Genes 2020, 12, 9. [Google Scholar] [CrossRef]
- Mudduluru, G.; Ceppi, P.; Kumarswamy, R.; Scagliotti, G.V.; Papotti, M.; Allgayer, H. Regulation of Axl receptor tyrosine kinase expression by miR-34a and miR-199a/b in solid cancer. Oncogene 2011, 30, 2888–2899. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Xu, J.; Wu, Y.; Chen, Q.; Zheng, W.; Lu, X.; Zhou, C.; Jiao, D. Identification of microRNA-93 as a functional dysregulated miRNA in triple-negative breast cancer. Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med. 2015, 36, 251–258. [Google Scholar] [CrossRef]
- An, G.; Lu, F.; Huang, S.; Bai, J.; He, L.; Liu, Y.; Hou, L. Effects of miR-93 on epithelial-to-mesenchymal transition and vasculogenic mimicry in triple-negative breast cancer cells. Mol. Med. Rep. 2021, 23, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Du, W.W.; Yang, W.; Rutnam, Z.J.; Peng, C.; Li, H.; O’Malley, Y.Q.; Askeland, R.W.; Sugg, S.; Liu, M.; et al. MiR-93 enhances angiogenesis and metastasis by targeting LATS2. Cell Cycle 2012, 11, 4352–4365. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy, V.; Ge, R.; Grazioli, A.; Xie, W.; Banach-Petrosky, W.; Kang, Y.; Lonning, S.; McPherson, J.; Yingling, J.M.; Biswas, S.; et al. Targeting the Transforming Growth Factor-beta pathway inhibits human basal-like breast cancer metastasis. Mol. Cancer 2010, 9, 122. [Google Scholar] [CrossRef]
- Herbertz, S.; Sawyer, J.S.; Stauber, A.J.; Gueorguieva, I.; Driscoll, K.E.; Estrem, S.T.; Cleverly, A.L.; Desaiah, D.; Guba, S.C.; Benhadji, K.A.; et al. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des. Dev. Ther. 2015, 9, 4479–4499. [Google Scholar] [CrossRef]
- Bhola, N.E.; Balko, J.M.; Dugger, T.C.; Kuba, M.G.; Sánchez, V.; Sanders, M.; Stanford, J.; Cook, R.S.; Arteaga, C.L. TGF-β inhibition enhances chemotherapy action against triple-negative breast cancer. J. Clin. Investig. 2013, 123, 1348–1358. [Google Scholar] [CrossRef] [PubMed]
- Martini, C.; Thompson, E.J.; Hyslop, S.R.; Cockshell, M.P.; Dale, B.J.; Ebert, L.M.; Woods, A.E.; Josefsson, E.C.; Bonder, C.S. Platelets disrupt vasculogenic mimicry by cancer cells. Sci. Rep. 2020, 10, 5869. [Google Scholar] [CrossRef]
- Taipale, M.; Krykbaeva, I.; Koeva, M.; Kayatekin, C.; Westover, K.D.; Karras, G.I.; Lindquist, S. Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. Cell 2012, 150, 987–1001. [Google Scholar] [CrossRef]
- Rohner, N.; Jarosz, D.F.; Kowalko, J.E.; Yoshizawa, M.; Jeffery, W.R.; Borowsky, R.L.; Lindquist, S.; Tabin, C.J. Cryptic variation in morphological evolution: HSP90 as a capacitor for loss of eyes in cavefish. Science 2013, 342, 1372–1375. [Google Scholar] [CrossRef]
- Neckers, L.; Trepel, J.B. Stressing the development of small molecules targeting HSP90. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 275–277. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Hong, H.M.; Chang, Y.Y.; Chang, W.W. Inhibition of heat shock protein (Hsp) 27 potentiates the suppressive effect of Hsp90 inhibitors in targeting breast cancer stem-like cells. Biochimie 2012, 94, 1382–1389. [Google Scholar] [CrossRef] [PubMed]
- Sims, J.D.; McCready, J.; Jay, D.G. Extracellular heat shock protein (Hsp)70 and Hsp90α assist in matrix metalloproteinase-2 activation and breast cancer cell migration and invasion. PLoS ONE 2011, 6, 18848. [Google Scholar] [CrossRef]




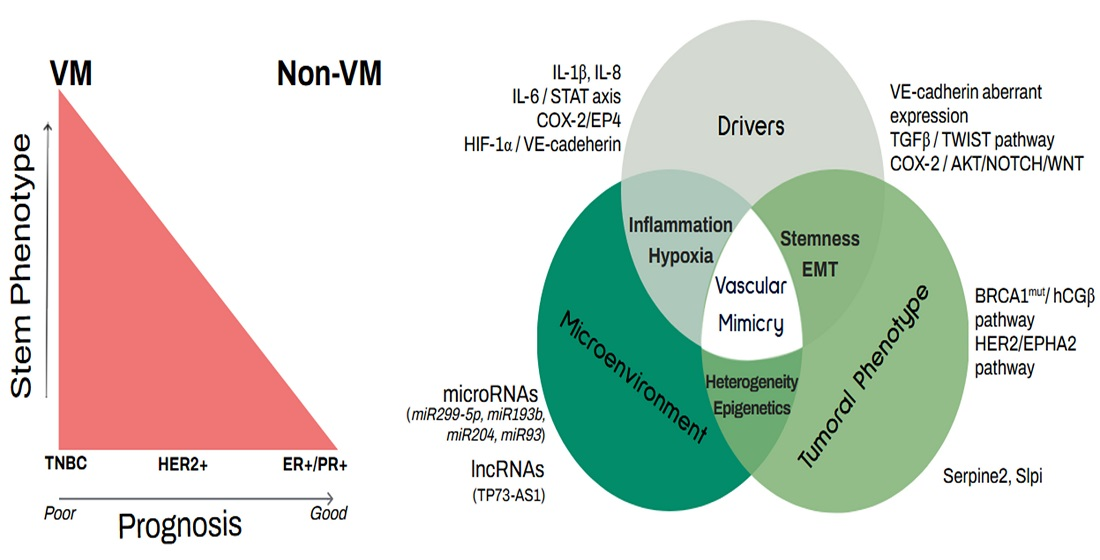{kind=link}
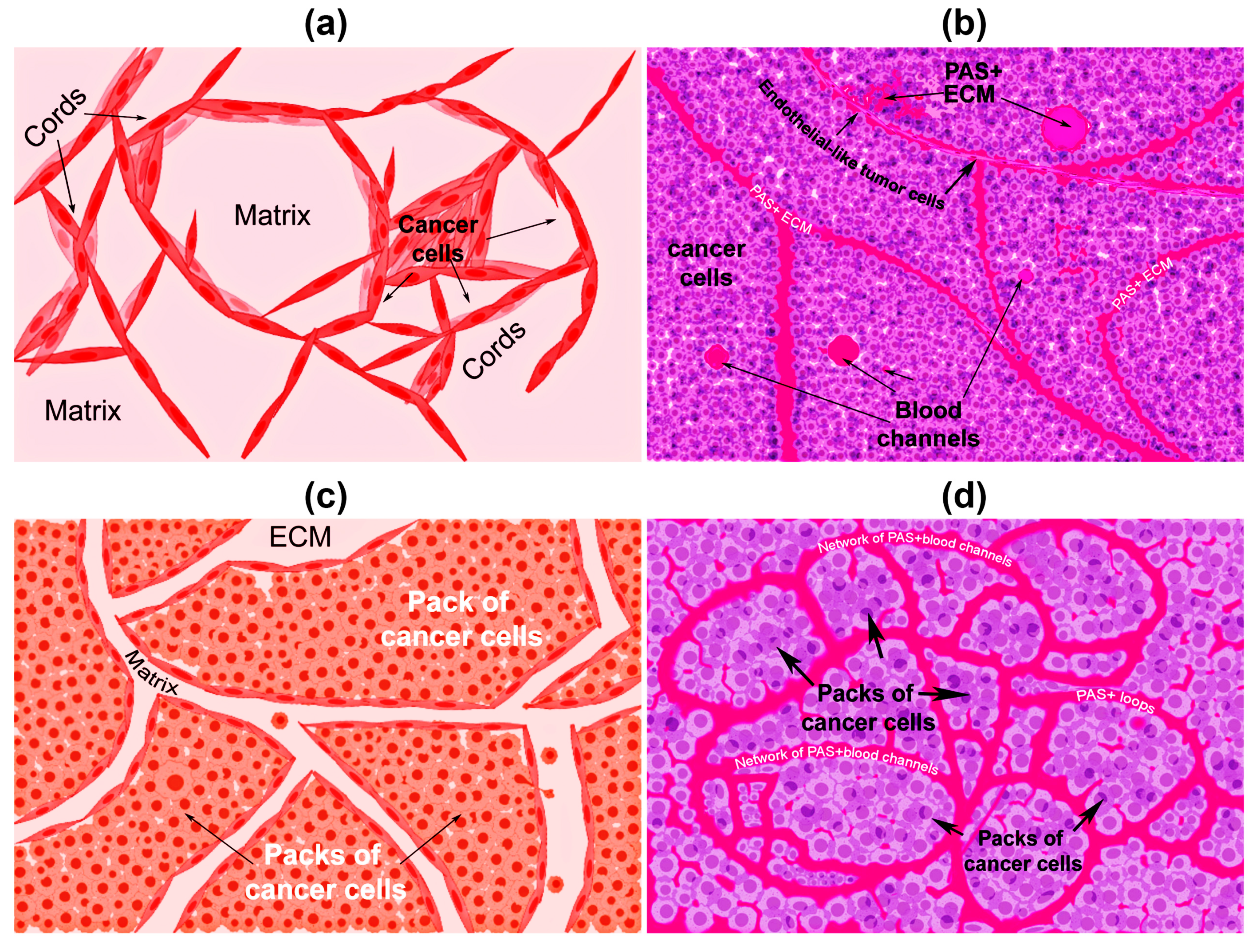{kind=link}
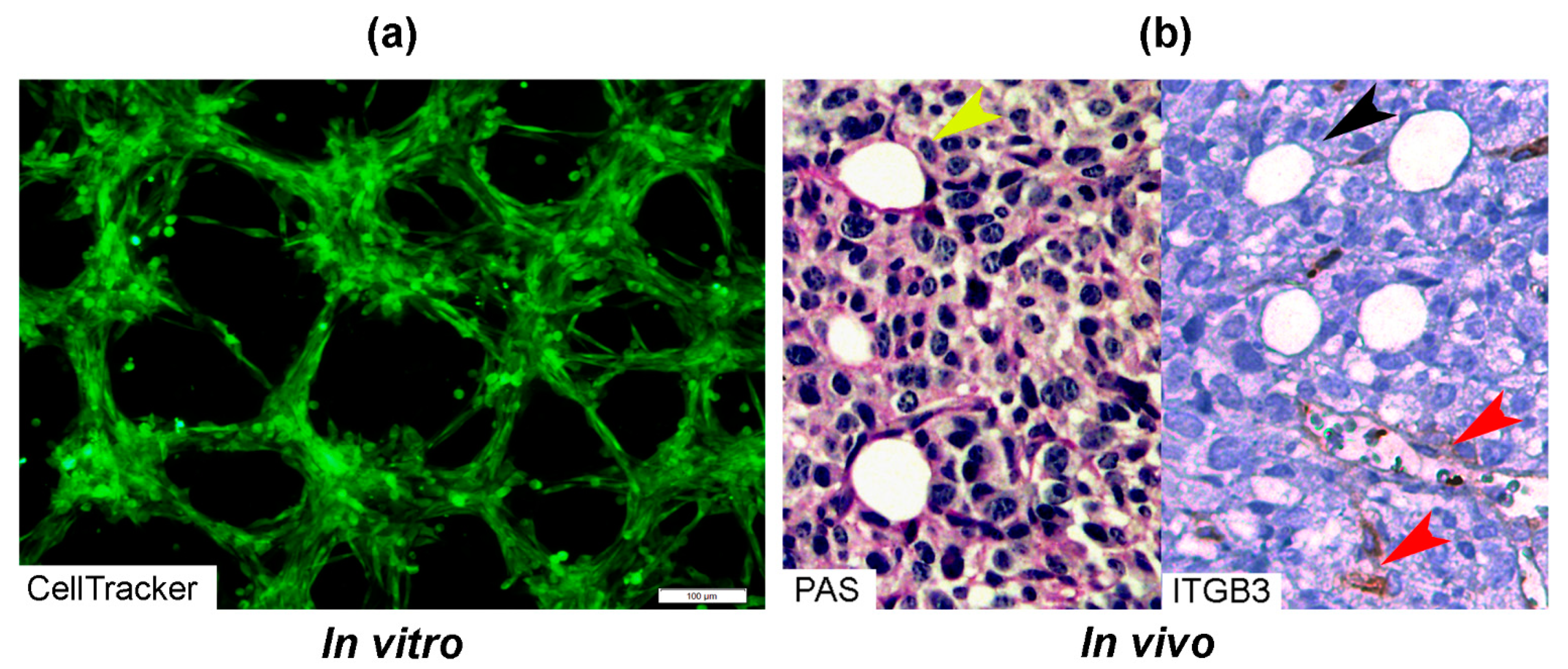{kind=link}
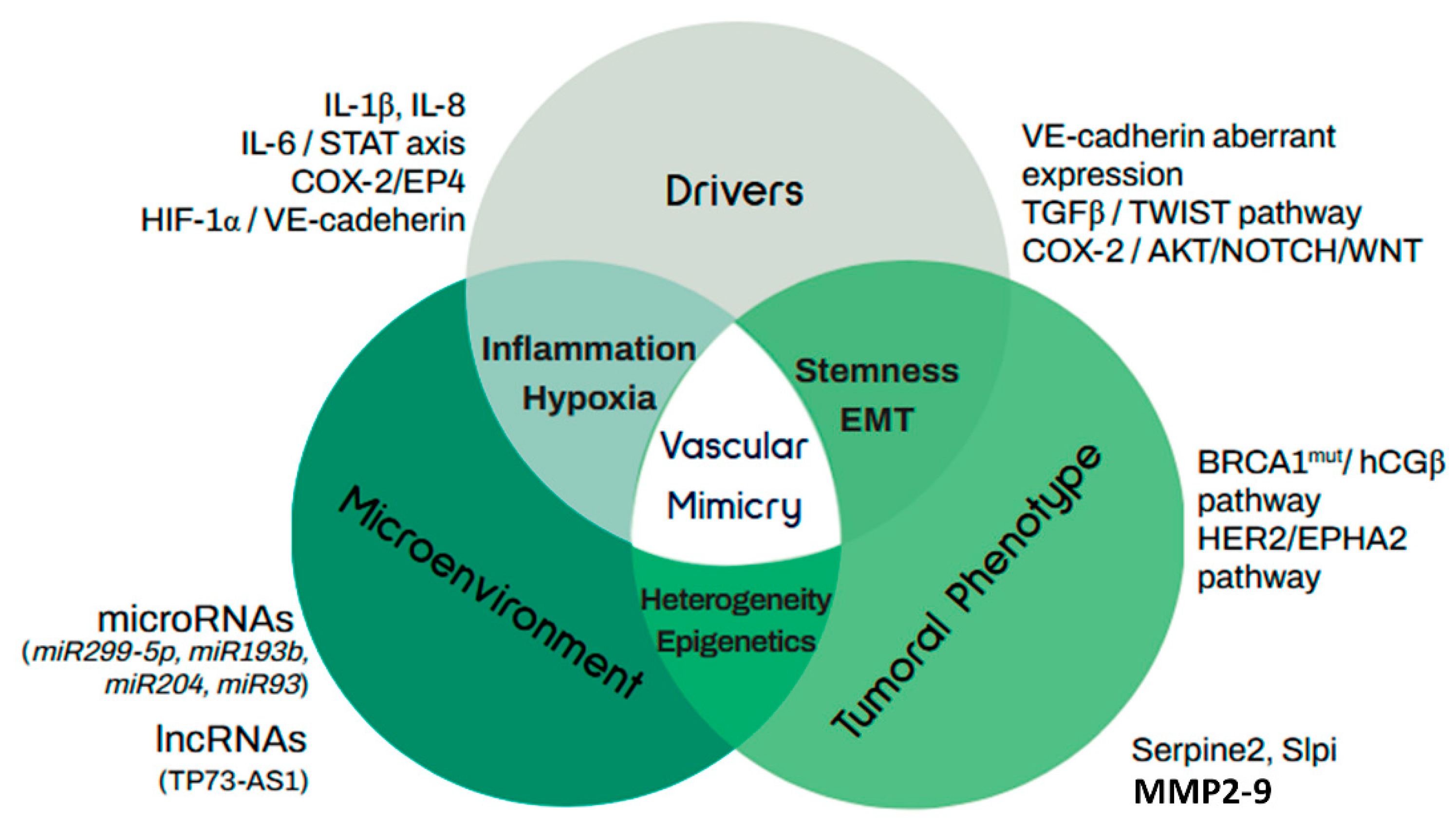{kind=link}
| Vasculogenic Mimicry | Angiogenesis | References |
|---|---|---|
| Formation of vascular channels from cancer stem cells (tumor cells). | Development of new blood vessels and capillaries from pre-existing ones. | [1,12] |
| Patterned networks of interconnected loops and cords formation | Formation by sprouting or intussusception | [1,7] |
| Formed by tumor cells and cancer stem cells | Formed by endothelial cells | [7] |
| Aberrant expression of VE-Cadherin | VE-Cadherin localization in cell membranes | [13] |
| PAS+, CD31−/low staining | PAS−/low, CD31+ staining | [7] |
| Factor VIII-related antigen negative or low | Factor VIII-related antigen highly positive | [7] |
| Unaffected by endostatin and other antiangiogenic factors | Inhibited by antiangiogenic factors | [14,15] |
| EPHA2, TIE1, LAMC2, overexpression | EPHA2, TIE1, LAMC2 generally negative. | [16] |
| Express stemness markers, e.g., CD133, ALDH1 | CD133 positivity mostly in endothelial precursor cells | [17,18,19,20] |
| More abundant in poorly differentiated tumors, such as HER2+ and TNBC | Present in embryogenesis, wound healing and tumor growth | [16,21] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morales-Guadarrama, G.; García-Becerra, R.; Méndez-Pérez, E.A.; García-Quiroz, J.; Avila, E.; Díaz, L. Vasculogenic Mimicry in Breast Cancer: Clinical Relevance and Drivers. Cells 2021, 10, 1758. https://doi.org/10.3390/cells10071758
Morales-Guadarrama G, García-Becerra R, Méndez-Pérez EA, García-Quiroz J, Avila E, Díaz L. Vasculogenic Mimicry in Breast Cancer: Clinical Relevance and Drivers. Cells. 2021; 10(7):1758. https://doi.org/10.3390/cells10071758
Chicago/Turabian StyleMorales-Guadarrama, Gabriela, Rocío García-Becerra, Edgar Armando Méndez-Pérez, Janice García-Quiroz, Euclides Avila, and Lorenza Díaz. 2021. "Vasculogenic Mimicry in Breast Cancer: Clinical Relevance and Drivers" Cells 10, no. 7: 1758. https://doi.org/10.3390/cells10071758
APA StyleMorales-Guadarrama, G., García-Becerra, R., Méndez-Pérez, E. A., García-Quiroz, J., Avila, E., & Díaz, L. (2021). Vasculogenic Mimicry in Breast Cancer: Clinical Relevance and Drivers. Cells, 10(7), 1758. https://doi.org/10.3390/cells10071758