
Overview on the Different Patterns of Tumor Vascularization



Abstract
1. Background
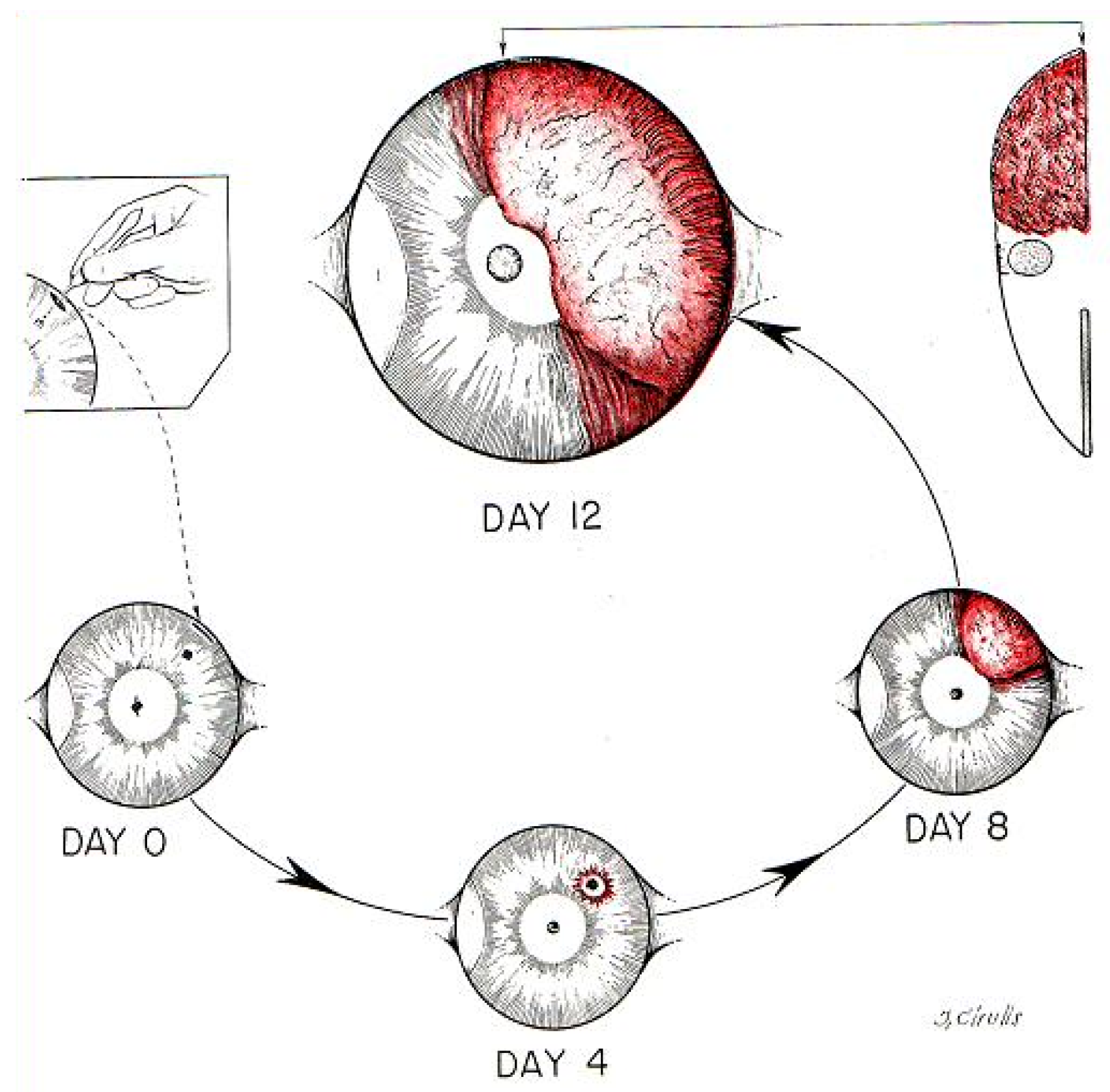
2. Isolation of the First Tumor Angiogenic Factor
3. The Avascular and Vascular Phases of Solid Tumor Growth
4. Tumor Angiogenesis in Human Tumor Progression as a Prognostic Indicator
5. Tumors Can Growth without Inducing Angiogenesis
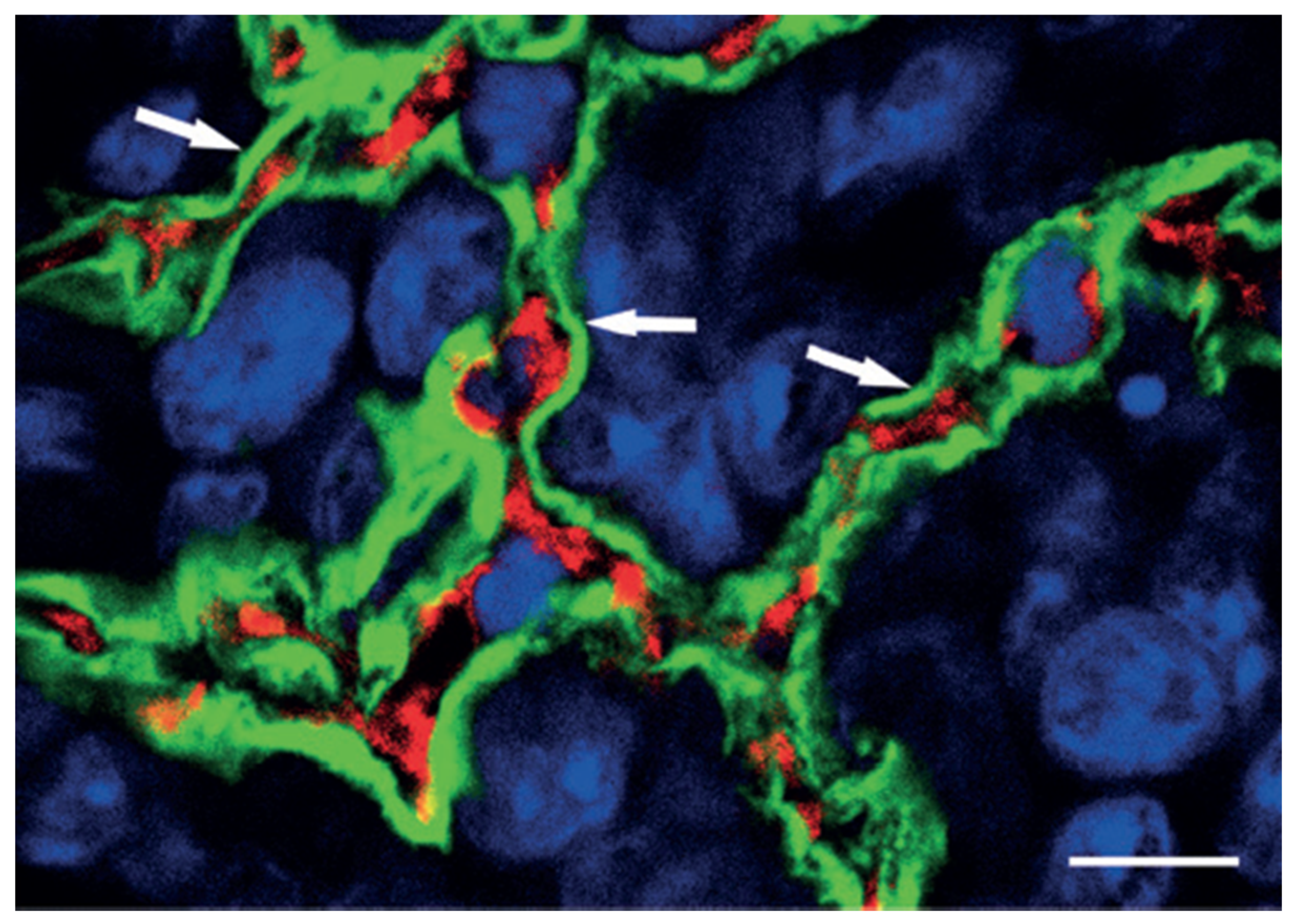
6. Vascular Co-Option
7. Vasculogenic Mimicry
8. Intussusceptive Angiogenesis
9. Concluding Remarks
Funding
Conflicts of Interest
References
- Hall, A.P. The role of angiogenesis in cancer. Comp. Clin. Pathol. 2005, 13, 95–99. [Google Scholar] [CrossRef]
- Lenzi, P.; Bocci, G.; Natale, G. John Hunter and the origin of the term “angiogenesis”. Angiogenesis 2016, 19, 255–256. [Google Scholar] [CrossRef]
- Natale, G.; Bocci, G.; Lenzi, P. Looking for the Word “Angiogenesis” in the History of Health Sciences: From Ancient Times to the First Decades of the Twentieth Century. World J. Surg. 2016, 41, 1625–1634. [Google Scholar] [CrossRef]
- Hunter, J.; Owen, R. Essays and Observations on Natural History, Anatomy, Physiology, Psychology, and Geology; John van Voorst: London, UK, 1861. [Google Scholar] [CrossRef]
- Flint, J.M. The angiology, angiogenesis, and organogenesis of the submaxillary gland. Am. J. Anat. 1903, 2, 417–444. [Google Scholar] [CrossRef]
- Shimkin, M.B. Contrary to Nature: Cancer. In For Sale by the Superintendent of Documents; 20401. DHEW Publication No (NIH); US Printing Office: Washington, DC, USA, 1979; pp. 76–720. [Google Scholar]
- Lytton, D.G.; Resuhr, L.M. Galen on Abnormal Swellings. J. Hist. Med. Allied Sci. 1978, XXXIII, 531–549. [Google Scholar] [CrossRef]
- Goldmann, E. The growth of malignant disease in man and the lower animals. Lancet 1907, 170, 1236–1240. [Google Scholar] [CrossRef]
- Folkman, J. What Is the Evidence That Tumors Are Angiogenesis Dependent? JNCI J. Natl. Cancer Inst. 1990, 82, 4–7. [Google Scholar] [CrossRef]
- Clark, E.R.; Clark, E.L. Microscopic observations on the growth of blood capillaries in the living mammal. Am. J. Anat. 1939, 64, 251–301. [Google Scholar] [CrossRef]
- Clark, E.R.; Hitschler, W.J.; Kirby-Smith, H.T.; Rex, R.O.; Smith, J.H. General observations on the ingrowth of new blood vessels into standardized chambers in the rabbit’s ear, and the subsequent changes in the newly grown vessels over a period of months. Anat. Rec. 1931, 50, 129–167. [Google Scholar] [CrossRef]
- Ide, A.G.; Baker, N.H.; Warren, S.L. Vascularization of the Brown Pearce rabbit epithelioma transplant as seen in the transparent ear chamber. Am. J. Roentgenol. 1939, 42, 891–899. [Google Scholar]
- Algire, G.H. An Adaptation of the Transparent-Chamber Technique to the Mouse. J. Natl. Cancer Inst. 1943, 4, 1–11. [Google Scholar]
- Algire, G.H. Microscopic Studies of the Early Growth of a Transplantable Melanoma of the Mouse, Using the Transparent-chamber Technique. J. Natl. Cancer Inst. 1943, 4, 13–20. [Google Scholar]
- Algire, G.H.; Chalkley, H.W.; Legallais, F.Y.; Park, H.D. Vasculae Reactions of Normal and Malignant Tissues in Vivo. I. Vascular Reactions of Mice to Wounds and to Normal and Neoplastic Transplants. JNCI J. Natl. Cancer Inst. 1945, 6, 73–85. [Google Scholar] [CrossRef]
- Ribatti, D.; Vacca, A.; Presta, M. An Italian pioneer in the study of tumor angiogenesis. Haematologica 2001, 86, 1234–1235. [Google Scholar] [PubMed]
- Rondoni, P. Il Cancro. In Istituzioni di Patologia Generale dei Tumori; Ambrosiana: Milano, Italy, 1945; p. XIV-860. [Google Scholar]
- Greenblatt, M.; Shubi, P. Tumor angiogenesis: Transfilter diffusion studies in the hamster by the transparent chamber technique. J. Natl Cancer Inst. 1968, 41, 111–124. [Google Scholar] [PubMed]
- McAuslan, B.R.; Hoffman, H. Endothelium stimulating factor from Walker carcinoma cells. Exp. Cell Res. 1979, 119, 181–190. [Google Scholar] [CrossRef]
- Fenselau, A.; Kaiser, D.; Wallis, K. Nucleoside requirements for the in vitro growth of bovine aortic endothelial cells. J. Cell Physiol. 1981, 108, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.B.; Brown, R.A.; Kumar, S.; Phillips, P. An angiogenic factor isolated from tumours: A potent low-molecular-weight compound. Br. J. Cancer 1979, 40, 493–496. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Folkman, J.; Merler, E.; Abernathy, C.; Williams, G. Isolation Of A Tumor Factor Responsible For Angiogenesis. J. Exp. Med. 1971, 133, 275–288. [Google Scholar] [CrossRef]
- Maciag, T.; Mehlman, T.; Friesel, R.; Schreiber, A. Heparin binds endothelial cell growth factor, the principal endothelial cell mitogen in bovine brain. Science 1984, 225, 932–935. [Google Scholar] [CrossRef]
- Shing, Y.; Folkman, J.; Sullivan, R.; Butterfield, C.; Murray, J.; Klagsbrun, M. Heparin affinity: Purification of a tumor-derived capillary endothelial cell growth factor. Science 1984, 223, 1296–1299. [Google Scholar] [CrossRef]
- Ferrara, N.; Henzel, W.J. Pituitary follicular cells secrete a novel heparin-binding growth factor specific for vascular endothelial cells. Biochem. Biophys. Res. Commun. 1989, 161, 851–858. [Google Scholar] [CrossRef]
- Plouët, J.; Schilling, J.; Gospodarowicz, D. Isolation and characterization of a newly identified endothelial cell mitogen produced by AtT-20 cells. Embo J. 1989, 8, 3801–3806. [Google Scholar] [CrossRef] [PubMed]
- Conn, G.; Bayne, M.L.; Soderman, D.D.; Kwok, P.W.; Sullivan, K.A.; Palisi, T.M.; Hope, D.A.; Thomas, K.A. Amino acid and cDNA sequences of a vascular endothelial cell mitogen that is homologous to platelet-derived growth factor. Proc. Natl. Acad. Sci. USA 1990, 87, 2628–2632. [Google Scholar] [CrossRef] [PubMed]
- Keck, P.; Hauser, S.; Krivi, G.; Sanzo, K.; Warren, T.; Feder, J.; Connolly, D. Vascular permeability factor, an endothelial cell mitogen related to PDGF. Science 1989, 246, 1309–1312. [Google Scholar] [CrossRef]
- Senger, D.R.; Galli, S.J.; Dvorak, A.M.; Perruzzi, C.A.; Harvey, V.S.; Dvorak, H.F. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science 1983, 219, 983–985. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J.; Long, D.M.; Becker, F.F. Growth and metastasis of tumor in organ culture. Cancer 1963, 16, 453–467. [Google Scholar] [CrossRef]
- Gimbrone, M.A.; Cotran, R.S.; Leapman, S.B.; Folkman, J. Tumor Growth and Neovascularization: An Experimental Model Using the Rabbit Cornea 2. JNCI J. Natl. Cancer Inst. 1974, 52, 413–427. [Google Scholar] [CrossRef] [PubMed]
- Gimbrone, M.A.; Leapman, S.B.; Cotran, R.S.; Folkman, J. Tumor dormancy in vivo by prevention of neovascularization. J. Exp. Med. 1972, 136, 261–276. [Google Scholar] [CrossRef]
- Brem, S.; Gullino, P.; Medina, D. Angiogenesis: A marker for neoplastic transformation of mammary papillary hyperplasia. Science 1977, 195, 880–882. [Google Scholar] [CrossRef]
- Brem, S.S.; Jensen, H.M.; Gullino, P.M. Angiogenesis as a marker of preneoplastic lesions of the human breast. Cancer 1978, 41, 239–244. [Google Scholar] [CrossRef]
- Maiorana, A.; Gullino, P.M. Acquisition of angiogenic capacity and neoplastic transformation in the rat mammary gland. Cancer Res. 1978, 38, 4409–4414. [Google Scholar] [PubMed]
- Hanahan, D.; Folkman, J. Patterns and Emerging Mechanisms of the Angiogenic Switch during Tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar] [CrossRef]
- Hanahan, D. Heritable formation of pancreatic β-cell tumours in transgenic mice expressing recombinant insulin/simian virus 40 oncogenes. Nature 1985, 315, 115–122. [Google Scholar] [CrossRef]
- Folkman, J.; Watson, K.; Ingber, D.; Hanahan, D. Induction of angiogenesis during the transition from hyperplasia to neoplasia. Nature 1989, 339, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Vacca, A.; Ribatti, D.; Roncali, L.; Ranieri, G.; Serio, G.; Silvestris, F.; Dammacco, F. Bone marrow angiogenesis and progression in multiple myeloma. Br. J. Haematol. 1994, 87, 503–508. [Google Scholar] [CrossRef]
- Ribatti, D.; Vacca, A.; Nico, B.; Fanelli, M.; Roncali, L.; Dammacco, F. Angiogenesis spectrum in the stroma of B-cell non-Hodgkin’s lymphomas. An immunohistochemical and ultrastructural study. Eur J. Haematol. 1996, 56, 45–53. [Google Scholar] [CrossRef]
- Perez-Atayde, A.R.; Sallan, S.E.; Tedrow, U.; Connors, S.; Allred, E.; Folkman, J. Spectrum of tumor angiogenesis in the bone marrow of children with acute lymphoblastic leukemia. Am. J. Pathol. 1997, 150, 815–821. [Google Scholar] [PubMed]
- Srivastava, A.; Laidler, P.; Davies, R.P.; Horgan, K.; Hughes, L.E. The prognostic significance of tumor vascularity in intermediate-thickness (0.76–4.0 mm thick) skin melanoma. A quantitative histologic study. Am. J. Pathol. 1988, 133, 419–423. [Google Scholar]
- Weidner, N.; Semple, J.P.; Welch, W.R.; Folkman, J. Tumor angiogenesis and metastasis--correlation in invasive breast carcinoma. N. Engl. J. Med. 1991, 324, 1–8. [Google Scholar] [CrossRef]
- Trivella, M.; Pezzella, F.; Pastorino, U.; Harris, A.L.; Altman, D.G.; Prognosis In Lung Cancer Collaborative Study, G. Microvessel density as a prognostic factor in non-small-cell lung carcinoma: A meta-analysis of individual patient data. Lancet Oncol. 2007, 8, 488–499. [Google Scholar] [CrossRef]
- Yoshii, Y.; Sugiyama, K. Intercapillary distance in the proliferating area of human glioma. Cancer Res. 1988, 48, 2938–2941. [Google Scholar]
- Mabjeesh, N.J.; Amir, S. Hypoxia-inducible factor (HIF) in human tumorigenesis. Histol. Histopathol. 2007, 22, 559–572. [Google Scholar] [CrossRef]
- Donnem, T.; Reynolds, A.R.; Kuczynski, E.A.; Gatter, K.; Vermeulen, P.B.; Kerbel, R.S.; Harris, A.L.; Pezzella, F. Non-angiogenic tumours and their influence on cancer biology. Nat. Rev. Cancer 2018, 18, 323–336. [Google Scholar] [CrossRef]
- Pezzella, F.; Pastorino, U.; Tagliabue, E.; Andreola, S.; Sozzi, G.; Gasparini, G.; Menard, S.; Gatter, K.C.; Harris, A.L.; Fox, S.; et al. Non-small-cell lung carcinoma tumor growth without morphological evidence of neo-angiogenesis. Am. J. Pathol. 1997, 151, 1417–1423. [Google Scholar] [PubMed]
- Pezzella, F.; Di Bacco, A.; Andreola, S.; Nicholson, A.G.; Pastorino, U.; Harris, A.L. Angiogenesis in primary lung cancer and lung secondaries. Eur. J. Cancer 1996, 32, 2494–2500. [Google Scholar] [CrossRef]
- Vermeulen, P.B.; Colpaert, C.; Salgado, R.; Royers, R.; Hellemans, H.; Van Den Heuvel, E.; Goovaerts, G.; Dirix, L.Y.; Van Marck, E. Liver metastases from colorectal adenocarcinomas grow in three patterns with different angiogenesis and desmoplasia. J. Pathol. 2001, 195, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Rubenstein, J.L.; Kim, J.; Ozawa, T.; Zhang, M.; Westphal, M.; Deen, D.F.; Shuman, M.A. Anti-VEGF antibody treatment of glioblastoma prolongs survival but results in increased vascular cooption. Neoplasia 2000, 2, 306–314. [Google Scholar] [CrossRef]
- Döme, B.; Paku, S.; Somlai, B.; Tímár, J. Vascularization of cutaneous melanoma involves vessel co-option and has clinical significance. J. Pathol. 2002, 197, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Holash, J. Vessel Cooption, Regression, and Growth in Tumors Mediated by Angiopoietins and VEGF. Science 1999, 284, 1994–1998. [Google Scholar] [CrossRef]
- Szabo, V.; Bugyik, E.; Dezso, K.; Ecker, N.; Nagy, P.; Timar, J.; Tovari, J.; Laszlo, V.; Bridgeman, V.L.; Wan, E.; et al. Mechanism of tumour vascularization in experimental lung metastases. J. Pathol. 2015, 235, 384–396. [Google Scholar] [CrossRef]
- Kolin, A.; Koutoulakis, T. Role of arterial occlusion in pulmonary scar cancers. Hum. Pathol. 1988, 19, 1161–1167. [Google Scholar] [CrossRef]
- Willis, R. The spread of tumours in the human body. Lond. J. A Churchill 1934, 133, 540. [Google Scholar]
- Moxon, W. Case of transplantation of epithelial cancer from the trachea to the pulmunary tissue, probably by desecent of cancer germs down the bronchial tubes. Trans. Pathol. Soc. Lond. 1869, 20, 28–29. [Google Scholar]
- Erichsen, J. Zwei Fälle von Carcinosis acuta miliaris. Arch. Für Pathol. Anat. Und Physiol. Und Für Klin. Med. 1861, 21, 465–479. [Google Scholar] [CrossRef][Green Version]
- Craig, J.; Peters, R.L.; Edmondson, H.A. Tumours of the Liver and intrahepatic ducts. Pathol. AfioEd. Bethesda Armed Forces Inst. Pathol. 1989. [Google Scholar] [CrossRef]
- Hayano, E. Reaction of the normal liver parenchyma to metastatic carcinoma. Acta Hepato Splenol. 1962, 9, 357–386. [Google Scholar]
- Stessels, F.; Van den Eynden, G.; Van der Auwera, I.; Salgado, R.; Van den Heuvel, E.; Harris, A.L.; Jackson, D.G.; Colpaert, C.G.; van Marck, E.A.; Dirix, L.Y.; et al. Breast adenocarcinoma liver metastases, in contrast to colorectal cancer liver metastases, display a non-angiogenic growth pattern that preserves the stroma and lacks hypoxia. Br. J. Cancer 2004, 90, 1429–1436. [Google Scholar] [CrossRef] [PubMed]
- Robbins, S.L. Pathological Basis of Disease; W.B. Saunders: Philadelphia, PA, USA, 1974. [Google Scholar]
- Ritchie, A.C. The Classification, Morphology, and Behaviour of Tumours. In General Pathology; Florey, H., Ed.; Lloyd-Luke (Medical Bokks) Ltd.: London, UK, 1962; pp. 551–597. [Google Scholar]
- Bald, T.; Quast, T.; Landsberg, J.; Rogava, M.; Glodde, N.; Lopez-Ramos, D.; Kohlmeyer, J.; Riesenberg, S.; van den Boorn-Konijnenberg, D.; Hömig-Hölzel, C.; et al. Ultraviolet-radiation-induced inflammation promotes angiotropism and metastasis in melanoma. Nature 2014, 507, 109–113. [Google Scholar] [CrossRef] [PubMed]
- García-Gómez, P.; Valiente, M. Vascular Co-Option. In Tumor Vascularization; Elsevier: Amsterdam, The Netherlands, 2020; pp. 33–47. [Google Scholar] [CrossRef]
- Kuczynski, E.A.; Vermeulen, P.B.; Pezzella, F.; Kerbel, R.S.; Reynolds, A.R. Vessel co-option in cancer. Nat. Rev. Clin. Oncol 2019, 16, 469–493. [Google Scholar] [CrossRef] [PubMed]
- Pastorino, U.; Andreola, S.; Tagliabue, E.; Pezzella, F.; Incarbone, M.; Sozzi, G.; Buyse, M.; Menard, S.; Pierotti, M.; Rilke, F. Immunocytochemical markers in stage I lung cancer: Relevance to prognosis. J. Clin. Oncol. 1997, 15, 2858–2865. [Google Scholar] [CrossRef] [PubMed]
- Sardari Nia, P.; Colpaert, C.; Blyweert, B.; Kui, B.; Vermeulen, P.; Ferguson, M.; Hendriks, J.; Weyler, J.; Pezzella, F.; Van Marck, E.; et al. Prognostic value of nonangiogenic and angiogenic growth patterns in non-small-cell lung cancer. Br. J. Cancer 2004, 91, 1293–1300. [Google Scholar] [CrossRef] [PubMed]
- Breast Cancer Progression Working Party. Evidence for novel non-angiogenic pathway in breast-cancer metastasis. Lancet 2000, 355, 1787–1788. [Google Scholar]
- Adighibe, O.; Micklem, K.; Campo, L.; Ferguson, M.; Harris, A.; Pozos, R.; Gatter, K.; Pezzella, F. Is nonangiogenesis a novel pathway for cancer progression? A study using 3-dimensional tumour reconstructions. Br. J. Cancer 2006, 94, 1176–1179. [Google Scholar] [CrossRef]
- Bentolila, L.A.; Prakash, R.; Mihic-Probst, D.; Wadehra, M.; Kleinman, H.K.; Carmichael, T.S.; Péault, B.; Barnhill, R.L.; Lugassy, C. Imaging of Angiotropism/Vascular Co-Option in a Murine Model of Brain Melanoma: Implications for Melanoma Progression along Extravascular Pathways. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Lugassy, C.; Kleinman, H.; Barnhill, R. Pericyte mimicry: An embryogenesis-derived program of extravascular tumor cell migration. In Tumor Vascularization; Elsevier: Amsterdam, The Netherlands, 2020; pp. 49–88. [Google Scholar] [CrossRef]
- Lugassy, C.; Péault, B.; Wadehra, M.; Kleinman, H.K.; Barnhill, R.L. Couldpericytic mimicryrepresent another type of melanoma cell plasticity with embryonic properties? Pigment. Cell Melanoma Res. 2013, 26, 746–754. [Google Scholar] [CrossRef]
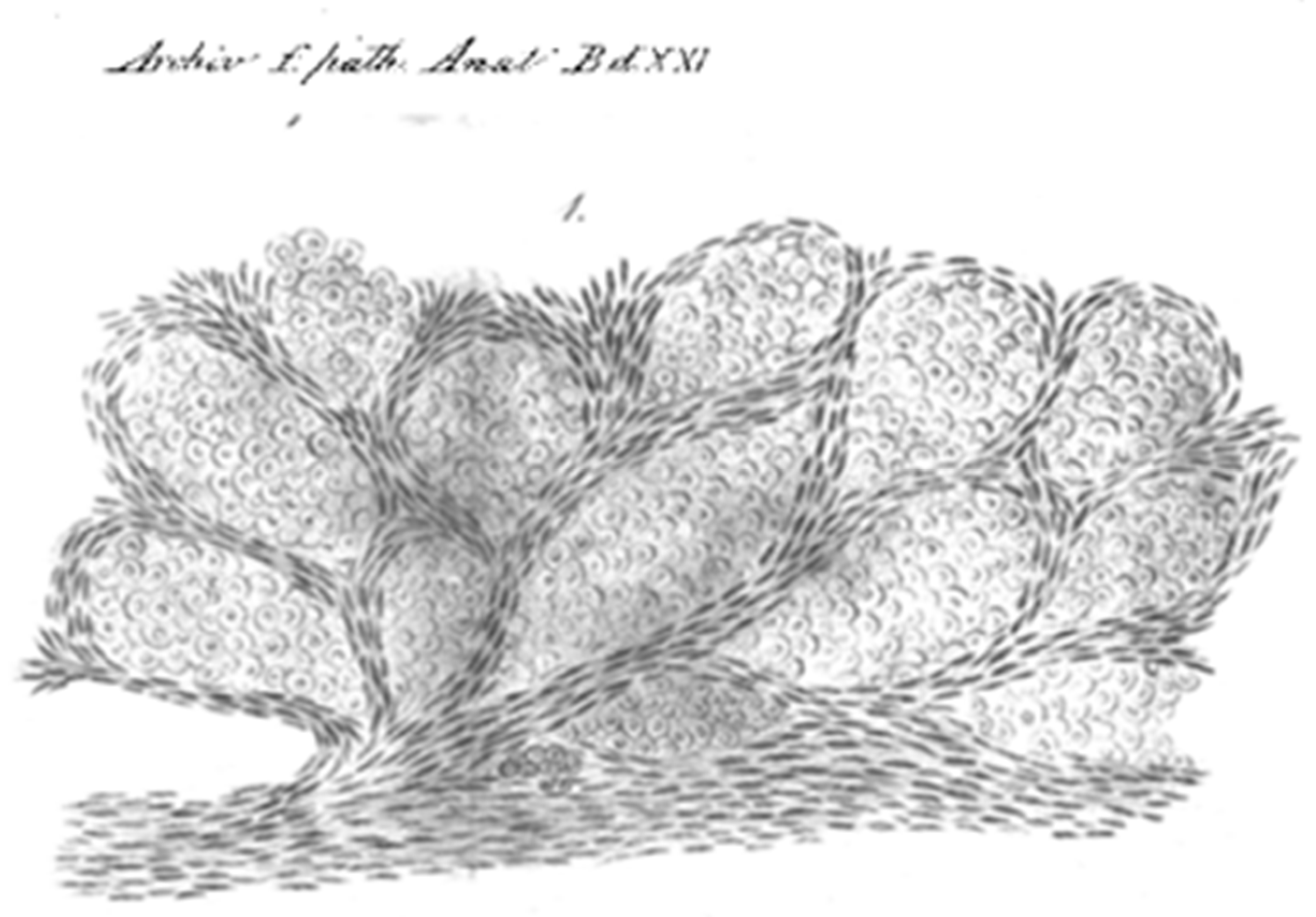
- Maniotis, A.J.; Folberg, R.; Hess, A.; Seftor, E.A.; Gardner, L.M.G.; Pe’er, J.; Trent, J.M.; Meltzer, P.S.; Hendrix, M.J.C. Vascular Channel Formation by Human Melanoma Cells in Vivo and in Vitro: Vasculogenic Mimicry. Am. J. Pathol. 1999, 155, 739–752. [Google Scholar] [CrossRef]
- Cao, Y.; Qian, C.-N. Vasculogenic mimicry. In Tumor Vascularization; Elsevier: Amsterdam, The Netherlands, 2020; pp. 89–100. [Google Scholar] [CrossRef]
- Seftor, R.E.; Seftor, E.A.; Koshikawa, N.; Meltzer, P.S.; Gardner, L.M.; Bilban, M.; Stetler-Stevenson, W.G.; Quaranta, V.; Hendrix, M.J. Cooperative interactions of laminin 5 gamma2 chain, matrix metalloproteinase-2, and membrane type-1-matrix/metalloproteinase are required for mimicry of embryonic vasculogenesis by aggressive melanoma. Cancer Res. 2001, 61, 6322–6327. [Google Scholar] [PubMed]
- Ribatti, D.; Djonov, V. Intussusceptive microvascular growth in tumors. Cancer Lett. 2012, 316, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Hlushchuk, R.; Makanya, A.N.; Djonov, V. Escape mechanisms after antiangiogenic treatment, or why are the tumors growing again? Int. J. Dev. Biol 2011, 55, 563–567. [Google Scholar] [CrossRef]
- Ribatti, D.; Vacca, A.; Nico, B.; Roncali, L.; Dammacco, F. Postnatal vasculogenesis. Mech. Dev. 2001, 100, 157–163. [Google Scholar] [CrossRef]
- Ribatti, D. The discovery of endothelial progenitor cells. An historical review. Leuk Res. 2007, 31, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Kopp, H.G.; Ramos, C.A.; Rafii, S. Contribution of endothelial progenitors and proangiogenic hematopoietic cells to vascularization of tumor and ischemic tissue. Curr. Opin. Hematol. 2006, 13, 175–181. [Google Scholar] [CrossRef]
- Ribatti, D. Inflammation and Angiogenesis; Springer International Publishing: Cham, Switzerland, 2017; pp. 25–26. [Google Scholar] [CrossRef]
- Li, S.; Meng, W.; Guan, Z.; Guo, Y.; Han, X. The hypoxia-related signaling pathways of vasculogenic mimicry in tumor treatment. Biomed. Pharm. 2016, 80, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.Z.; Sun, W. Molecular regulation of vasculogenic mimicry in tumors and potential tumor-target therapy. World J. Gastrointest. Surg. 2010, 2, 117–127. [Google Scholar] [CrossRef]
- di Tomaso, E.; Snuderl, M.; Kamoun, W.S.; Duda, D.G.; Auluck, P.K.; Fazlollahi, L.; Andronesi, O.C.; Frosch, M.P.; Wen, P.Y.; Plotkin, S.R.; et al. Glioblastoma recurrence after cediranib therapy in patients: Lack of “rebound” revascularization as mode of escape. Cancer Res. 2011, 71, 19–28. [Google Scholar] [CrossRef] [PubMed]
- de Groot, J.F.; Fuller, G.; Kumar, A.J.; Piao, Y.; Eterovic, K.; Ji, Y.; Conrad, C.A. Tumor invasion after treatment of glioblastoma with bevacizumab: Radiographic and pathologic correlation in humans and mice. Neuro. Oncol. 2010, 12, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Carbonell, W.S.; DeLay, M.; Jahangiri, A.; Park, C.C.; Aghi, M.K. beta1 integrin targeting potentiates antiangiogenic therapy and inhibits the growth of bevacizumab-resistant glioblastoma. Cancer Res. 2013, 73, 3145–3154. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1787 | Hunter firstly used the term “angiogenesis”. |
| 1907 | Goldmann visualized angiogenesis. |
| 1934 | Detailed description of intra-alveolar growth of lung metastatic tumors spreading from one alveolus to another through the alveolar pores. |
| 1939 | Vascularization of the Brown Pearce rabbit epithelioma transplant in the transparent ear chamber in rabbits. |
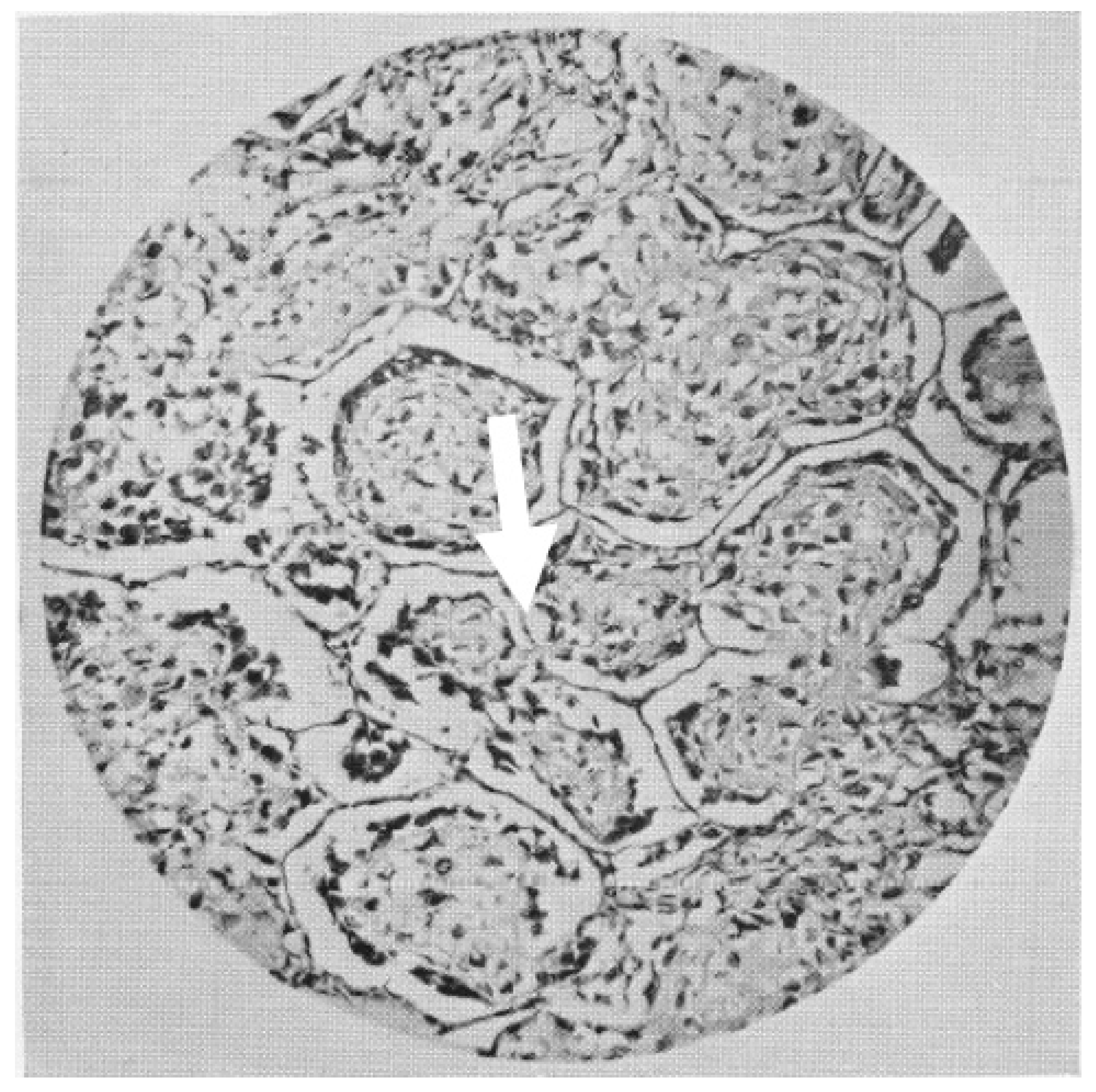
| 1956 | Proliferation of cancer cells alongside hepatocellular trabeculae with a replacement pattern. |
| 1962 | Florey’s General Pathology textbook states that sometime a tumor supplement or replace the stroma using of pre-existing structures like the alveolar walls. |
| 1971 | Folkman introduces the hypothesis that tumor growth is dependent on angiogenesis. |
| 1974 | Robbins’ textbook describes that sometime metastatic cancer cells will grow in the lung without destruction of the lung parenchyma in a fashion resembling bacterial pneumonia. |
| 1980 | Demonstration that inhibition of angiogenesis suppresses tumor growth in animal models. |
| 1988 | Trabecular growth of hepatocellular carcinoma and intra-alveolar growth of primary lung carcinomas. |
| 1996 | First description of non-angiogenic tumors. |
| 1999 | First description of vascular co-option in a mouse model. |
| 1999 | First description of vasculogenic mimicry. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ribatti, D.; Pezzella, F. Overview on the Different Patterns of Tumor Vascularization. Cells 2021, 10, 639. https://doi.org/10.3390/cells10030639
Ribatti D, Pezzella F. Overview on the Different Patterns of Tumor Vascularization. Cells. 2021; 10(3):639. https://doi.org/10.3390/cells10030639
Chicago/Turabian StyleRibatti, Domenico, and Francesco Pezzella. 2021. "Overview on the Different Patterns of Tumor Vascularization" Cells 10, no. 3: 639. https://doi.org/10.3390/cells10030639
APA StyleRibatti, D., & Pezzella, F. (2021). Overview on the Different Patterns of Tumor Vascularization. Cells, 10(3), 639. https://doi.org/10.3390/cells10030639