
Tuning IgE: IgE-Associating Molecules and Their Effects on IgE-Dependent Mast Cell Reactions


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
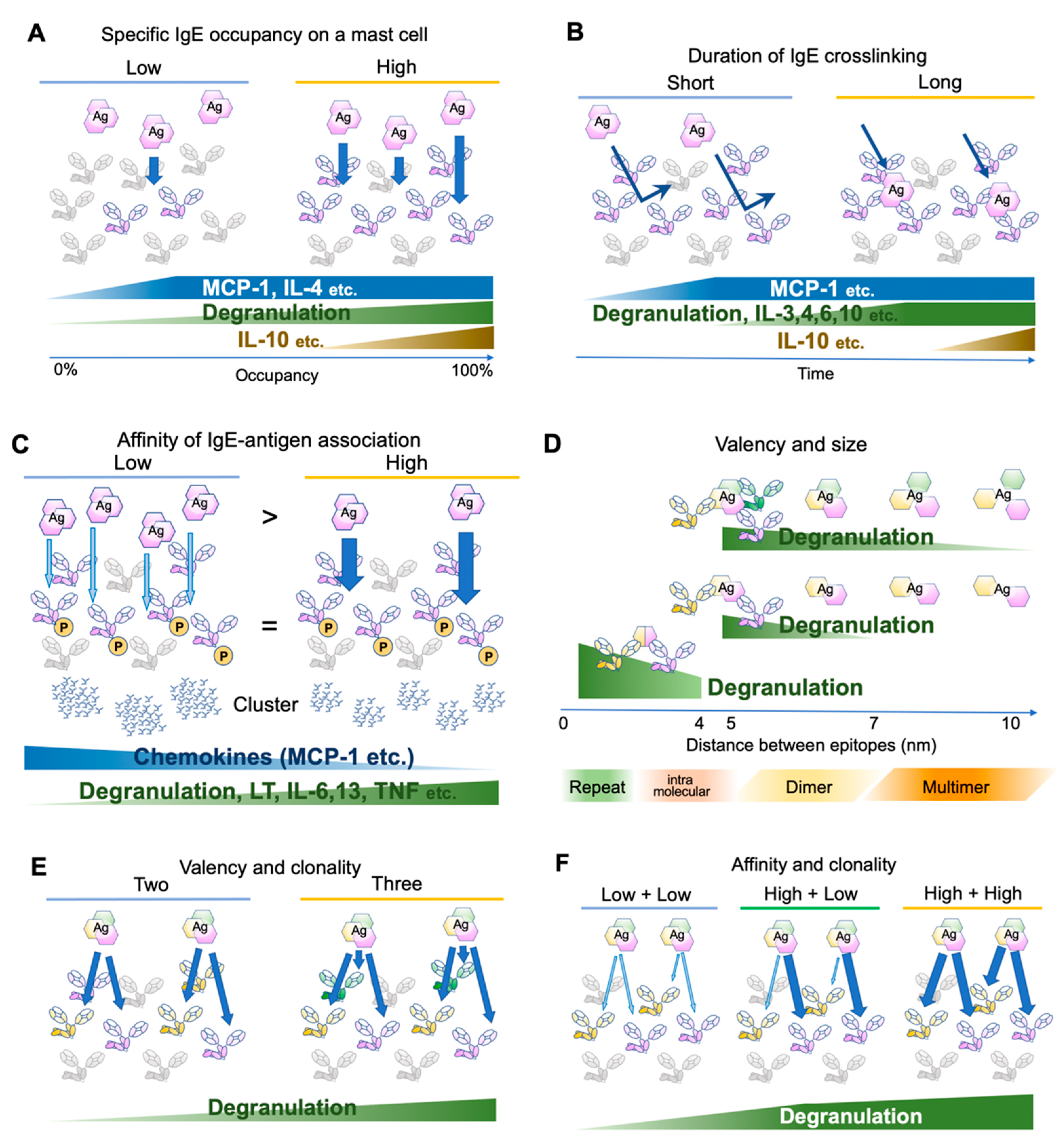
2. FcεRI Receptor Dynamics and Antigen Properties
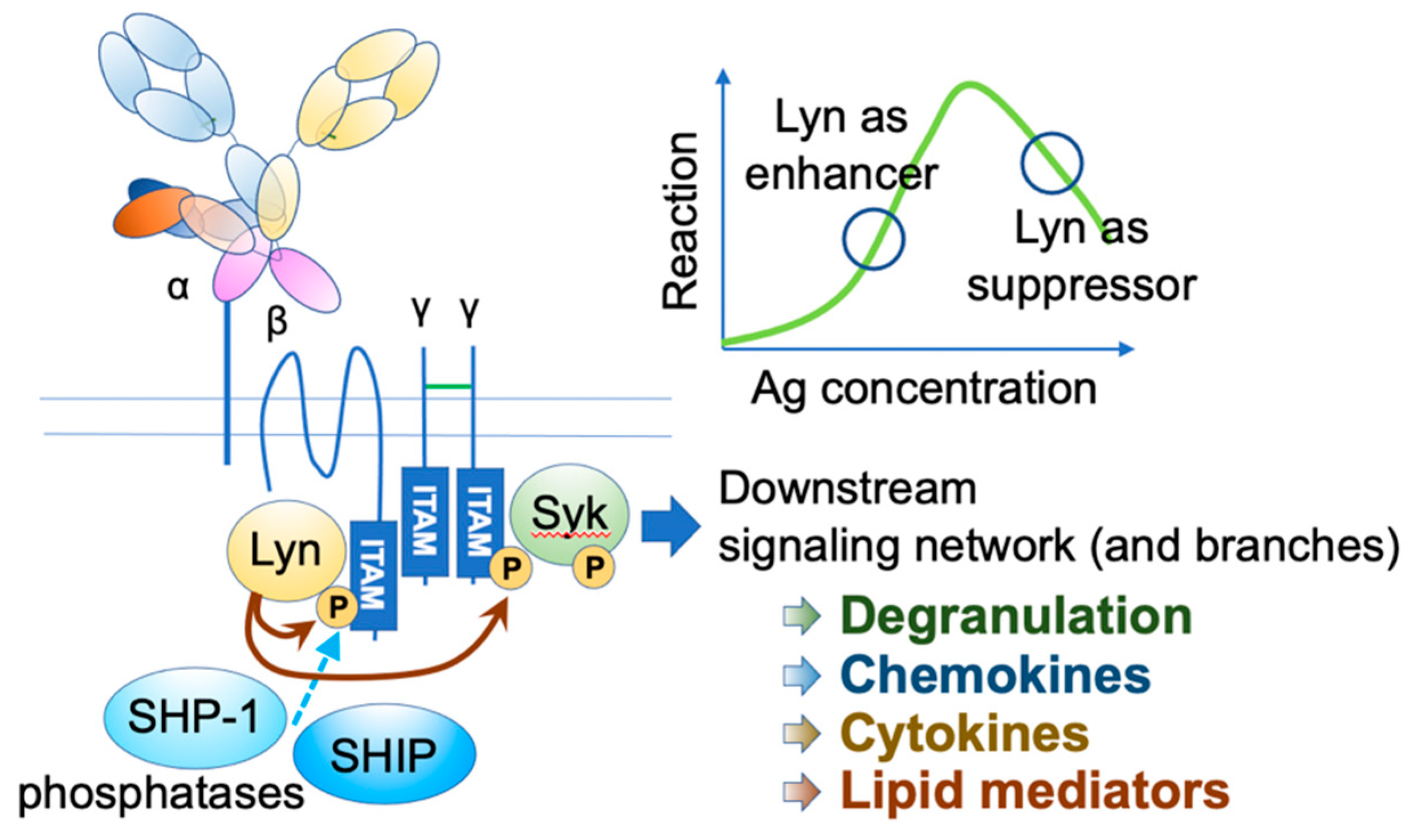
3. Histamine-Releasing Factor (HRF)
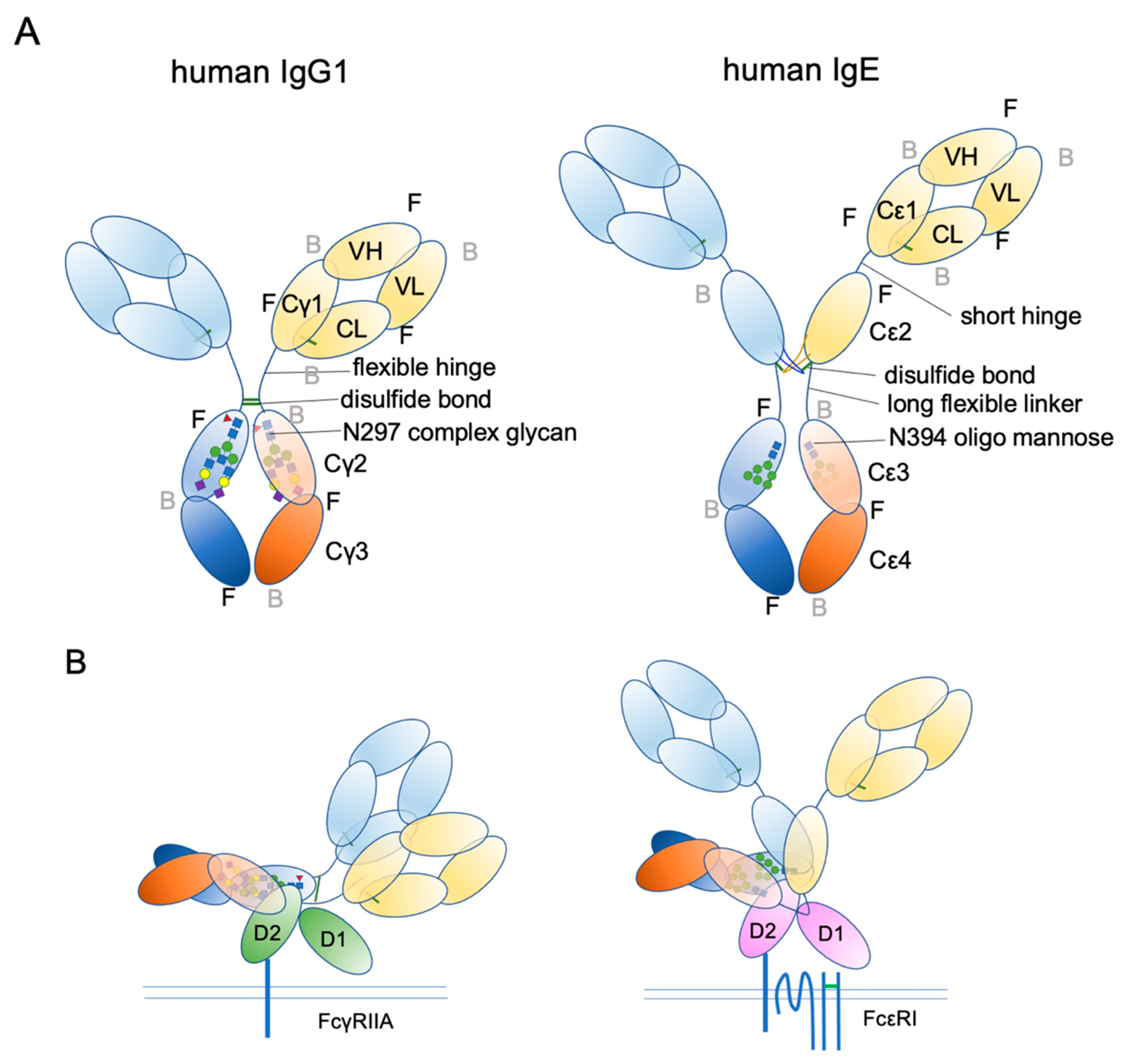
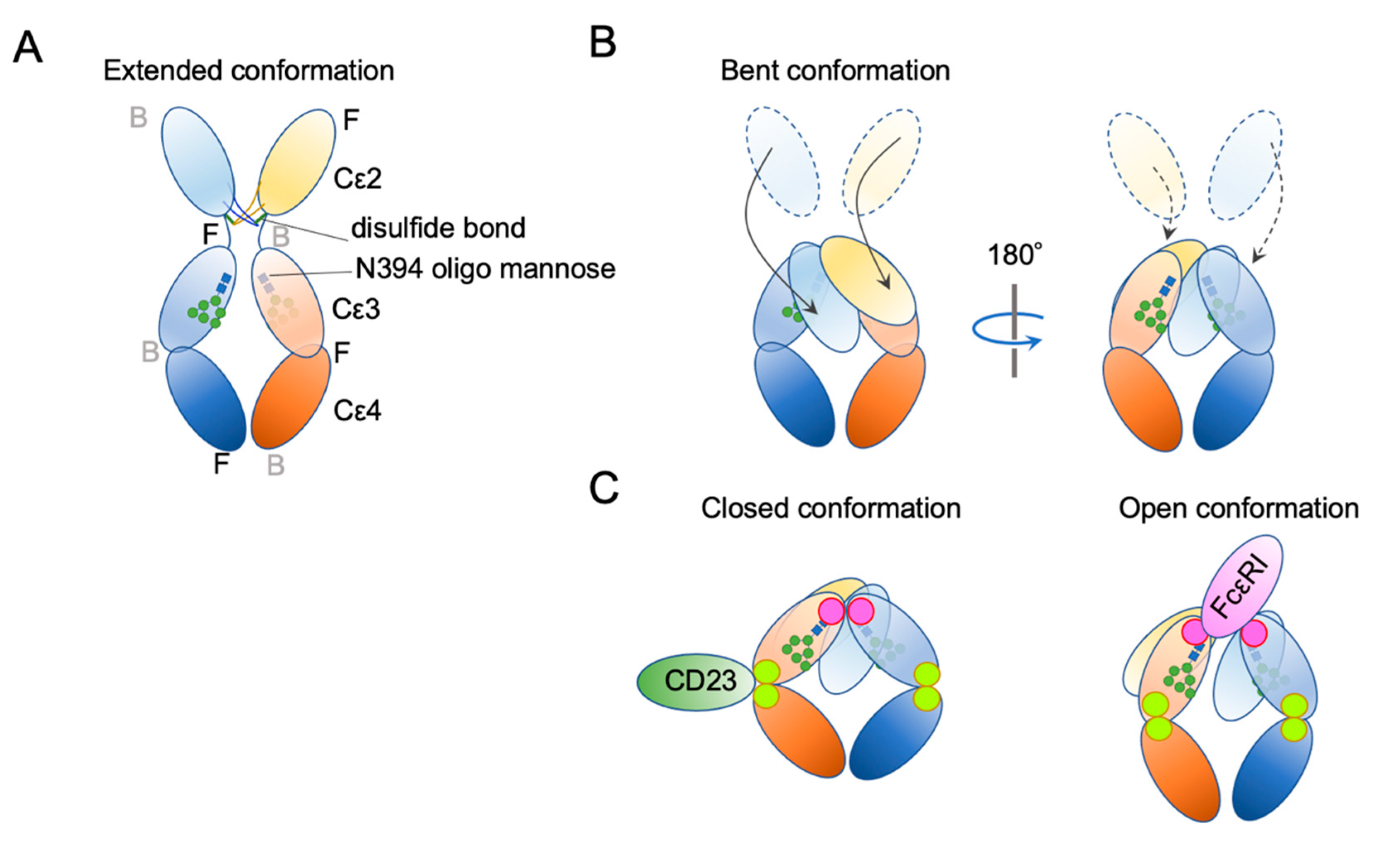
4. IgE Structural Conformation
5. IgE-Binding Therapeutics
6. Glycosylation
7. Galectins
8. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Starkl, P.; Marichal, T.; Gaudenzio, N.; Reber, L.L.; Sibilano, R.; Tsai, M.; Galli, S.J. IgE antibodies, FcepsilonRIalpha, and IgE-mediated local anaphylaxis can limit snake venom toxicity. J. Allergy Clin. Immunol. 2016, 137, 246–257.e211. [Google Scholar] [CrossRef] [Green Version]
- Martin, R.K.; Damle, S.R.; Valentine, Y.A.; Zellner, M.P.; James, B.N.; Lownik, J.C.; Luker, A.J.; Davis, E.H.; DeMeules, M.M.; Khandjian, L.M.; et al. B1 Cell IgE Impedes Mast Cell-Mediated Enhancement of Parasite Expulsion through B2 IgE Blockade. Cell Rep. 2018, 22, 1824–1834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starkl, P.; Watzenboeck, M.L.; Popov, L.M.; Zahalka, S.; Hladik, A.; Lakovits, K.; Radhouani, M.; Haschemi, A.; Marichal, T.; Reber, L.L.; et al. IgE Effector Mechanisms, in Concert with Mast Cells, Contribute to Acquired Host Defense against Staphylococcus aureus. Immunity 2020, 53, 1333. [Google Scholar] [CrossRef]
- Kinet, J.P. The high-affinity IgE receptor (Fc epsilon RI): From physiology to pathology. Annu. Rev. Immunol. 1999, 17, 931–972. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, T.; Galli, S.J. Regulation of mast-cell and basophil function and survival by IgE. Nat. Rev. Immunol. 2002, 2, 773–786. [Google Scholar] [CrossRef] [PubMed]
- Blank, U.; Huang, H.; Kawakami, T. The high affinity IgE receptor: A signaling update. Curr. Opin. Immunol. 2021, 72, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Shamji, M.H.; Valenta, R.; Jardetzky, T.; Verhasselt, V.; Durham, S.R.; Wurtzen, P.A.; van Neerven, R.J.J. The role of allergen-specific IgE, IgG and IgA in allergic disease. Allergy 2021. [Google Scholar] [CrossRef]
- Miyake, K.; Shibata, S.; Yoshikawa, S.; Karasuyama, H. Basophils and their effector molecules in allergic disorders. Allergy 2021, 76, 1693–1706. [Google Scholar] [CrossRef] [PubMed]
- Michelet, M.; Balbino, B.; Guilleminault, L.; Reber, L.L. IgE in the pathophysiology and therapy of food allergy. Eur. J. Immunol. 2021, 51, 531–543. [Google Scholar] [CrossRef]
- Charles, N. Autoimmunity, IgE and FcepsilonRI-bearing cells. Curr. Opin. Immunol. 2021, 72, 43–50. [Google Scholar] [CrossRef]
- Engeroff, P.; Vogel, M. The role of CD23 in the regulation of allergic responses. Allergy 2020. [Google Scholar] [CrossRef]
- Maurer, M.; Altrichter, S.; Schmetzer, O.; Scheffel, J.; Church, M.K.; Metz, M. Immunoglobulin E-Mediated Autoimmunity. Front. Immunol. 2018, 9, 689. [Google Scholar] [CrossRef] [Green Version]
- Mukai, K.; Tsai, M.; Starkl, P.; Marichal, T.; Galli, S.J. IgE and mast cells in host defense against parasites and venoms. Semin. Immunopathol. 2016, 38, 581–603. [Google Scholar] [CrossRef]
- Sallmann, E.; Reininger, B.; Brandt, S.; Duschek, N.; Hoflehner, E.; Garner-Spitzer, E.; Platzer, B.; Dehlink, E.; Hammer, M.; Holcmann, M.; et al. High-affinity IgE receptors on dendritic cells exacerbate Th2-dependent inflammation. J. Immunol. 2011, 187, 164–171. [Google Scholar] [CrossRef] [Green Version]
- Maurer, D.; Fiebiger, S.; Ebner, C.; Reininger, B.; Fischer, G.F.; Wichlas, S.; Jouvin, M.H.; Schmitt-Egenolf, M.; Kraft, D.; Kinet, J.P.; et al. Peripheral blood dendritic cells express Fc epsilon RI as a complex composed of Fc epsilon RI alpha- and Fc epsilon RI gamma-chains and can use this receptor for IgE-mediated allergen presentation. J. Immunol. 1996, 157, 607–616. [Google Scholar]
- Lin, S.; Cicala, C.; Scharenberg, A.M.; Kinet, J.P. The Fc(epsilon)RIbeta subunit functions as an amplifier of Fc(epsilon)RIgamma-mediated cell activation signals. Cell 1996, 85, 985–995. [Google Scholar] [CrossRef] [Green Version]
- Xiao, W.; Nishimoto, H.; Hong, H.; Kitaura, J.; Nunomura, S.; Maeda-Yamamoto, M.; Kawakami, Y.; Lowell, C.A.; Ra, C.; Kawakami, T. Positive and negative regulation of mast cell activation by Lyn via the FcepsilonRI. J. Immunol. 2005, 175, 6885–6892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, W.; Kashiwakura, J.; Hong, H.; Yasudo, H.; Ando, T.; Maeda-Yamamoto, M.; Wu, D.; Kawakami, Y.; Kawakami, T. Phospholipase C-beta3 regulates FcvarepsilonRI-mediated mast cell activation by recruiting the protein phosphatase SHP-1. Immunity 2011, 34, 893–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conrad, D.H.; Bazin, H.; Sehon, A.H.; Froese, A. Binding parameters of the interaction between rat IgE and rat mast cell receptors. J. Immunol. 1975, 114, 1688–1691. [Google Scholar] [PubMed]
- DeLisi, C.; Siraganian, R.P. Receptor cross-linking and histamine release. I. The quantitative dependence of basophil degranulation on the number of receptor doublets. J. Immunol. 1979, 122, 2286–2292. [Google Scholar] [PubMed]
- Schweitzer-Stenner, R.; Pecht, I. Parameters determining the stimulatory capacity of the type I Fc epsilon-receptor. Immunol. Lett. 1999, 68, 59–69. [Google Scholar] [CrossRef]
- Hjort, C.; Schiotz, P.O.; Ohlin, M.; Wurtzen, P.A.; Christensen, L.H.; Hoffmann, H.J. The number and affinity of productive IgE pairs determine allergen activation of mast cells. J. Allergy Clin. Immunol. 2017, 140, 1167–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Espinosa, C.; Odom, S.; Olivera, A.; Hobson, J.P.; Martinez, M.E.; Oliveira-Dos-Santos, A.; Barra, L.; Spiegel, S.; Penninger, J.M.; Rivera, J. Preferential signaling and induction of allergy-promoting lymphokines upon weak stimulation of the high affinity IgE receptor on mast cells. J. Exp. Med. 2003, 197, 1453–1465. [Google Scholar] [CrossRef] [PubMed]
- Cabanillas, B.; Jappe, U.; Novak, N. Allergy to Peanut, Soybean, and Other Legumes: Recent Advances in Allergen Characterization, Stability to Processing and IgE Cross-Reactivity. Mol. Nutr. Food Res. 2018, 62. [Google Scholar] [CrossRef]
- Hils, M.; Wolbing, F.; Hilger, C.; Fischer, J.; Hoffard, N.; Biedermann, T. The History of Carbohydrates in Type I Allergy. Front. Immunol. 2020, 11, 586924. [Google Scholar] [CrossRef] [PubMed]
- Kashiwakura, J.; Okayama, Y.; Furue, M.; Kabashima, K.; Shimada, S.; Ra, C.; Siraganian, R.P.; Kawakami, Y.; Kawakami, T. Most Highly Cytokinergic IgEs Have Polyreactivity to Autoantigens. Allergy Asthma Immunol. Res. 2012, 4, 332–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen-Jarolim, E.; Vogel, M.; Zavazal, V.; Stadler, B.M. Nonspecific binding of IgE to allergens. Allergy 1997, 52, 844–852. [Google Scholar] [CrossRef] [PubMed]
- Pomes, A. Relevant B cell epitopes in allergic disease. Int. Arch. Allergy Immunol. 2010, 152, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pali-Scholl, I.; Jensen-Jarolim, E. The concept of allergen-associated molecular patterns (AAMP). Curr. Opin. Immunol. 2016, 42, 113–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurer, M.; Eyerich, K.; Eyerich, S.; Ferrer, M.; Gutermuth, J.; Hartmann, K.; Jakob, T.; Kapp, A.; Kolkhir, P.; Larenas-Linnemann, D.; et al. Urticaria: Collegium Internationale Allergologicum (CIA) Update 2020. Int. Arch. Allergy Immunol. 2020, 181, 321–333. [Google Scholar] [CrossRef]
- Torigoe, C.; Inman, J.K.; Metzger, H. An unusual mechanism for ligand antagonism. Science 1998, 281, 568–572. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, R.; Leach, S.; Liu, W.; Ralston, E.; Scheffel, J.; Zhang, W.; Lowell, C.A.; Rivera, J. Molecular editing of cellular responses by the high-affinity receptor for IgE. Science 2014, 343, 1021–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, N.L.; Pfeiffer, J.R.; Martinez, A.M.; Haaland, D.M.; Davis, R.W.; Kawakami, T.; Oliver, J.M.; Wilson, B.S.; Lidke, D.S. Small, mobile FcepsilonRI receptor aggregates are signaling competent. Immunity 2009, 31, 469–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakefield, D.L.; Holowka, D.; Baird, B. The FcepsilonRI Signaling Cascade and Integrin Trafficking Converge at Patterned Ligand Surfaces. Mol. Biol. Cell 2017. [Google Scholar] [CrossRef] [Green Version]
- Sil, D.; Lee, J.B.; Luo, D.; Holowka, D.; Baird, B. Trivalent ligands with rigid DNA spacers reveal structural requirements for IgE receptor signaling in RBL mast cells. ACS Chem. Biol. 2007, 2, 674–684. [Google Scholar] [CrossRef] [Green Version]
- Paar, J.M.; Harris, N.T.; Holowka, D.; Baird, B. Bivalent ligands with rigid double-stranded DNA spacers reveal structural constraints on signaling by Fc epsilon RI. J. Immunol. 2002, 169, 856–864. [Google Scholar] [CrossRef] [Green Version]
- Paolini, R.; Jouvin, M.H.; Kinet, J.P. Phosphorylation and dephosphorylation of the high-affinity receptor for immunoglobulin E immediately after receptor engagement and disengagement. Nature 1991, 353, 855–858. [Google Scholar] [CrossRef]
- Felce, J.H.; Sezgin, E.; Wane, M.; Brouwer, H.; Dustin, M.L.; Eggeling, C.; Davis, S.J. CD45 exclusion- and cross-linking-based receptor signaling together broaden FcepsilonRI reactivity. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Kato, Y.; Abbott, R.K.; Freeman, B.L.; Haupt, S.; Groschel, B.; Silva, M.; Menis, S.; Irvine, D.J.; Schief, W.R.; Crotty, S. Multifaceted Effects of Antigen Valency on B Cell Response Composition and Differentiation In Vivo. Immunity 2020, 53, 548–563. [Google Scholar] [CrossRef]
- Battais, F.; Mothes, T.; Moneret-Vautrin, D.A.; Pineau, F.; Kanny, G.; Popineau, Y.; Bodinier, M.; Denery-Papini, S. Identification of IgE-binding epitopes on gliadins for patients with food allergy to wheat. Allergy 2005, 60, 815–821. [Google Scholar] [CrossRef]
- Niemi, M.H.; Rytkonen-Nissinen, M.; Miettinen, I.; Janis, J.; Virtanen, T.; Rouvinen, J. Dimerization of lipocalin allergens. Sci. Rep. 2015, 5, 13841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gieras, A.; Linhart, B.; Roux, K.H.; Dutta, M.; Khodoun, M.; Zafred, D.; Cabauatan, C.R.; Lupinek, C.; Weber, M.; Focke-Tejkl, M.; et al. IgE epitope proximity determines immune complex shape and effector cell activation capacity. J. Allergy Clin. Immunol. 2016, 137, 1557–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawakami, Y.; Kasakura, K.; Kawakami, T. Histamine-Releasing Factor, a New Therapeutic Target in Allergic Diseases. Cells 2019, 8, 1515. [Google Scholar] [CrossRef] [Green Version]
- MacDonald, S.M.; Rafnar, T.; Langdon, J.; Lichtenstein, L.M. Molecular identification of an IgE-dependent histamine-releasing factor. Science 1995, 269, 688–690. [Google Scholar] [CrossRef] [PubMed]
- Bommer, U.A.; Telerman, A. Dysregulation of TCTP in Biological Processes and Diseases. Cells 2020, 9, 1632. [Google Scholar] [CrossRef]
- Amson, R.; Pece, S.; Marine, J.C.; Di Fiore, P.P.; Telerman, A. TPT1/ TCTP-regulated pathways in phenotypic reprogramming. Trends Cell Biol. 2013, 23, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Pinkaew, D.; Fujise, K. Fortilin: A Potential Target for the Prevention and Treatment of Human Diseases. Adv. Clin. Chem. 2017, 82, 265–300. [Google Scholar] [CrossRef]
- Schroeder, J.T.; Lichtenstein, L.M.; MacDonald, S.M. Recombinant histamine-releasing factor enhances IgE-dependent IL-4 and IL-13 secretion by human basophils. J. Immunol. 1997, 159, 447–452. [Google Scholar]
- Schroeder, J.T.; Lichtenstein, L.M.; MacDonald, S.M. An immunoglobulin E-dependent recombinant histamine-releasing factor induces interleukin-4 secretion from human basophils. J. Exp. Med. 1996, 183, 1265–1270. [Google Scholar] [CrossRef] [Green Version]
- MacDonald, S.M.; Lichtenstein, L.M.; Proud, D.; Plaut, M.; Naclerio, R.M.; MacGlashan, D.W.; Kagey-Sobotka, A. Studies of IgE-dependent histamine releasing factors: Heterogeneity of IgE. J. Immunol. 1987, 139, 506–512. [Google Scholar]
- Wantke, F.; MacGlashan, D.W.; Langdon, J.M.; MacDonald, S.M. The human recombinant histamine releasing factor: Functional evidence that it does not bind to the IgE molecule. J. Allergy Clin. Immunol. 1999, 103, 642–648. [Google Scholar] [CrossRef]
- Kashiwakura, J.C.; Ando, T.; Matsumoto, K.; Kimura, M.; Kitaura, J.; Matho, M.H.; Zajonc, D.M.; Ozeki, T.; Ra, C.; MacDonald, S.M.; et al. Histamine-releasing factor has a proinflammatory role in mouse models of asthma and allergy. J. Clin. Investig. 2012, 122, 218–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dore, K.A.; Kashiwakura, J.I.; McDonnell, J.M.; Gould, H.J.; Kawakami, T.; Sutton, B.J.; Davies, A.M. Crystal structures of murine and human Histamine-Releasing Factor (HRF/TCTP) and a model for HRF dimerisation in mast cell activation. Mol. Immunol. 2018, 93, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Ando, T.; Kashiwakura, J.I.; Itoh-Nagato, N.; Yamashita, H.; Baba, M.; Kawakami, Y.; Tsai, S.H.; Inagaki, N.; Takeda, K.; Iwata, T.; et al. Histamine-releasing factor enhances food allergy. J. Clin. Investig. 2017, 127, 4541–4553. [Google Scholar] [CrossRef] [Green Version]
- Kawakami, Y.; Sielski, R.; Kawakami, T. Mouse Body Temperature Measurement Using Infrared Thermometer During Passive Systemic Anaphylaxis and Food Allergy Evaluation. J. Vis. Exp. 2018. [Google Scholar] [CrossRef]
- Huang, X.; Li, Z.; Sun, R. Synergistic Actions of Histamine-Releasing Factor and Histamine Releasing Factor-Reactive IgE in Chronic Urticaria. Int. Arch. Allergy Immunol. 2017, 172, 27–32. [Google Scholar] [CrossRef]
- Ulambayar, B.; Lee, H.; Yang, E.M.; Park, H.S.; Lee, K.; Ye, Y.M. Dimerized, Not Monomeric, Translationally Controlled Tumor Protein Induces Basophil Activation and Mast Cell Degranulation in Chronic Urticaria. Immune Netw. 2019, 19, e20. [Google Scholar] [CrossRef]
- Ferrer, E.; Dunmore, B.J.; Hassan, D.; Ormiston, M.L.; Moore, S.; Deighton, J.; Long, L.; Yang, X.D.; Stewart, D.J.; Morrell, N.W. A Potential Role for Exosomal Translationally Controlled Tumor Protein Export in Vascular Remodeling in Pulmonary Arterial Hypertension. Am. J. Respir. Cell Mol. Biol. 2018, 59, 467–478. [Google Scholar] [CrossRef]
- Lavoie, J.R.; Ormiston, M.L.; Perez-Iratxeta, C.; Courtman, D.W.; Jiang, B.; Ferrer, E.; Caruso, P.; Southwood, M.; Foster, W.S.; Morrell, N.W.; et al. Proteomic analysis implicates translationally controlled tumor protein as a novel mediator of occlusive vascular remodeling in pulmonary arterial hypertension. Circulation 2014, 129, 2125–2135. [Google Scholar] [CrossRef]
- Lin, Z.B.; Yang, P.J.; Zhang, X.; Wang, J.L.; Liu, K.; Dou, K.F. Translationally controlled tumor protein exerts a proinflammatory role in acute rejection after liver transplantation. Hepatobiliary Pancreat Dis. Int. 2020, 19, 235–243. [Google Scholar] [CrossRef]
- Lee, H.; Kim, M.S.; Lee, J.S.; Cho, H.; Park, J.; Hae Shin, D.; Lee, K. Flexible loop and helix 2 domains of TCTP are the functional domains of dimerized TCTP. Sci. Rep. 2020, 10, 197. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Min, H.J.; Won, H.Y.; Park, H.; Lee, J.C.; Park, H.W.; Chung, J.; Hwang, E.S.; Lee, K. Dimerization of translationally controlled tumor protein is essential for its cytokine-like activity. PLoS ONE 2009, 4, e6464. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhang, L.; Tong, H.; Peng, B.; Rames, M.J.; Zhang, S.; Ren, G. 3D Structural Fluctuation of IgG1 Antibody Revealed by Individual Particle Electron Tomography. Sci. Rep. 2015, 5, 9803. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.K.; Jabs, F.; Miehe, M.; Molgaard, B.; Pfutzner, W.; Mobs, C.; Spillner, E.; Andersen, G.R. Structure of intact IgE and the mechanism of ligelizumab revealed by electron microscopy. Allergy 2020, 75, 1956–1965. [Google Scholar] [CrossRef]
- Wan, T.; Beavil, R.L.; Fabiane, S.M.; Beavil, A.J.; Sohi, M.K.; Keown, M.; Young, R.J.; Henry, A.J.; Owens, R.J.; Gould, H.J.; et al. The crystal structure of IgE Fc reveals an asymmetrically bent conformation. Nat. Immunol. 2002, 3, 681–686. [Google Scholar] [CrossRef]
- Kikhney, A.G.; Svergun, D.I. A practical guide to small angle X-ray scattering (SAXS) of flexible and intrinsically disordered proteins. FEBS Lett. 2015, 589, 2570–2577. [Google Scholar] [CrossRef] [Green Version]
- Drinkwater, N.; Cossins, B.; Keeble, A.H.; Wright, M.; Cain, K.; Hailu, H.; Oxbrow, A.; Delgado, J.; Shuttleworth, L.K.; Kao, M.W.; et al. Human immunoglobulin E flexes between acutely bent and extended conformations. Nat. Struct. Mol. Biol. 2014, 21, 397–404. [Google Scholar] [CrossRef] [Green Version]
- Holdom, M.D.; Davies, A.M.; Nettleship, J.E.; Bagby, S.C.; Dhaliwal, B.; Girardi, E.; Hunt, J.; Gould, H.J.; Beavil, A.J.; McDonnell, J.M.; et al. Conformational changes in IgE contribute to its uniquely slow dissociation rate from receptor FcvarepsilonRI. Nat. Struct. Mol. Biol. 2011, 18, 571–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, A.M.; Allan, E.G.; Keeble, A.H.; Delgado, J.; Cossins, B.P.; Mitropoulou, A.N.; Pang, M.O.Y.; Ceska, T.; Beavil, A.J.; Craggs, G.; et al. Allosteric mechanism of action of the therapeutic anti-IgE antibody omalizumab. J. Biol. Chem. 2017, 292, 9975–9987. [Google Scholar] [CrossRef] [Green Version]
- Garman, S.C.; Wurzburg, B.A.; Tarchevskaya, S.S.; Kinet, J.P.; Jardetzky, T.S. Structure of the Fc fragment of human IgE bound to its high-affinity receptor Fc epsilonRI alpha. Nature 2000, 406, 259–266. [Google Scholar] [CrossRef]
- Miller, L.; Blank, U.; Metzger, H.; Kinet, J.P. Expression of high-affinity binding of human immunoglobulin E by transfected cells. Science 1989, 244, 334–337. [Google Scholar] [CrossRef]
- McDonnell, J.M.; Calvert, R.; Beavil, R.L.; Beavil, A.J.; Henry, A.J.; Sutton, B.J.; Gould, H.J.; Cowburn, D. The structure of the IgE Cepsilon2 domain and its role in stabilizing the complex with its high-affinity receptor FcepsilonRIalpha. Nat. Struct. Biol. 2001, 8, 437–441. [Google Scholar] [CrossRef]
- Dehlink, E.; Platzer, B.; Baker, A.H.; Larosa, J.; Pardo, M.; Dwyer, P.; Yen, E.H.; Szepfalusi, Z.; Nurko, S.; Fiebiger, E. A soluble form of the high affinity IgE receptor, Fc-epsilon-RI, circulates in human serum. PLoS ONE 2011, 6, e19098. [Google Scholar] [CrossRef]
- Seminario, M.C.; Saini, S.S.; MacGlashan, D.W., Jr.; Bochner, B.S. Intracellular expression and release of Fc epsilon RI alpha by human eosinophils. J. Immunol. 1999, 162, 6893–6900. [Google Scholar]
- Monino-Romero, S.; Lexmond, W.S.; Singer, J.; Bannert, C.; Amoah, A.S.; Yazdanbakhsh, M.; Boakye, D.A.; Jensen-Jarolim, E.; Fiebiger, E.; Szepfalusi, Z. Soluble FcvarepsilonRI: A biomarker for IgE-mediated diseases. Allergy 2019, 74, 1381–1384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weskamp, G.; Ford, J.W.; Sturgill, J.; Martin, S.; Docherty, A.J.; Swendeman, S.; Broadway, N.; Hartmann, D.; Saftig, P.; Umland, S.; et al. ADAM10 is a principal ‘sheddase’ of the low-affinity immunoglobulin E receptor CD23. Nat. Immunol. 2006, 7, 1293–1298. [Google Scholar] [CrossRef] [PubMed]
- Gibb, D.R.; El Shikh, M.; Kang, D.J.; Rowe, W.J.; El Sayed, R.; Cichy, J.; Yagita, H.; Tew, J.G.; Dempsey, P.J.; Crawford, H.C.; et al. ADAM10 is essential for Notch2-dependent marginal zone B cell development and CD23 cleavage in vivo. J. Exp. Med. 2010, 207, 623–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platzer, B.; Ruiter, F.; van der Mee, J.; Fiebiger, E. Soluble IgE receptors--elements of the IgE network. Immunol. Lett. 2011, 141, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Strunk, R.C.; Bloomberg, G.R. Omalizumab for asthma. N. Engl. J. Med. 2006, 354, 2689–2695. [Google Scholar] [CrossRef] [Green Version]
- Busse, W.; Corren, J.; Lanier, B.Q.; McAlary, M.; Fowler-Taylor, A.; Cioppa, G.D.; van As, A.; Gupta, N. Omalizumab, anti-IgE recombinant humanized monoclonal antibody, for the treatment of severe allergic asthma. J. Allergy Clin. Immunol. 2001, 108, 184–190. [Google Scholar] [CrossRef]
- Holgate, S.T.; Chuchalin, A.G.; Hebert, J.; Lotvall, J.; Persson, G.B.; Chung, K.F.; Bousquet, J.; Kerstjens, H.A.; Fox, H.; Thirlwell, J.; et al. Efficacy and safety of a recombinant anti-immunoglobulin E antibody (omalizumab) in severe allergic asthma. Clin. Exp. Allergy 2004, 34, 632–638. [Google Scholar] [CrossRef]
- Soler, M.; Matz, J.; Townley, R.; Buhl, R.; O’Brien, J.; Fox, H.; Thirlwell, J.; Gupta, N.; Della Cioppa, G. The anti-IgE antibody omalizumab reduces exacerbations and steroid requirement in allergic asthmatics. Eur. Respir. J. 2001, 18, 254–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milgrom, H.; Berger, W.; Nayak, A.; Gupta, N.; Pollard, S.; McAlary, M.; Taylor, A.F.; Rohane, P. Treatment of childhood asthma with anti-immunoglobulin E antibody (omalizumab). Pediatrics 2001, 108, E36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saini, S.; Rosen, K.E.; Hsieh, H.J.; Wong, D.A.; Conner, E.; Kaplan, A.; Spector, S.; Maurer, M. A randomized, placebo-controlled, dose-ranging study of single-dose omalizumab in patients with H1-antihistamine-refractory chronic idiopathic urticaria. J. Allergy Clin. Immunol. 2011, 128, 567–573. [Google Scholar] [CrossRef]
- Maurer, M.; Rosen, K.; Hsieh, H.J.; Saini, S.; Grattan, C.; Gimenez-Arnau, A.; Agarwal, S.; Doyle, R.; Canvin, J.; Kaplan, A.; et al. Omalizumab for the treatment of chronic idiopathic or spontaneous urticaria. N. Engl. J. Med. 2013, 368, 924–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gevaert, P.; Calus, L.; Van Zele, T.; Blomme, K.; De Ruyck, N.; Bauters, W.; Hellings, P.; Brusselle, G.; De Bacquer, D.; van Cauwenberge, P.; et al. Omalizumab is effective in allergic and nonallergic patients with nasal polyps and asthma. J. Allergy Clin. Immunol. 2013, 131, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Gevaert, P.; Omachi, T.A.; Corren, J.; Mullol, J.; Han, J.; Lee, S.E.; Kaufman, D.; Ligueros-Saylan, M.; Howard, M.; Zhu, R.; et al. Efficacy and safety of omalizumab in nasal polyposis: 2 randomized phase 3 trials. J. Allergy Clin. Immunol. 2020, 146, 595–605. [Google Scholar] [CrossRef]
- Okubo, K.; Okano, M.; Sato, N.; Tamaki, Y.; Suzuki, H.; Uddin, A.; Fogel, R. Add-On Omalizumab for Inadequately Controlled Severe Pollinosis Despite Standard-of-Care: A Randomized Study. J. Allergy Clin. Immunol. Pract. 2020, 8, 3130–3140. [Google Scholar] [CrossRef]
- Dantzer, J.A.; Wood, R.A. The use of omalizumab in allergen immunotherapy. Clin. Exp. Allergy 2018, 48, 232–240. [Google Scholar] [CrossRef]
- Beck, L.A.; Marcotte, G.V.; MacGlashan, D.; Togias, A.; Saini, S. Omalizumab-induced reductions in mast cell Fce psilon RI expression and function. J. Allergy Clin. Immunol. 2004, 114, 527–530. [Google Scholar] [CrossRef]
- Kaplan, A.P.; Gimenez-Arnau, A.M.; Saini, S.S. Mechanisms of action that contribute to efficacy of omalizumab in chronic spontaneous urticaria. Allergy 2017, 72, 519–533. [Google Scholar] [CrossRef] [Green Version]
- Pennington, L.F.; Tarchevskaya, S.; Brigger, D.; Sathiyamoorthy, K.; Graham, M.T.; Nadeau, K.C.; Eggel, A.; Jardetzky, T.S. Structural basis of omalizumab therapy and omalizumab-mediated IgE exchange. Nat. Commun. 2016, 7, 11610. [Google Scholar] [CrossRef] [Green Version]
- Eggel, A.; Baravalle, G.; Hobi, G.; Kim, B.; Buschor, P.; Forrer, P.; Shin, J.S.; Vogel, M.; Stadler, B.M.; Dahinden, C.A.; et al. Accelerated dissociation of IgE-FcepsilonRI complexes by disruptive inhibitors actively desensitizes allergic effector cells. J. Allergy Clin. Immunol. 2014, 133, 1709–1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, E.S.; Dobson, C.L.; Kack, H.; Wang, B.; Sims, D.A.; Lloyd, C.O.; England, E.; Rees, D.G.; Guo, H.; Karagiannis, S.N.; et al. A novel IgE-neutralizing antibody for the treatment of severe uncontrolled asthma. MAbs 2014, 6, 756–764. [Google Scholar] [CrossRef]
- Sheldon, E.; Schwickart, M.; Li, J.; Kim, K.; Crouch, S.; Parveen, S.; Kell, C.; Birrell, C. Pharmacokinetics, Pharmacodynamics, and Safety of MEDI4212, an Anti-IgE Monoclonal Antibody, in Subjects with Atopy: A Phase I Study. Adv. Ther. 2016, 33, 225–251. [Google Scholar] [CrossRef] [PubMed]
- Trischler, J.; Bottoli, I.; Janocha, R.; Heusser, C.; Jaumont, X.; Lowe, P.; Gautier, A.; Pethe, A.; Woessner, R.; Zerwes, H.G.; et al. Ligelizumab treatment for severe asthma: Learnings from the clinical development programme. Clin. Transl. Immunol. 2021, 10, e1255. [Google Scholar] [CrossRef] [PubMed]
- Maurer, M.; Gimenez-Arnau, A.M.; Sussman, G.; Metz, M.; Baker, D.R.; Bauer, A.; Bernstein, J.A.; Brehler, R.; Chu, C.Y.; Chung, W.H.; et al. Ligelizumab for Chronic Spontaneous Urticaria. N. Engl. J. Med. 2019, 381, 1321–1332. [Google Scholar] [CrossRef] [Green Version]
- Gauvreau, G.M.; Arm, J.P.; Boulet, L.P.; Leigh, R.; Cockcroft, D.W.; Davis, B.E.; Mayers, I.; FitzGerald, J.M.; Dahlen, B.; Killian, K.J.; et al. Efficacy and safety of multiple doses of QGE031 (ligelizumab) versus omalizumab and placebo in inhibiting allergen-induced early asthmatic responses. J. Allergy Clin. Immunol. 2016, 138, 1051–1059. [Google Scholar] [CrossRef] [Green Version]
- Gasser, P.; Tarchevskaya, S.S.; Guntern, P.; Brigger, D.; Ruppli, R.; Zbaren, N.; Kleinboelting, S.; Heusser, C.; Jardetzky, T.S.; Eggel, A. The mechanistic and functional profile of the therapeutic anti-IgE antibody ligelizumab differs from omalizumab. Nat. Commun. 2020, 11, 165. [Google Scholar] [CrossRef] [Green Version]
- Jabs, F.; Plum, M.; Laursen, N.S.; Jensen, R.K.; Molgaard, B.; Miehe, M.; Mandolesi, M.; Rauber, M.M.; Pfutzner, W.; Jakob, T.; et al. Trapping IgE in a closed conformation by mimicking CD23 binding prevents and disrupts FcepsilonRI interaction. Nat. Commun. 2018, 9, 7. [Google Scholar] [CrossRef] [Green Version]
- Caputi, A.P.; Navarra, P. Beyond antibodies: Ankyrins and DARPins. From basic research to drug approval. Curr. Opin. Pharmacol. 2020, 51, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Baumann, M.J.; Eggel, A.; Amstutz, P.; Stadler, B.M.; Vogel, M. DARPins against a functional IgE epitope. Immunol. Lett. 2010, 133, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Eggel, A.; Tarchevskaya, S.S.; Vogel, M.; Prinz, H.; Jardetzky, T.S. Accelerated disassembly of IgE-receptor complexes by a disruptive macromolecular inhibitor. Nature 2012, 491, 613–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prinz, H.; Striessnig, J. Ligand-induced accelerated dissociation of (+)-cis-diltiazem from L-type Ca2+ channels is simply explained by competition for individual attachment points. J. Biol. Chem. 1993, 268, 18580–18585. [Google Scholar] [CrossRef]
- Erbas, A.; Marko, J.F. How do DNA-bound proteins leave their binding sites? The role of facilitated dissociation. Curr. Opin. Chem. Biol. 2019, 53, 118–124. [Google Scholar] [CrossRef]
- Bernasconi, C.F. Relaxation Kinetics; Academic Press, Inc.: New York, NY, USA, 1976. [Google Scholar]
- Hirano, T.; Koyanagi, A.; Kotoshiba, K.; Shinkai, Y.; Kasai, M.; Ando, T.; Kaitani, A.; Okumura, K.; Kitaura, J. The Fab fragment of anti-IgE Cepsilon2 domain prevents allergic reactions through interacting with IgE-FcepsilonRIalpha complex on rat mast cells. Sci. Rep. 2018, 8, 14237. [Google Scholar] [CrossRef] [Green Version]
- Harris, J.M.; Cabanski, C.R.; Scheerens, H.; Samineni, D.; Bradley, M.S.; Cochran, C.; Staubach, P.; Metz, M.; Sussman, G.; Maurer, M. A randomized trial of quilizumab in adults with refractory chronic spontaneous urticaria. J. Allergy Clin. Immunol. 2016, 138, 1730–1732. [Google Scholar] [CrossRef] [Green Version]
- Harris, J.M.; Maciuca, R.; Bradley, M.S.; Cabanski, C.R.; Scheerens, H.; Lim, J.; Cai, F.; Kishnani, M.; Liao, X.C.; Samineni, D.; et al. A randomized trial of the efficacy and safety of quilizumab in adults with inadequately controlled allergic asthma. Respir. Res. 2016, 17, 29. [Google Scholar] [CrossRef] [Green Version]
- Gauvreau, G.M.; Harris, J.M.; Boulet, L.P.; Scheerens, H.; Fitzgerald, J.M.; Putnam, W.S.; Cockcroft, D.W.; Davis, B.E.; Leigh, R.; Zheng, Y.; et al. Targeting membrane-expressed IgE B cell receptor with an antibody to the M1 prime epitope reduces IgE production. Sci. Transl. Med. 2014, 6, 243ra285. [Google Scholar] [CrossRef]
- Chu, S.Y.; Horton, H.M.; Pong, E.; Leung, I.W.; Chen, H.; Nguyen, D.H.; Bautista, C.; Muchhal, U.S.; Bernett, M.J.; Moore, G.L.; et al. Reduction of total IgE by targeted coengagement of IgE B-cell receptor and FcgammaRIIb with Fc-engineered antibody. J. Allergy Clin. Immunol. 2012, 129, 1102–1115. [Google Scholar] [CrossRef]
- Muta, T.; Kurosaki, T.; Misulovin, Z.; Sanchez, M.; Nussenzweig, M.C.; Ravetch, J.V. A 13-amino-acid motif in the cytoplasmic domain of Fc gamma RIIB modulates B-cell receptor signalling. Nature 1994, 368, 70–73. [Google Scholar] [CrossRef]
- Kirak, O.; Riethmuller, G. A novel, nonanaphylactogenic, bispecific IgE-CD3 antibody eliminates IgE(+) B cells. J. Allergy Clin. Immunol. 2015, 136, 800–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bargou, R.; Leo, E.; Zugmaier, G.; Klinger, M.; Goebeler, M.; Knop, S.; Noppeney, R.; Viardot, A.; Hess, G.; Schuler, M.; et al. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science 2008, 321, 974–977. [Google Scholar] [CrossRef] [PubMed]
- Foa, R.; Bassan, R.; Vitale, A.; Elia, L.; Piciocchi, A.; Puzzolo, M.C.; Canichella, M.; Viero, P.; Ferrara, F.; Lunghi, M.; et al. Dasatinib-Blinatumomab for Ph-Positive Acute Lymphoblastic Leukemia in Adults. N. Engl. J. Med. 2020, 383, 1613–1623. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.; Stein, A.; Gokbuget, N.; Fielding, A.K.; Schuh, A.C.; Ribera, J.M.; Wei, A.; Dombret, H.; Foa, R.; Bassan, R.; et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2017, 376, 836–847. [Google Scholar] [CrossRef]
- Rudolf, M.P.; Zuercher, A.W.; Nechansky, A.; Ruf, C.; Vogel, M.; Miescher, S.M.; Stadler, B.M.; Kricek, F. Molecular basis for nonanaphylactogenicity of a monoclonal anti-IgE antibody. J. Immunol. 2000, 165, 813–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keefe, A.D.; Pai, S.; Ellington, A. Aptamers as therapeutics. Nat. Rev. Drug Discov. 2010, 9, 537–550. [Google Scholar] [CrossRef] [PubMed]
- Wiegand, T.W.; Williams, P.B.; Dreskin, S.C.; Jouvin, M.H.; Kinet, J.P.; Tasset, D. High-affinity oligonucleotide ligands to human IgE inhibit binding to Fc epsilon receptor I. J. Immunol. 1996, 157, 221–230. [Google Scholar]
- Mendonsa, S.D.; Bowser, M.T. In vitro selection of high-affinity DNA ligands for human IgE using capillary electrophoresis. Anal. Chem. 2004, 76, 5387–5392. [Google Scholar] [CrossRef] [PubMed]
- Poongavanam, M.V.; Kisley, L.; Kourentzi, K.; Landes, C.F.; Willson, R.C. Ensemble and single-molecule biophysical characterization of D17.4 DNA aptamer-IgE interactions. Biochim. Biophys. Acta 2016, 1864, 154–164. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Hitchen, P.G.; Panico, M.; North, S.J.; Barbouche, M.R.; Binet, D.; Morris, H.R.; Dell, A.; Haslam, S.M. Glycoproteomic studies of IgE from a novel hyper IgE syndrome linked to PGM3 mutation. Glycoconj. J. 2016, 33, 447–456. [Google Scholar] [CrossRef] [Green Version]
- Shade, K.T.; Platzer, B.; Washburn, N.; Mani, V.; Bartsch, Y.C.; Conroy, M.; Pagan, J.D.; Bosques, C.; Mempel, T.R.; Fiebiger, E.; et al. A single glycan on IgE is indispensable for initiation of anaphylaxis. J. Exp. Med. 2015, 212, 457–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayers, I.; Cain, S.A.; Swan, J.R.; Pickett, M.A.; Watt, P.J.; Holgate, S.T.; Padlan, E.A.; Schuck, P.; Helm, B.A. Amino acid residues that influence Fc epsilon RI-mediated effector functions of human immunoglobulin E. Biochemistry 1998, 37, 16152–16164. [Google Scholar] [CrossRef] [PubMed]
- Sondermann, P.; Huber, R.; Oosthuizen, V.; Jacob, U. The 3.2-A crystal structure of the human IgG1 Fc fragment-Fc gammaRIII complex. Nature 2000, 406, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Chu, J.; Zou, Z.; Hamacher, N.B.; Rixon, M.W.; Sun, P.D. Structure of FcgammaRI in complex with Fc reveals the importance of glycan recognition for high-affinity IgG binding. Proc. Natl Acad Sci. USA 2015, 112, 833–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shade, K.T.; Conroy, M.E.; Anthony, R.M. IgE Glycosylation in Health and Disease. Curr. Top. Microbiol. Immunol. 2019, 423, 77–93. [Google Scholar] [CrossRef] [PubMed]
- Bjorklund, J.E.; Karlsson, T.; Magnusson, C.G. N-glycosylation influences epitope expression and receptor binding structures in human IgE. Mol. Immunol. 1999, 36, 213–221. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Barb, A.W. A synopsis of recent developments defining how N-glycosylation impacts immunoglobulin G structure and function. Glycobiology 2020, 30, 214–225. [Google Scholar] [CrossRef]
- Shade, K.C.; Conroy, M.E.; Washburn, N.; Kitaoka, M.; Huynh, D.J.; Laprise, E.; Patil, S.U.; Shreffler, W.G.; Anthony, R.M. Sialylation of immunoglobulin E is a determinant of allergic pathogenicity. Nature 2020, 582, 265–270. [Google Scholar] [CrossRef]
- Nabi, I.R.; Shankar, J.; Dennis, J.W. The galectin lattice at a glance. J. Cell Sci. 2015, 128, 2213–2219. [Google Scholar] [CrossRef] [Green Version]
- Gao, P.; Simpson, J.L.; Zhang, J.; Gibson, P.G. Galectin-3: Its role in asthma and potential as an anti-inflammatory target. Respir. Res. 2013, 14, 136. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Liu, L.; Lin, X.; Wang, Y.; Li, Y.; Guo, Q.; Li, S.; Sun, Y.; Tao, X.; Zhang, D.; et al. A Translocation Pathway for Vesicle-Mediated Unconventional Protein Secretion. Cell 2020, 181, 637–652. [Google Scholar] [CrossRef]
- Liu, F.T.; Albrandt, K.; Mendel, E.; Kulczycki, A., Jr.; Orida, N.K. Identification of an IgE-binding protein by molecular cloning. Proc. Natl Acad Sci. USA 1985, 82, 4100–4104. [Google Scholar] [CrossRef] [Green Version]
- Ho, M.K.; Springer, T.A. Mac-2, a novel 32,000 Mr mouse macrophage subpopulation-specific antigen defined by monoclonal antibodies. J. Immunol. 1982, 128, 1221–1228. [Google Scholar]
- Schroeder, J.T.; Adeosun, A.A.; Do, D.; Bieneman, A.P. Galectin-3 is essential for IgE-dependent activation of human basophils by A549 lung epithelial cells. J. Allergy Clin. Immunol. 2019, 144, 312–315. [Google Scholar] [CrossRef] [Green Version]
- Nieminen, J.; Kuno, A.; Hirabayashi, J.; Sato, S. Visualization of galectin-3 oligomerization on the surface of neutrophils and endothelial cells using fluorescence resonance energy transfer. J. Biol. Chem. 2007, 282, 1374–1383. [Google Scholar] [CrossRef] [Green Version]
- Tsai, H.F.; Wu, C.S.; Chen, Y.L.; Liao, H.J.; Chyuan, I.T.; Hsu, P.N. Galectin-3 suppresses mucosal inflammation and reduces disease severity in experimental colitis. J. Mol. Med. 2016, 94, 545–556. [Google Scholar] [CrossRef] [PubMed]
- del Pozo, V.; Rojo, M.; Rubio, M.L.; Cortegano, I.; Cardaba, B.; Gallardo, S.; Ortega, M.; Civantos, E.; Lopez, E.; Martin-Mosquero, C.; et al. Gene therapy with galectin-3 inhibits bronchial obstruction and inflammation in antigen-challenged rats through interleukin-5 gene downregulation. Am. J. Respir. Crit Care Med. 2002, 166, 732–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuberi, R.I.; Hsu, D.K.; Kalayci, O.; Chen, H.Y.; Sheldon, H.K.; Yu, L.; Apgar, J.R.; Kawakami, T.; Lilly, C.M.; Liu, F.T. Critical role for galectin-3 in airway inflammation and bronchial hyperresponsiveness in a murine model of asthma. Am. J. Pathol. 2004, 165, 2045–2053. [Google Scholar] [CrossRef] [Green Version]
- Peters, K.; Peters, M. The Role of Lectin Receptors and Their Ligands in Controlling Allergic Inflammation. Front. Immunol. 2021, 12, 635411. [Google Scholar] [CrossRef] [PubMed]
- Niki, T.; Tsutsui, S.; Hirose, S.; Aradono, S.; Sugimoto, Y.; Takeshita, K.; Nishi, N.; Hirashima, M. Galectin-9 is a high affinity IgE-binding lectin with anti-allergic effect by blocking IgE-antigen complex formation. J. Biol. Chem. 2009, 284, 32344–32352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engeroff, P.; Caviezel, F.; Mueller, D.; Thoms, F.; Bachmann, M.F.; Vogel, M. CD23 provides a noninflammatory pathway for IgE-allergen complexes. J. Allergy Clin. Immunol. 2020, 145, 301–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
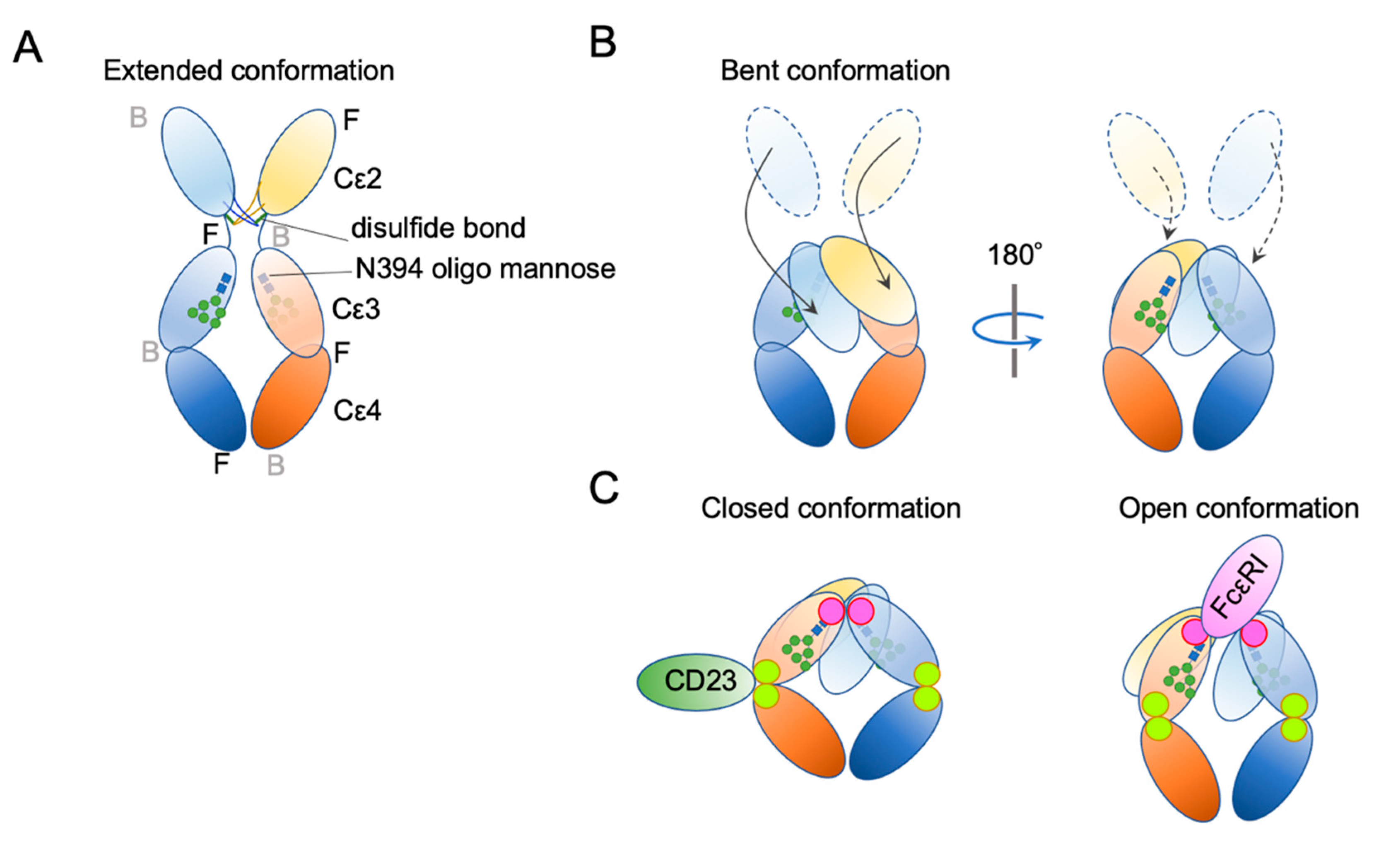
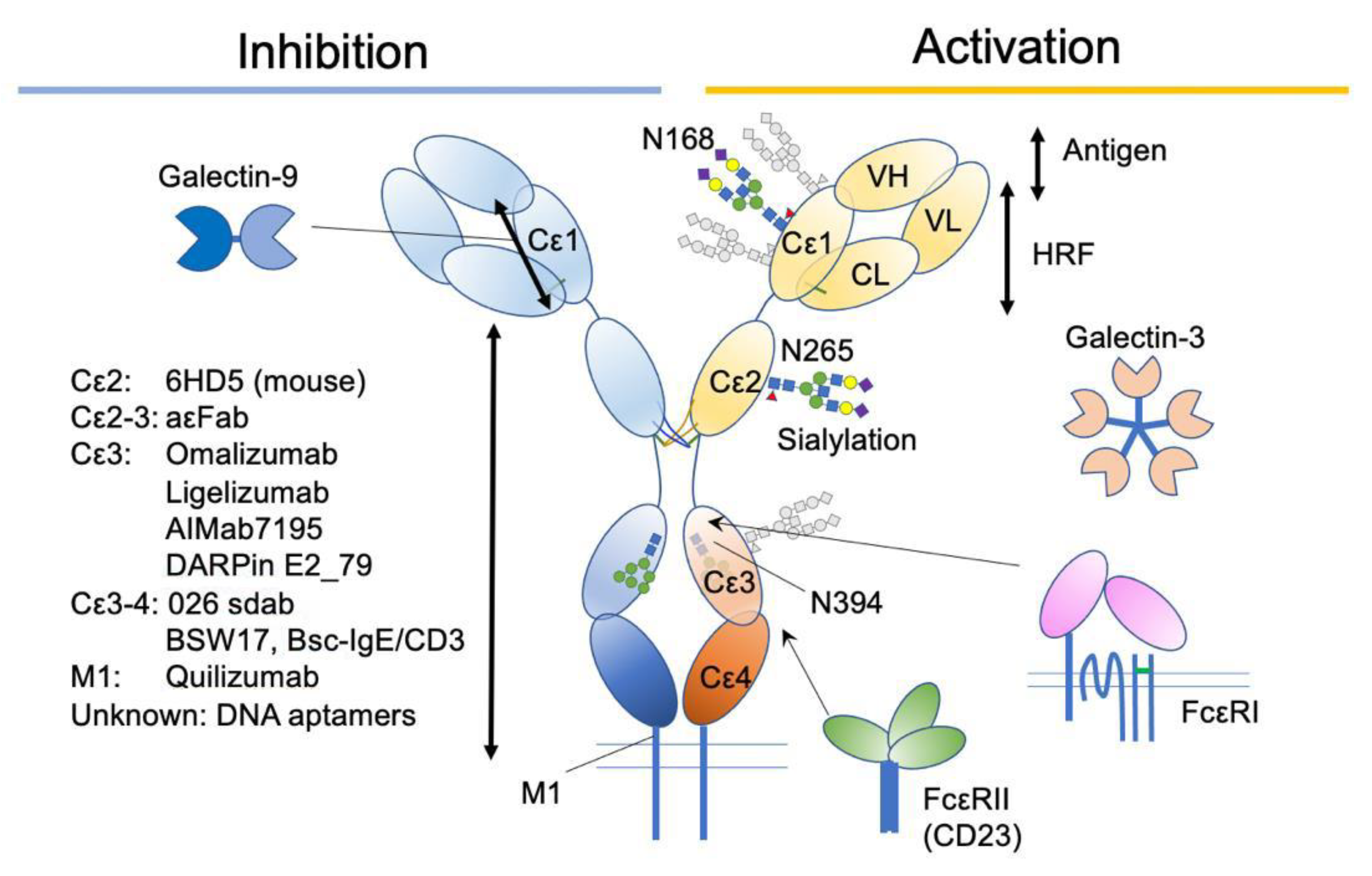
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ando, T.; Kitaura, J. Tuning IgE: IgE-Associating Molecules and Their Effects on IgE-Dependent Mast Cell Reactions. Cells 2021, 10, 1697. https://doi.org/10.3390/cells10071697
Ando T, Kitaura J. Tuning IgE: IgE-Associating Molecules and Their Effects on IgE-Dependent Mast Cell Reactions. Cells. 2021; 10(7):1697. https://doi.org/10.3390/cells10071697
Chicago/Turabian StyleAndo, Tomoaki, and Jiro Kitaura. 2021. "Tuning IgE: IgE-Associating Molecules and Their Effects on IgE-Dependent Mast Cell Reactions" Cells 10, no. 7: 1697. https://doi.org/10.3390/cells10071697
APA StyleAndo, T., & Kitaura, J. (2021). Tuning IgE: IgE-Associating Molecules and Their Effects on IgE-Dependent Mast Cell Reactions. Cells, 10(7), 1697. https://doi.org/10.3390/cells10071697