
Neutrophil Extracellular Traps Affecting Cardiovascular Health in Infectious and Inflammatory Diseases

 ,
,
Abstract
1. Introduction
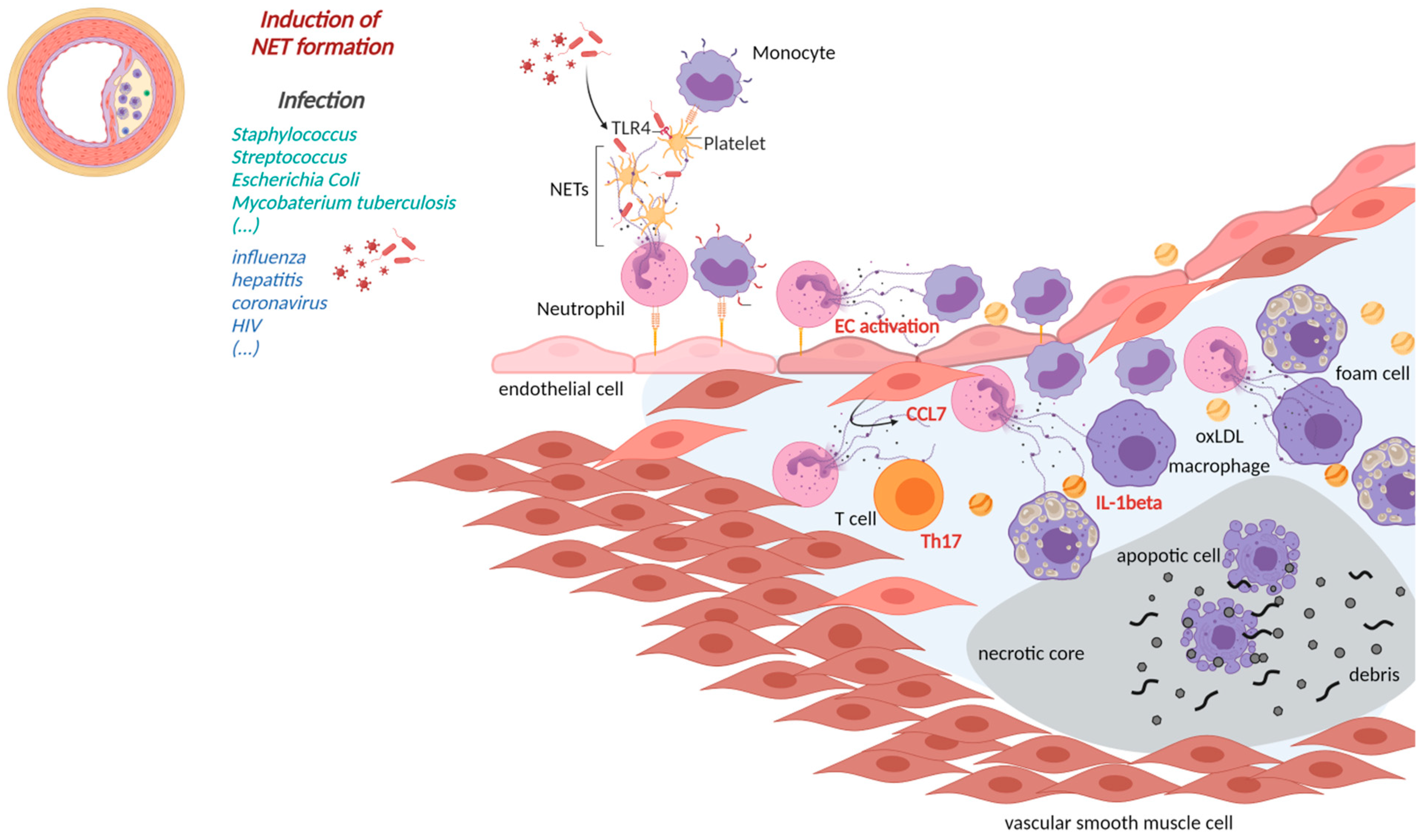
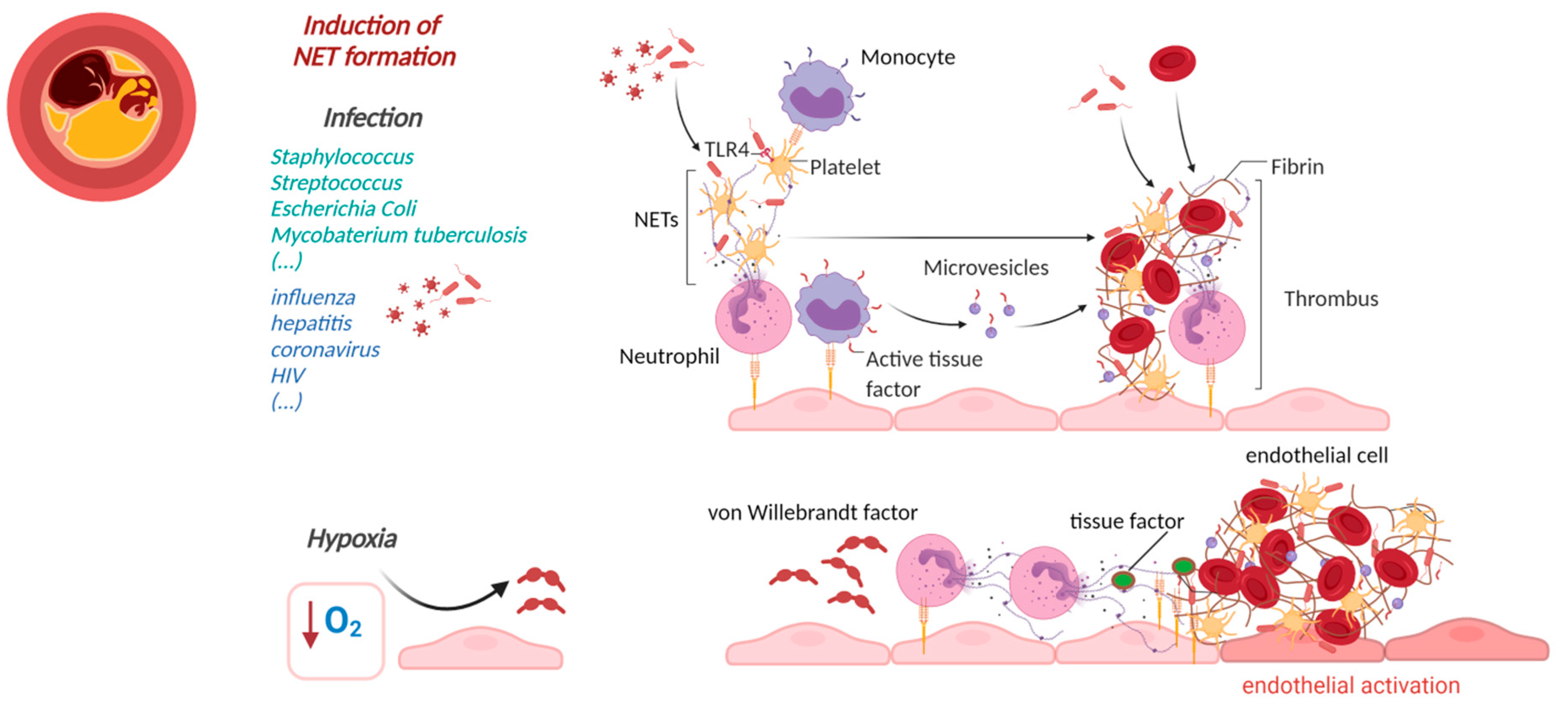
2. NETs Affecting Cardiovascular Health in Infectious Diseases
2.1. Cardiovascular Manifestations of Bacterial NETs Induction
2.2. Cardiovascular Manifestations of Viral NET Induction
3. Implications of NETs Formation in CVD
3.1. NETs in Atherosclerosis

3.2. NETs-Induced Thrombosis

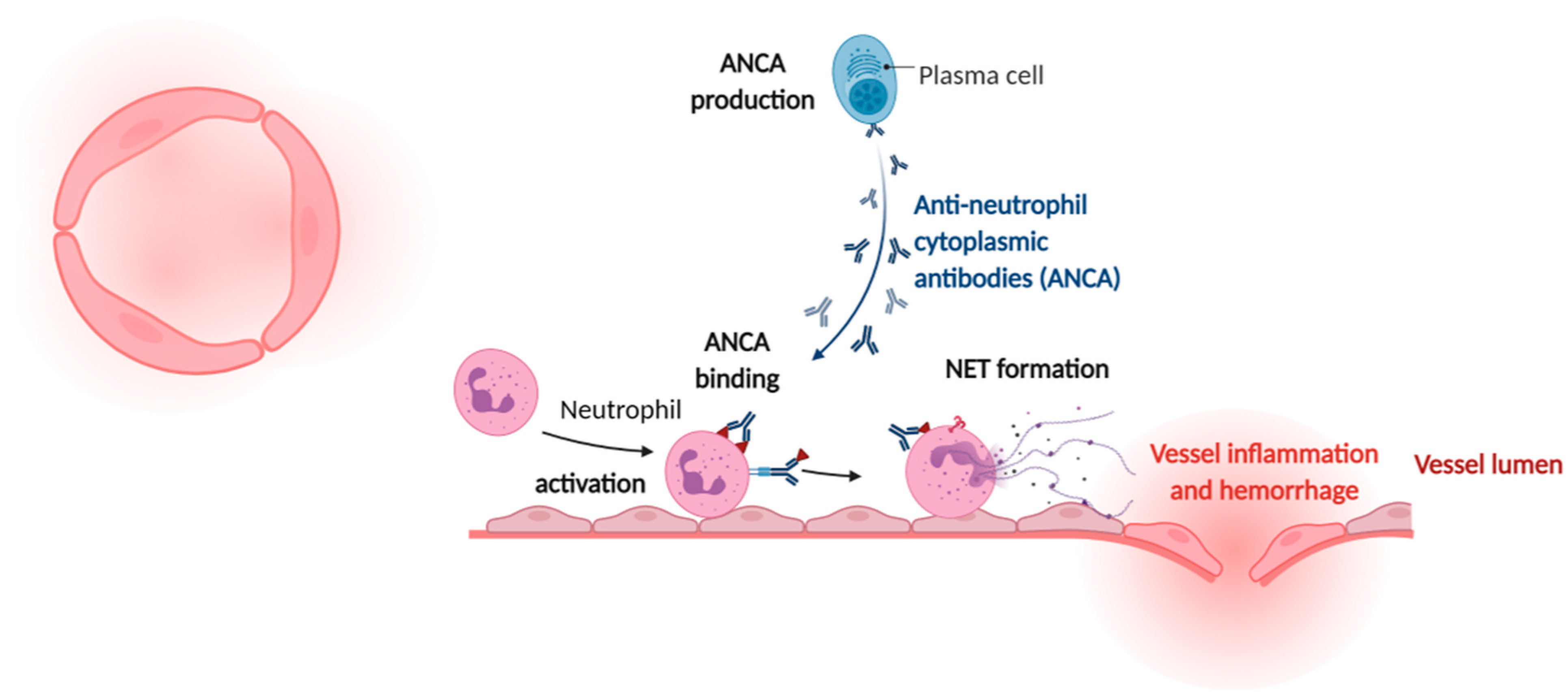
4. Cardiovascular Manifestations of Sterile NETs Induction
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Klopf, J.; Brostjan, C.; Eilenberg, W.; Neumayer, C. Neutrophil Extracellular Traps and Their Implications in Cardiovascular and Inflammatory Disease. Int. J. Mol. Sci. 2021, 22, 559. [Google Scholar] [CrossRef] [PubMed]
- Leppkes, M.; Schick, M.; Hohberger, B.; Mahajan, A.; Knopf, J.; Schett, G.; Munoz, L.E.; Herrmann, M. Updates on NET formation in health and disease. Semin. Arthritis Rheum. 2019, 49, S43–S48. [Google Scholar] [CrossRef] [PubMed]
- Doring, Y.; Libby, P.; Soehnlein, O. Neutrophil Extracellular Traps Participate in Cardiovascular Diseases: Recent Experimental and Clinical Insights. Circ. Res. 2020, 126, 1228–1241. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Czaikoski, P.G.; Mota, J.M.; Nascimento, D.C.; Sonego, F.; Castanheira, F.V.; Melo, P.H.; Scortegagna, G.T.; Silva, R.L.; Barroso-Sousa, R.; Souto, F.O.; et al. Neutrophil Extracellular Traps Induce Organ Damage during Experimental and Clinical Sepsis. PLoS ONE 2016, 11, e0148142. [Google Scholar] [CrossRef]
- Tanaka, K.; Koike, Y.; Shimura, T.; Okigami, M.; Ide, S.; Toiyama, Y.; Okugawa, Y.; Inoue, Y.; Araki, T.; Uchida, K.; et al. In vivo characterization of neutrophil extracellular traps in various organs of a murine sepsis model. PLoS ONE 2014, 9, e111888. [Google Scholar] [CrossRef]
- Kolaczkowska, E.; Jenne, C.N.; Surewaard, B.G.; Thanabalasuriar, A.; Lee, W.Y.; Sanz, M.J.; Mowen, K.; Opdenakker, G.; Kubes, P. Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature. Nat. Commun. 2015, 6, 6673. [Google Scholar] [CrossRef]
- Meng, W.; Paunel-Gorgulu, A.; Flohe, S.; Hoffmann, A.; Witte, I.; MacKenzie, C.; Baldus, S.E.; Windolf, J.; Logters, T.T. Depletion of neutrophil extracellular traps in vivo results in hypersusceptibility to polymicrobial sepsis in mice. Crit. Care 2012, 16, R137. [Google Scholar] [CrossRef]
- Dalager-Pedersen, M.; Sogaard, M.; Schonheyder, H.C.; Nielsen, H.; Thomsen, R.W. Risk for myocardial infarction and stroke after community-acquired bacteremia: A 20-year population-based cohort study. Circulation 2014, 129, 1387–1396. [Google Scholar] [CrossRef]
- Corrales-Medina, V.F.; Alvarez, K.N.; Weissfeld, L.A.; Angus, D.C.; Chirinos, J.A.; Chang, C.C.; Newman, A.; Loehr, L.; Folsom, A.R.; Elkind, M.S.; et al. Association between hospitalization for pneumonia and subsequent risk of cardiovascular disease. JAMA 2015, 313, 264–274. [Google Scholar] [CrossRef]
- Mori, Y.; Yamaguchi, M.; Terao, Y.; Hamada, S.; Ooshima, T.; Kawabata, S. alpha-Enolase of Streptococcus pneumoniae induces formation of neutrophil extracellular traps. J. Biol. Chem. 2012, 287, 10472–10481. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Francis, R.J.; Butler, R.E.; Stewart, G.R. Mycobacterium tuberculosis ESAT-6 is a leukocidin causing Ca2+ influx, necrosis and neutrophil extracellular trap formation. Cell Death Dis. 2014, 5, e1474. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.C.; Hsu, R.B.; Ohniwa, R.L.; Chen, J.W.; Yuan, C.T.; Chia, J.S.; Jung, C.J. Neutrophil Extracellular Traps Enhance Staphylococcus Aureus Vegetation Formation through Interaction with Platelets in Infective Endocarditis. Thromb. Haemost. 2019, 119, 786–796. [Google Scholar] [CrossRef]
- Huaman, M.A.; Ticona, E.; Miranda, G.; Kryscio, R.J.; Mugruza, R.; Aranda, E.; Rondan, P.L.; Henson, D.; Ticona, C.; Sterling, T.R.; et al. The Relationship Between Latent Tuberculosis Infection and Acute Myocardial Infarction. Clin. Infect. Dis. 2018, 66, 886–892. [Google Scholar] [CrossRef] [PubMed]
- Schumski, A.; Ortega-Gomez, A.; Wichapong, K.; Winter, C.; Lemnitzer, P.; Viola, J.R.; Pinilla-Vera, M.; Folco, E.; Solis-Mezarino, V.; Volker-Albert, M.; et al. Endotoxinemia Accelerates Atherosclerosis Through Electrostatic Charge-Mediated Monocyte Adhesion. Circulation 2021, 143, 254–266. [Google Scholar] [CrossRef]
- McDonald, B.; Davis, R.P.; Kim, S.J.; Tse, M.; Esmon, C.T.; Kolaczkowska, E.; Jenne, C.N. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood 2017, 129, 1357–1367. [Google Scholar] [CrossRef]
- Sass, L.A.; Ziemba, K.J.; Heiser, E.A.; Mauriello, C.T.; Werner, A.L.; Aguiar, M.A.; Nyalwidhe, J.O.; Cunnion, K.M. A 1-Year-Old with Mycobacterium tuberculosis Endocarditis with Mass Spectrometry Analysis of Cardiac Vegetation Composition. J. Pediatr. Infect. Dis. Soc. 2016, 5, 85–88. [Google Scholar] [CrossRef]
- Arcanjo, A.; Logullo, J.; Menezes, C.C.B.; de Souza Carvalho Giangiarulo, T.C.; Dos Reis, M.C.; de Castro, G.M.M.; da Silva Fontes, Y.; Todeschini, A.R.; Freire-de-Lima, L.; Decote-Ricardo, D.; et al. The emerging role of neutrophil extracellular traps in severe acute respiratory syndrome coronavirus 2 (COVID-19). Sci. Rep. 2020, 10, 19630. [Google Scholar] [CrossRef] [PubMed]
- Chicca, I.J.; Milward, M.R.; Chapple, I.L.C.; Griffiths, G.; Benson, R.; Dietrich, T.; Cooper, P.R. Development and Application of High-Content Biological Screening for Modulators of NET Production. Front. Immunol. 2018, 9, 337. [Google Scholar] [CrossRef]
- Barnes, B.J.; Adrover, J.M.; Baxter-Stoltzfus, A.; Borczuk, A.; Cools-Lartigue, J.; Crawford, J.M.; Dassler-Plenker, J.; Guerci, P.; Huynh, C.; Knight, J.S.; et al. Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [PubMed]
- Nicolai, L.; Leunig, A.; Brambs, S.; Kaiser, R.; Weinberger, T.; Weigand, M.; Muenchhoff, M.; Hellmuth, J.C.; Ledderose, S.; Schulz, H.; et al. Immunothrombotic Dysregulation in COVID-19 Pneumonia Is Associated With Respiratory Failure and Coagulopathy. Circulation 2020, 142, 1176–1189. [Google Scholar] [CrossRef] [PubMed]
- Warnatsch, A.; Ioannou, M.; Wang, Q.; Papayannopoulos, V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 2015, 349, 316–320. [Google Scholar] [CrossRef] [PubMed]
- An, Z.; Li, J.; Yu, J.; Wang, X.; Gao, H.; Zhang, W.; Wei, Z.; Zhang, J.; Zhang, Y.; Zhao, J.; et al. Neutrophil extracellular traps induced by IL-8 aggravate atherosclerosis via activation NF-kappaB signaling in macrophages. Cell Cycle 2019, 18, 2928–2938. [Google Scholar] [CrossRef] [PubMed]
- Obama, T.; Ohinata, H.; Takaki, T.; Iwamoto, S.; Sawada, N.; Aiuchi, T.; Kato, R.; Itabe, H. Cooperative Action of Oxidized Low-Density Lipoproteins and Neutrophils on Endothelial Inflammatory Responses Through Neutrophil Extracellular Trap Formation. Front. Immunol. 2019, 10, 1899. [Google Scholar] [CrossRef] [PubMed]
- da Silva, R.F.; Baptista, D.; Roth, A.; Miteva, K.; Burger, F.; Vuilleumier, N.; Carbone, F.; Montecucco, F.; Mach, F.; Brandt, K.J. Anti-Apolipoprotein A-1 IgG Influences Neutrophil Extracellular Trap Content at Distinct Regions of Human Carotid Plaques. Int. J. Mol. Sci. 2020, 21, 7721. [Google Scholar] [CrossRef]
- Gao, H.; Wang, X.; Lin, C.; An, Z.; Yu, J.; Cao, H.; Fan, Y.; Liang, X. Exosomal MALAT1 derived from ox-LDL-treated endothelial cells induce neutrophil extracellular traps to aggravate atherosclerosis. Biol. Chem. 2020, 401, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Quillard, T.; Araujo, H.A.; Franck, G.; Shvartz, E.; Sukhova, G.; Libby, P. TLR2 and neutrophils potentiate endothelial stress, apoptosis and detachment: Implications for superficial erosion. Eur. Heart J. 2015, 36, 1394–1404. [Google Scholar] [CrossRef]
- Silvestre-Roig, C.; Braster, Q.; Wichapong, K.; Lee, E.Y.; Teulon, J.M.; Berrebeh, N.; Winter, J.; Adrover, J.M.; Santos, G.S.; Froese, A.; et al. Externalized histone H4 orchestrates chronic inflammation by inducing lytic cell death. Nature 2019, 569, 236–240. [Google Scholar] [CrossRef]
- Westerterp, M.; Fotakis, P.; Ouimet, M.; Bochem, A.E.; Zhang, H.; Molusky, M.M.; Wang, W.; Abramowicz, S.; la Bastide-van Gemert, S.; Wang, N.; et al. Cholesterol Efflux Pathways Suppress Inflammasome Activation, NETosis, and Atherogenesis. Circulation 2018, 138, 898–912. [Google Scholar] [CrossRef]
- Josefs, T.; Barrett, T.J.; Brown, E.J.; Quezada, A.; Wu, X.; Voisin, M.; Amengual, J.; Fisher, E.A. Neutrophil extracellular traps promote macrophage inflammation and impair atherosclerosis resolution in diabetic mice. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Knight, J.S.; Luo, W.; O’Dell, A.A.; Yalavarthi, S.; Zhao, W.; Subramanian, V.; Guo, C.; Grenn, R.C.; Thompson, P.R.; Eitzman, D.T.; et al. Peptidylarginine deiminase inhibition reduces vascular damage and modulates innate immune responses in murine models of atherosclerosis. Circ. Res. 2014, 114, 947–956. [Google Scholar] [CrossRef]
- Kim, S.W.; Lee, J.K. Role of HMGB1 in the Interplay between NETosis and Thrombosis in Ischemic Stroke: A Review. Cells 2020, 9, 1794. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Tan, J.; Diamond, S.L. Hemodynamic force triggers rapid NETosis within sterile thrombotic occlusions. J. Thromb. Haemost. 2018, 16, 316–329. [Google Scholar] [CrossRef] [PubMed]
- Gaertner, F.; Massberg, S. Blood coagulation in immunothrombosis-At the frontline of intravascular immunity. Semin. Immunol. 2016, 28, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, B.; Massberg, S. Thrombosis as an intravascular effector of innate immunity. Nat. Rev. Immunol. 2013, 13, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Mangold, A.; Alias, S.; Scherz, T.; Hofbauer, M.; Jakowitsch, J.; Panzenbock, A.; Simon, D.; Laimer, D.; Bangert, C.; Kammerlander, A.; et al. Coronary neutrophil extracellular trap burden and deoxyribonuclease activity in ST-elevation acute coronary syndrome are predictors of ST-segment resolution and infarct size. Circ. Res. 2015, 116, 1182–1192. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, E.; Yalavarthi, S.; Berthier, C.C.; Hodgin, J.B.; Khandpur, R.; Lin, A.M.; Rubin, C.J.; Zhao, W.; Olsen, S.H.; Klinker, M.; et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J. Immunol. 2011, 187, 538–552. [Google Scholar] [CrossRef]
- Lande, R.; Ganguly, D.; Facchinetti, V.; Frasca, L.; Conrad, C.; Gregorio, J.; Meller, S.; Chamilos, G.; Sebasigari, R.; Riccieri, V.; et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci. Transl. Med. 2011, 3, 73ra19. [Google Scholar] [CrossRef]
- Carmona-Rivera, C.; Zhao, W.; Yalavarthi, S.; Kaplan, M.J. Neutrophil extracellular traps induce endothelial dysfunction in systemic lupus erythematosus through the activation of matrix metalloproteinase-2. Ann. Rheum. Dis. 2015, 74, 1417–1424. [Google Scholar] [CrossRef]
- Davignon, J.; Ganz, P. Role of endothelial dysfunction in atherosclerosis. Circulation 2004, 109, III27–III32. [Google Scholar] [CrossRef]
- Smith, C.K.; Vivekanandan-Giri, A.; Tang, C.; Knight, J.S.; Mathew, A.; Padilla, R.L.; Gillespie, B.W.; Carmona-Rivera, C.; Liu, X.; Subramanian, V.; et al. Neutrophil extracellular trap-derived enzymes oxidize high-density lipoprotein: An additional proatherogenic mechanism in systemic lupus erythematosus. Arthritis Rheumatol. 2014, 66, 2532–2544. [Google Scholar] [CrossRef]
- Pilsczek, F.H.; Salina, D.; Poon, K.K.; Fahey, C.; Yipp, B.G.; Sibley, C.D.; Robbins, S.M.; Green, F.H.; Surette, M.G.; Sugai, M.; et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J. Immunol. 2010, 185, 7413–7425. [Google Scholar] [CrossRef]
- Thammavongsa, V.; Missiakas, D.M.; Schneewind, O. Staphylococcus aureus degrades neutrophil extracellular traps to promote immune cell death. Science 2013, 342, 863–866. [Google Scholar] [CrossRef]
- Brown, A.O.; Mann, B.; Gao, G.; Hankins, J.S.; Humann, J.; Giardina, J.; Faverio, P.; Restrepo, M.I.; Halade, G.V.; Mortensen, E.M.; et al. Streptococcus pneumoniae translocates into the myocardium and forms unique microlesions that disrupt cardiac function. PLoS Pathog. 2014, 10, e1004383. [Google Scholar] [CrossRef] [PubMed]
- Croxen, M.A.; Finlay, B.B. Molecular mechanisms of Escherichia coli pathogenicity. Nat. Rev. Microbiol. 2010, 8, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Kambas, K.; Mitroulis, I.; Apostolidou, E.; Girod, A.; Chrysanthopoulou, A.; Pneumatikos, I.; Skendros, P.; Kourtzelis, I.; Koffa, M.; Kotsianidis, I.; et al. Autophagy mediates the delivery of thrombogenic tissue factor to neutrophil extracellular traps in human sepsis. PLoS ONE 2012, 7, e45427. [Google Scholar] [CrossRef] [PubMed]
- Noubouossie, D.F.; Whelihan, M.F.; Yu, Y.B.; Sparkenbaugh, E.; Pawlinski, R.; Monroe, D.M.; Key, N.S. In vitro activation of coagulation by human neutrophil DNA and histone proteins but not neutrophil extracellular traps. Blood 2017, 129, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Kichik, V.; Mondragon-Flores, R.; Mondragon-Castelan, M.; Gonzalez-Pozos, S.; Muniz-Hernandez, S.; Rojas-Espinosa, O.; Chacon-Salinas, R.; Estrada-Parra, S.; Estrada-Garcia, I. Neutrophil extracellular traps are induced by Mycobacterium tuberculosis. Tuberculosis 2009, 89, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Sultan, F.A.; Fatimi, S.; Jamil, B.; Moustafa, S.E.; Mookadam, F. Tuberculous endocarditis: Valvular and right atrial involvement. Eur. J. Echocardiogr. 2010, 11, E13. [Google Scholar] [CrossRef]
- Sogabe, O.; Ohya, T. A case of tuberculous endocarditis with acute aortic valve insufficiency and annular subvalvular left ventricular aneurysm. Gen. Thorac. Cardiovasc. Surg. 2007, 55, 61–64. [Google Scholar] [CrossRef]
- Hu, S.; Liu, X.; Gao, Y.; Zhou, R.; Wei, M.; Dong, J.; Yan, H.; Zhao, Y. Hepatitis B Virus Inhibits Neutrophil Extracellular Trap Release by Modulating Reactive Oxygen Species Production and Autophagy. J. Immunol. 2019, 202, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Jaresko, G.S. Etiology of neutropenia in HIV-infected patients. Am. J. Health Syst. Pharm. 1999, 56 (Suppl. 5), S5–S8. [Google Scholar] [CrossRef]
- Cloke, T.; Munder, M.; Bergin, P.; Herath, S.; Modolell, M.; Taylor, G.; Muller, I.; Kropf, P. Phenotypic alteration of neutrophils in the blood of HIV seropositive patients. PLoS ONE 2013, 8, e72034. [Google Scholar] [CrossRef] [PubMed]
- Vecchiarelli, A.; Monari, C.; Palazzetti, B.; Bistoni, F.; Casadevall, A. Dysregulation in IL-12 secretion by neutrophils from HIV-infected patients. Clin. Exp. Immunol. 2000, 121, 311–319. [Google Scholar] [CrossRef]
- Saitoh, T.; Komano, J.; Saitoh, Y.; Misawa, T.; Takahama, M.; Kozaki, T.; Uehata, T.; Iwasaki, H.; Omori, H.; Yamaoka, S.; et al. Neutrophil extracellular traps mediate a host defense response to human immunodeficiency virus-1. Cell Host Microbe 2012, 12, 109–116. [Google Scholar] [CrossRef]
- Hsu, D.H.; de Waal Malefyt, R.; Fiorentino, D.F.; Dang, M.N.; Vieira, P.; de Vries, J.; Spits, H.; Mosmann, T.R.; Moore, K.W. Expression of interleukin-10 activity by Epstein-Barr virus protein BCRF1. Science 1990, 250, 830–832. [Google Scholar] [CrossRef] [PubMed]
- Kotenko, S.V.; Saccani, S.; Izotova, L.S.; Mirochnitchenko, O.V.; Pestka, S. Human cytomegalovirus harbors its own unique IL-10 homolog (cmvIL-10). Proc. Natl. Acad. Sci. USA 2000, 97, 1695–1700. [Google Scholar] [CrossRef]
- Raftery, M.J.; Wieland, D.; Gronewald, S.; Kraus, A.A.; Giese, T.; Schonrich, G. Shaping phenotype, function, and survival of dendritic cells by cytomegalovirus-encoded IL-10. J. Immunol. 2004, 173, 3383–3391. [Google Scholar] [CrossRef]
- Field, T.S.; Zhu, H.; Tarrant, M.; Mitchell, J.R.; Hill, M.D. Relationship between supra-annual trends in influenza rates and stroke occurrence. Neuroepidemiology 2004, 23, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Soltero, I.; Liu, K.; Cooper, R.; Stamler, J.; Garside, D. Trends in mortality from cerebrovascular diseases in the United States, 1960 to 1975. Stroke 1978, 9, 549–558. [Google Scholar] [CrossRef]
- Spodick, D.H.; Flessas, A.P.; Johnson, M.M. Association of acute respiratory symptoms with onset of acute myocardial infarction: Prospective investigation of 150 consecutive patients and matched control patients. Am. J. Cardiol. 1984, 53, 481–482. [Google Scholar] [CrossRef]
- Tang, B.M.; Shojaei, M.; Teoh, S.; Meyers, A.; Ho, J.; Ball, T.B.; Keynan, Y.; Pisipati, A.; Kumar, A.; Eisen, D.P.; et al. Neutrophils-related host factors associated with severe disease and fatality in patients with influenza infection. Nat. Commun. 2019, 10, 3422. [Google Scholar] [CrossRef]
- Lo Sasso, G.; Schlage, W.K.; Boue, S.; Veljkovic, E.; Peitsch, M.C.; Hoeng, J. The Apoe(−/−) mouse model: A suitable model to study cardiovascular and respiratory diseases in the context of cigarette smoke exposure and harm reduction. J. Transl. Med. 2016, 14, 146. [Google Scholar] [CrossRef]
- Naghavi, M.; Wyde, P.; Litovsky, S.; Madjid, M.; Akhtar, A.; Naguib, S.; Siadaty, M.S.; Sanati, S.; Casscells, W. Influenza infection exerts prominent inflammatory and thrombotic effects on the atherosclerotic plaques of apolipoprotein E-deficient mice. Circulation 2003, 107, 762–768. [Google Scholar] [CrossRef] [PubMed]
- Madjid, M.; Awan, I.; Ali, M.; Frazier, L.; Casscells, W. Influenza and atherosclerosis: Vaccination for cardiovascular disease prevention. Expert Opin. Biol. Ther. 2005, 5, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Colden-Stanfield, M.; Ratcliffe, D.; Cramer, E.B.; Gallin, E.K. Characterization of influenza virus-induced leukocyte adherence to human umbilical vein endothelial cell monolayers. J. Immunol. 1993, 151, 310–321. [Google Scholar] [PubMed]
- Haidari, M.; Wyde, P.R.; Litovsky, S.; Vela, D.; Ali, M.; Casscells, S.W.; Madjid, M. Influenza virus directly infects, inflames, and resides in the arteries of atherosclerotic and normal mice. Atherosclerosis 2010, 208, 90–96. [Google Scholar] [CrossRef]
- Ishiguro, N.; Takada, A.; Yoshioka, M.; Ma, X.; Kikuta, H.; Kida, H.; Kobayashi, K. Induction of interferon-inducible protein-10 and monokine induced by interferon-gamma from human endothelial cells infected with Influenza A virus. Arch. Virol. 2004, 149, 17–34. [Google Scholar] [CrossRef]
- Wang, S.; Le, T.Q.; Kurihara, N.; Chida, J.; Cisse, Y.; Yano, M.; Kido, H. Influenza virus-cytokine-protease cycle in the pathogenesis of vascular hyperpermeability in severe influenza. J. Infect. Dis. 2010, 202, 991–1001. [Google Scholar] [CrossRef]
- Saffarzadeh, M.; Juenemann, C.; Queisser, M.A.; Lochnit, G.; Barreto, G.; Galuska, S.P.; Lohmeyer, J.; Preissner, K.T. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: A predominant role of histones. PLoS ONE 2012, 7, e32366. [Google Scholar] [CrossRef] [PubMed]
- Narasaraju, T.; Yang, E.; Samy, R.P.; Ng, H.H.; Poh, W.P.; Liew, A.A.; Phoon, M.C.; van Rooijen, N.; Chow, V.T. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am. J. Pathol. 2011, 179, 199–210. [Google Scholar] [CrossRef]
- Zhu, L.; Liu, L.; Zhang, Y.; Pu, L.; Liu, J.; Li, X.; Chen, Z.; Hao, Y.; Wang, B.; Han, J.; et al. High Level of Neutrophil Extracellular Traps Correlates With Poor Prognosis of Severe Influenza A Infection. J. Infect. Dis. 2018, 217, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA 2020, 323, 1061–1069. [Google Scholar] [CrossRef]
- Magro, C.; Mulvey, J.J.; Berlin, D.; Nuovo, G.; Salvatore, S.; Harp, J.; Baxter-Stoltzfus, A.; Laurence, J. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl. Res. 2020, 220, 1–13. [Google Scholar] [CrossRef]
- Bourgonje, A.R.; Abdulle, A.E.; Timens, W.; Hillebrands, J.L.; Navis, G.J.; Gordijn, S.J.; Bolling, M.C.; Dijkstra, G.; Voors, A.A.; Osterhaus, A.D.; et al. Angiotensin-converting enzyme 2 (ACE2), SARS-CoV-2 and the pathophysiology of coronavirus disease 2019 (COVID-19). J. Pathol. 2020, 251, 228–248. [Google Scholar] [CrossRef]
- Kalkeri, R.; Goebel, S.; Sharma, G.D. SARS-CoV-2 Shedding from Asymptomatic Patients: Contribution of Potential Extrapulmonary Tissue Reservoirs. Am. J. Trop. Med. Hyg. 2020, 103, 18–21. [Google Scholar] [CrossRef]
- Connors, J.M.; Levy, J.H. COVID-19 and its implications for thrombosis and anticoagulation. Blood 2020, 135, 2033–2040. [Google Scholar] [CrossRef] [PubMed]
- Silvin, A.; Chapuis, N.; Dunsmore, G.; Goubet, A.G.; Dubuisson, A.; Derosa, L.; Almire, C.; Henon, C.; Kosmider, O.; Droin, N.; et al. Elevated Calprotectin and Abnormal Myeloid Cell Subsets Discriminate Severe from Mild COVID-19. Cell 2020, 182, 1401–1418. [Google Scholar] [CrossRef] [PubMed]
- Schulte-Schrepping, J.; Reusch, N.; Paclik, D.; Bassler, K.; Schlickeiser, S.; Zhang, B.; Kramer, B.; Krammer, T.; Brumhard, S.; Bonaguro, L.; et al. Severe COVID-19 Is Marked by a Dysregulated Myeloid Cell Compartment. Cell 2020, 182, 1419–1440. [Google Scholar] [CrossRef]
- Sreeramkumar, V.; Adrover, J.M.; Ballesteros, I.; Cuartero, M.I.; Rossaint, J.; Bilbao, I.; Nacher, M.; Pitaval, C.; Radovanovic, I.; Fukui, Y.; et al. Neutrophils scan for activated platelets to initiate inflammation. Science 2014, 346, 1234–1238. [Google Scholar] [CrossRef]
- Hidalgo, A.; Chang, J.; Jang, J.E.; Peired, A.J.; Chiang, E.Y.; Frenette, P.S. Heterotypic interactions enabled by polarized neutrophil microdomains mediate thromboinflammatory injury. Nat. Med. 2009, 15, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Middleton, E.A.; He, X.Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood 2020, 136, 1169–1179. [Google Scholar] [CrossRef] [PubMed]
- Manne, B.K.; Denorme, F.; Middleton, E.A.; Portier, I.; Rowley, J.W.; Stubben, C.; Petrey, A.C.; Tolley, N.D.; Guo, L.; Cody, M.; et al. Platelet gene expression and function in patients with COVID-19. Blood 2020, 136, 1317–1329. [Google Scholar] [CrossRef] [PubMed]
- Skendros, P.; Mitsios, A.; Chrysanthopoulou, A.; Mastellos, D.C.; Metallidis, S.; Rafailidis, P.; Ntinopoulou, M.; Sertaridou, E.; Tsironidou, V.; Tsigalou, C.; et al. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J. Clin. Investig. 2020, 130, 6151–6157. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Zuo, M.; Yalavarthi, S.; Gockman, K.; Madison, J.A.; Shi, H.; Woodard, W.; Lezak, S.P.; Lugogo, N.L.; Knight, J.S.; et al. Neutrophil extracellular traps and thrombosis in COVID-19. medRxiv 2020. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Weber, A.; Barnes, B.J.; Egeblad, M.; et al. Neutrophil extracellular traps (NETs) as markers of disease severity in COVID-19. medRxiv 2020. [Google Scholar] [CrossRef]
- Martinod, K.; Witsch, T.; Erpenbeck, L.; Savchenko, A.; Hayashi, H.; Cherpokova, D.; Gallant, M.; Mauler, M.; Cifuni, S.M.; Wagner, D.D. Peptidylarginine deiminase 4 promotes age-related organ fibrosis. J. Exp. Med. 2017, 214, 439–458. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, W.; Wang, N.; Tall, A.R.; Tabas, I. Mitochondrial Oxidative Stress Promotes Atherosclerosis and Neutrophil Extracellular Traps in Aged Mice. Arterioscler. Thromb. Vasc. Biol. 2017, 37, e99–e107. [Google Scholar] [CrossRef]
- Megens, R.T.; Vijayan, S.; Lievens, D.; Doring, Y.; van Zandvoort, M.A.; Grommes, J.; Weber, C.; Soehnlein, O. Presence of luminal neutrophil extracellular traps in atherosclerosis. Thromb. Haemost. 2012, 107, 597–598. [Google Scholar] [CrossRef] [PubMed]
- Helseth, R.; Shetelig, C.; Andersen, G.O.; Langseth, M.S.; Limalanathan, S.; Opstad, T.B.; Arnesen, H.; Hoffmann, P.; Eritsland, J.; Seljeflot, I. Neutrophil Extracellular Trap Components Associate with Infarct Size, Ventricular Function, and Clinical Outcome in STEMI. Mediat. Inflamm. 2019, 2019, 7816491. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D., Jr.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef]
- Robbins, C.S.; Hilgendorf, I.; Weber, G.F.; Theurl, I.; Iwamoto, Y.; Figueiredo, J.L.; Gorbatov, R.; Sukhova, G.K.; Gerhardt, L.M.; Smyth, D.; et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat. Med. 2013, 19, 1166–1172. [Google Scholar] [CrossRef]
- Ionita, M.G.; van den Borne, P.; Catanzariti, L.M.; Moll, F.L.; de Vries, J.P.; Pasterkamp, G.; Vink, A.; de Kleijn, D.P. High neutrophil numbers in human carotid atherosclerotic plaques are associated with characteristics of rupture-prone lesions. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1842–1848. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sjoberg, S.; Tang, T.T.; Oorni, K.; Wu, W.; Liu, C.; Secco, B.; Tia, V.; Sukhova, G.K.; Fernandes, C.; et al. Cathepsin G activity lowers plasma LDL and reduces atherosclerosis. Biochim. Biophys. Acta 2014, 1842, 2174–2183. [Google Scholar] [CrossRef] [PubMed]
- Wantha, S.; Alard, J.E.; Megens, R.T.; van der Does, A.M.; Doring, Y.; Drechsler, M.; Pham, C.T.; Wang, M.W.; Wang, J.M.; Gallo, R.L.; et al. Neutrophil-derived cathelicidin promotes adhesion of classical monocytes. Circ. Res. 2013, 112, 792–801. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Bobryshev, Y.V.; Orekhov, A.N. Neutrophil’s weapons in atherosclerosis. Exp. Mol. Pathol. 2015, 99, 663–671. [Google Scholar] [CrossRef]
- Yamamoto, K.; Yamada, H.; Wakana, N.; Kikai, M.; Terada, K.; Wada, N.; Motoyama, S.; Saburi, M.; Sugimoto, T.; Kami, D.; et al. Augmented neutrophil extracellular traps formation promotes atherosclerosis development in socially defeated apoE(−/−) mice. Biochem. Biophys. Res. Commun. 2018, 500, 490–496. [Google Scholar] [CrossRef]
- Franck, G.; Mawson, T.L.; Folco, E.J.; Molinaro, R.; Ruvkun, V.; Engelbertsen, D.; Liu, X.; Tesmenitsky, Y.; Shvartz, E.; Sukhova, G.K.; et al. Roles of PAD4 and NETosis in Experimental Atherosclerosis and Arterial Injury: Implications for Superficial Erosion. Circ. Res. 2018, 123, 33–42. [Google Scholar] [CrossRef]
- Liu, Y.; Carmona-Rivera, C.; Moore, E.; Seto, N.L.; Knight, J.S.; Pryor, M.; Yang, Z.H.; Hemmers, S.; Remaley, A.T.; Mowen, K.A.; et al. Myeloid-Specific Deletion of Peptidylarginine Deiminase 4 Mitigates Atherosclerosis. Front. Immunol. 2018, 9, 1680. [Google Scholar] [CrossRef]
- Molinaro, R.; Yu, M.; Sausen, G.; Bichsel, C.A.; Corbo, C.; Folco, E.J.; Lee, G.Y.; Liu, Y.; Tesmenitsky, Y.; Shvartz, E.; et al. Targeted delivery of Protein Arginine Deiminase-4 inhibitors to limit arterial intimal NETosis and preserve endothelial integrity. Cardiovasc. Res. 2021. [Google Scholar] [CrossRef]
- Folco, E.J.; Mawson, T.L.; Vromman, A.; Bernardes-Souza, B.; Franck, G.; Persson, O.; Nakamura, M.; Newton, G.; Luscinskas, F.W.; Libby, P. Neutrophil Extracellular Traps Induce Endothelial Cell Activation and Tissue Factor Production Through Interleukin-1alpha and Cathepsin G. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1901–1912. [Google Scholar] [CrossRef]
- Brill, A.; Fuchs, T.A.; Chauhan, A.K.; Yang, J.J.; De Meyer, S.F.; Kollnberger, M.; Wakefield, T.W.; Lammle, B.; Massberg, S.; Wagner, D.D. von Willebrand factor-mediated platelet adhesion is critical for deep vein thrombosis in mouse models. Blood 2011, 117, 1400–1407. [Google Scholar] [CrossRef]
- Etulain, J.; Martinod, K.; Wong, S.L.; Cifuni, S.M.; Schattner, M.; Wagner, D.D. P-selectin promotes neutrophil extracellular trap formation in mice. Blood 2015, 126, 242–246. [Google Scholar] [CrossRef]
- Stakos, D.A.; Kambas, K.; Konstantinidis, T.; Mitroulis, I.; Apostolidou, E.; Arelaki, S.; Tsironidou, V.; Giatromanolaki, A.; Skendros, P.; Konstantinides, S.; et al. Expression of functional tissue factor by neutrophil extracellular traps in culprit artery of acute myocardial infarction. Eur. Heart J. 2015, 36, 1405–1414. [Google Scholar] [CrossRef] [PubMed]
- Frangou, E.; Chrysanthopoulou, A.; Mitsios, A.; Kambas, K.; Arelaki, S.; Angelidou, I.; Arampatzioglou, A.; Gakiopoulou, H.; Bertsias, G.K.; Verginis, P.; et al. REDD1/autophagy pathway promotes thromboinflammation and fibrosis in human systemic lupus erythematosus (SLE) through NETs decorated with tissue factor (TF) and interleukin-17A (IL-17A). Ann. Rheum. Dis. 2019, 78, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Mackman, N.; Tilley, R.E.; Key, N.S. Role of the extrinsic pathway of blood coagulation in hemostasis and thrombosis. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1687–1693. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, X.; Pelayo, R.; Monestier, M.; Ammollo, C.T.; Semeraro, F.; Taylor, F.B.; Esmon, N.L.; Lupu, F.; Esmon, C.T. Extracellular histones are major mediators of death in sepsis. Nat. Med. 2009, 15, 1318–1321. [Google Scholar] [CrossRef] [PubMed]
- Ammollo, C.T.; Semeraro, F.; Xu, J.; Esmon, N.L.; Esmon, C.T. Extracellular histones increase plasma thrombin generation by impairing thrombomodulin-dependent protein C activation. J. Thromb. Haemost. 2011, 9, 1795–1803. [Google Scholar] [CrossRef] [PubMed]
- Massberg, S.; Grahl, L.; von Bruehl, M.L.; Manukyan, D.; Pfeiler, S.; Goosmann, C.; Brinkmann, V.; Lorenz, M.; Bidzhekov, K.; Khandagale, A.B.; et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat. Med. 2010, 16, 887–896. [Google Scholar] [CrossRef] [PubMed]
- von Bruhl, M.L.; Stark, K.; Steinhart, A.; Chandraratne, S.; Konrad, I.; Lorenz, M.; Khandoga, A.; Tirniceriu, A.; Coletti, R.; Kollnberger, M.; et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J. Exp. Med. 2012, 209, 819–835. [Google Scholar] [CrossRef]
- Wang, Y.; Luo, L.; Braun, O.O.; Westman, J.; Madhi, R.; Herwald, H.; Morgelin, M.; Thorlacius, H. Neutrophil extracellular trap-microparticle complexes enhance thrombin generation via the intrinsic pathway of coagulation in mice. Sci. Rep. 2018, 8, 4020. [Google Scholar] [CrossRef]
- Jimenez-Alcazar, M.; Rangaswamy, C.; Panda, R.; Bitterling, J.; Simsek, Y.J.; Long, A.T.; Bilyy, R.; Krenn, V.; Renne, C.; Renne, T.; et al. Host DNases prevent vascular occlusion by neutrophil extracellular traps. Science 2017, 358, 1202–1206. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Alcazar, M.; Napirei, M.; Panda, R.; Kohler, E.C.; Kremer Hovinga, J.A.; Mannherz, H.G.; Peine, S.; Renne, T.; Lammle, B.; Fuchs, T.A. Impaired DNase1-mediated degradation of neutrophil extracellular traps is associated with acute thrombotic microangiopathies. J. Thromb. Haemost. 2015, 13, 732–742. [Google Scholar] [CrossRef]
- Perdomo, J.; Leung, H.H.L.; Ahmadi, Z.; Yan, F.; Chong, J.J.H.; Passam, F.H.; Chong, B.H. Neutrophil activation and NETosis are the major drivers of thrombosis in heparin-induced thrombocytopenia. Nat. Commun. 2019, 10, 1322. [Google Scholar] [CrossRef]
- Brill, A.; Fuchs, T.A.; Savchenko, A.S.; Thomas, G.M.; Martinod, K.; De Meyer, S.F.; Bhandari, A.A.; Wagner, D.D. Neutrophil extracellular traps promote deep vein thrombosis in mice. J. Thromb. Haemost. 2012, 10, 136–144. [Google Scholar] [CrossRef]
- Savchenko, A.S.; Martinod, K.; Seidman, M.A.; Wong, S.L.; Borissoff, J.I.; Piazza, G.; Libby, P.; Goldhaber, S.Z.; Mitchell, R.N.; Wagner, D.D. Neutrophil extracellular traps form predominantly during the organizing stage of human venous thromboembolism development. J. Thromb. Haemost. 2014, 12, 860–870. [Google Scholar] [CrossRef]
- de Boer, O.J.; Li, X.; Teeling, P.; Mackaay, C.; Ploegmakers, H.J.; van der Loos, C.M.; Daemen, M.J.; de Winter, R.J.; van der Wal, A.C. Neutrophils, neutrophil extracellular traps and interleukin-17 associate with the organisation of thrombi in acute myocardial infarction. Thromb. Haemost. 2013, 109, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Riegger, J.; Byrne, R.A.; Joner, M.; Chandraratne, S.; Gershlick, A.H.; Ten Berg, J.M.; Adriaenssens, T.; Guagliumi, G.; Godschalk, T.C.; Neumann, F.J.; et al. Histopathological evaluation of thrombus in patients presenting with stent thrombosis. A multicenter European study: A report of the prevention of late stent thrombosis by an interdisciplinary global European effort consortium. Eur. Heart J. 2016, 37, 1538–1549. [Google Scholar] [CrossRef]
- Laridan, E.; Denorme, F.; Desender, L.; Francois, O.; Andersson, T.; Deckmyn, H.; Vanhoorelbeke, K.; De Meyer, S.F. Neutrophil extracellular traps in ischemic stroke thrombi. Ann. Neurol. 2017, 82, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Hellesen, A.; Bratland, E.; Husebye, E.S. Autoimmune Addison’s disease-An update on pathogenesis. Ann. Endocrinol. 2018, 79, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Granger, V.; Peyneau, M.; Chollet-Martin, S.; de Chaisemartin, L. Neutrophil Extracellular Traps in Autoimmunity and Allergy: Immune Complexes at Work. Front. Immunol. 2019, 10, 2824. [Google Scholar] [CrossRef] [PubMed]
- Frieri, M.; Stampfl, H. Systemic lupus erythematosus and atherosclerosis: Review of the literature. Autoimmun. Rev. 2016, 15, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Denny, M.F.; Yalavarthi, S.; Zhao, W.; Thacker, S.G.; Anderson, M.; Sandy, A.R.; McCune, W.J.; Kaplan, M.J. A distinct subset of proinflammatory neutrophils isolated from patients with systemic lupus erythematosus induces vascular damage and synthesizes type I IFNs. J. Immunol. 2010, 184, 3284–3297. [Google Scholar] [CrossRef] [PubMed]
- Brandt, L.; Hedberg, H. Impaired phagocytosis by peripheral blood granulocytes in systemic lupus erythematosus. Scand. J. Haematol. 1969, 6, 348–353. [Google Scholar] [CrossRef] [PubMed]
- Cairns, A.P.; Crockard, A.D.; McConnell, J.R.; Courtney, P.A.; Bell, A.L. Reduced expression of CD44 on monocytes and neutrophils in systemic lupus erythematosus: Relations with apoptotic neutrophils and disease activity. Ann. Rheum. Dis. 2001, 60, 950–955. [Google Scholar] [CrossRef]
- Donnelly, S.; Roake, W.; Brown, S.; Young, P.; Naik, H.; Wordsworth, P.; Isenberg, D.A.; Reid, K.B.; Eggleton, P. Impaired recognition of apoptotic neutrophils by the C1q/calreticulin and CD91 pathway in systemic lupus erythematosus. Arthritis Rheum. 2006, 54, 1543–1556. [Google Scholar] [CrossRef] [PubMed]
- Pieterse, E.; van der Vlag, J. Breaking immunological tolerance in systemic lupus erythematosus. Front. Immunol. 2014, 5, 164. [Google Scholar] [CrossRef]
- Kaplan, M.J. Neutrophils in the pathogenesis and manifestations of SLE. Nat. Rev. Rheumatol. 2011, 7, 691–699. [Google Scholar] [CrossRef]
- Hacbarth, E.; Kajdacsy-Balla, A. Low density neutrophils in patients with systemic lupus erythematosus, rheumatoid arthritis, and acute rheumatic fever. Arthritis Rheum. 1986, 29, 1334–1342. [Google Scholar] [CrossRef]
- Hakkim, A.; Furnrohr, B.G.; Amann, K.; Laube, B.; Abed, U.A.; Brinkmann, V.; Herrmann, M.; Voll, R.E.; Zychlinsky, A. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc. Natl. Acad. Sci. USA 2010, 107, 9813–9818. [Google Scholar] [CrossRef]
- Kahlenberg, J.M.; Carmona-Rivera, C.; Smith, C.K.; Kaplan, M.J. Neutrophil extracellular trap-associated protein activation of the NLRP3 inflammasome is enhanced in lupus macrophages. J. Immunol. 2013, 190, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Kahlenberg, J.M.; Kaplan, M.J. Mechanisms of premature atherosclerosis in rheumatoid arthritis and lupus. Annu. Rev. Med. 2013, 64, 249–263. [Google Scholar] [CrossRef]
- Knight, J.S.; Zhao, W.; Luo, W.; Subramanian, V.; O’Dell, A.A.; Yalavarthi, S.; Hodgin, J.B.; Eitzman, D.T.; Thompson, P.R.; Kaplan, M.J. Peptidylarginine deiminase inhibition is immunomodulatory and vasculoprotective in murine lupus. J. Clin. Investig. 2013, 123, 2981–2993. [Google Scholar] [CrossRef]
- Khandpur, R.; Carmona-Rivera, C.; Vivekanandan-Giri, A.; Gizinski, A.; Yalavarthi, S.; Knight, J.S.; Friday, S.; Li, S.; Patel, R.M.; Subramanian, V.; et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci. Transl. Med. 2013, 5, 178ra140. [Google Scholar] [CrossRef] [PubMed]
- Warrington, K.J.; Kent, P.D.; Frye, R.L.; Lymp, J.F.; Kopecky, S.L.; Goronzy, J.J.; Weyand, C.M. Rheumatoid arthritis is an independent risk factor for multi-vessel coronary artery disease: A case control study. Arthritis Res. Ther. 2005, 7, R984–R991. [Google Scholar] [CrossRef] [PubMed]
- Doring, Y.; Manthey, H.D.; Drechsler, M.; Lievens, D.; Megens, R.T.; Soehnlein, O.; Busch, M.; Manca, M.; Koenen, R.R.; Pelisek, J.; et al. Auto-antigenic protein-DNA complexes stimulate plasmacytoid dendritic cells to promote atherosclerosis. Circulation 2012, 125, 1673–1683. [Google Scholar] [CrossRef] [PubMed]
- Sangaletti, S.; Tripodo, C.; Chiodoni, C.; Guarnotta, C.; Cappetti, B.; Casalini, P.; Piconese, S.; Parenza, M.; Guiducci, C.; Vitali, C.; et al. Neutrophil extracellular traps mediate transfer of cytoplasmic neutrophil antigens to myeloid dendritic cells toward ANCA induction and associated autoimmunity. Blood 2012, 120, 3007–3018. [Google Scholar] [CrossRef]
- Houben, E.; Penne, E.L.; Voskuyl, A.E.; van der Heijden, J.W.; Otten, R.H.J.; Boers, M.; Hoekstra, T. Cardiovascular events in anti-neutrophil cytoplasmic antibody-associated vasculitis: A meta-analysis of observational studies. Rheumatology 2018, 57, 555–562. [Google Scholar] [CrossRef]
- Imamoto, T.; Nakazawa, D.; Shida, H.; Suzuki, A.; Otsuka, N.; Tomaru, U.; Ishizu, A. Possible linkage between microscopic polyangiitis and thrombosis via neutrophil extracellular traps. Clin. Exp. Rheumatol. 2014, 32, 149–150. [Google Scholar]
- Nakazawa, D.; Tomaru, U.; Yamamoto, C.; Jodo, S.; Ishizu, A. Abundant neutrophil extracellular traps in thrombus of patient with microscopic polyangiitis. Front. Immunol. 2012, 3, 333. [Google Scholar] [CrossRef]
- Bonaventura, A.; Liberale, L.; Montecucco, F. Aspirin in primary prevention for patients with diabetes: Still a matter of debate. Eur. J. Clin. Investig. 2018, 48, e13001. [Google Scholar] [CrossRef]
- Wellen, K.E.; Hotamisligil, G.S. Inflammation, stress, and diabetes. J. Clin. Investig. 2005, 115, 1111–1119. [Google Scholar] [CrossRef]
- Harsunen, M.H.; Puff, R.; D’Orlando, O.; Giannopoulou, E.; Lachmann, L.; Beyerlein, A.; von Meyer, A.; Ziegler, A.G. Reduced blood leukocyte and neutrophil numbers in the pathogenesis of type 1 diabetes. Horm. Metab. Res. 2013, 45, 467–470. [Google Scholar] [CrossRef]
- Valle, A.; Giamporcaro, G.M.; Scavini, M.; Stabilini, A.; Grogan, P.; Bianconi, E.; Sebastiani, G.; Masini, M.; Maugeri, N.; Porretti, L.; et al. Reduction of circulating neutrophils precedes and accompanies type 1 diabetes. Diabetes 2013, 62, 2072–2077. [Google Scholar] [CrossRef]
- Carestia, A.; Frechtel, G.; Cerrone, G.; Linari, M.A.; Gonzalez, C.D.; Casais, P.; Schattner, M. NETosis before and after Hyperglycemic Control in Type 2 Diabetes Mellitus Patients. PLoS ONE 2016, 11, e0168647. [Google Scholar] [CrossRef]
- Vecchio, F.; Lo Buono, N.; Stabilini, A.; Nigi, L.; Dufort, M.J.; Geyer, S.; Rancoita, P.M.; Cugnata, F.; Mandelli, A.; Valle, A.; et al. Abnormal neutrophil signature in the blood and pancreas of presymptomatic and symptomatic type 1 diabetes. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Fu, S.; Speake, C.; Greenbaum, C.J.; Odegard, J.M. NETosis-associated serum biomarkers are reduced in type 1 diabetes in association with neutrophil count. Clin. Exp. Immunol. 2016, 184, 318–322. [Google Scholar] [CrossRef]
- Wang, Y.; Xiao, Y.; Zhong, L.; Ye, D.; Zhang, J.; Tu, Y.; Bornstein, S.R.; Zhou, Z.; Lam, K.S.; Xu, A. Increased neutrophil elastase and proteinase 3 and augmented NETosis are closely associated with beta-cell autoimmunity in patients with type 1 diabetes. Diabetes 2014, 63, 4239–4248. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.L.; Demers, M.; Martinod, K.; Gallant, M.; Wang, Y.; Goldfine, A.B.; Kahn, C.R.; Wagner, D.D. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat. Med. 2015, 21, 815–819. [Google Scholar] [CrossRef] [PubMed]
- de Vries, J.J.; Hoppenbrouwers, T.; Martinez-Torres, C.; Majied, R.; Ozcan, B.; van Hoek, M.; Leebeek, F.W.G.; Rijken, D.C.; Koenderink, G.H.; de Maat, M.P.M. Effects of Diabetes Mellitus on Fibrin Clot Structure and Mechanics in a Model of Acute Neutrophil Extracellular Traps (NETs) Formation. Int. J. Mol. Sci. 2020, 21, 7107. [Google Scholar] [CrossRef]
- Yu, T.; Robotham, J.L.; Yoon, Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2006, 103, 2653–2658. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhou, X.; Yin, Y.; Mai, Y.; Wang, D.; Zhang, X. Hyperglycemia Induces Neutrophil Extracellular Traps Formation Through an NADPH Oxidase-Dependent Pathway in Diabetic Retinopathy. Front. Immunol. 2018, 9, 3076. [Google Scholar] [CrossRef]
- Berezin, A. Neutrophil extracellular traps: The core player in vascular complications of diabetes mellitus. Diabetes Metab. Syndr. 2019, 13, 3017–3023. [Google Scholar] [CrossRef]
- Karima, M.; Kantarci, A.; Ohira, T.; Hasturk, H.; Jones, V.L.; Nam, B.H.; Malabanan, A.; Trackman, P.C.; Badwey, J.A.; Van Dyke, T.E. Enhanced superoxide release and elevated protein kinase C activity in neutrophils from diabetic patients: Association with periodontitis. J. Leukoc. Biol. 2005, 78, 862–870. [Google Scholar] [CrossRef] [PubMed]
- Menegazzo, L.; Ciciliot, S.; Poncina, N.; Mazzucato, M.; Persano, M.; Bonora, B.; Albiero, M.; Vigili de Kreutzenberg, S.; Avogaro, A.; Fadini, G.P. NETosis is induced by high glucose and associated with type 2 diabetes. Acta Diabetol. 2015, 52, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Joshi, M.B.; Lad, A.; Bharath Prasad, A.S.; Balakrishnan, A.; Ramachandra, L.; Satyamoorthy, K. High glucose modulates IL-6 mediated immune homeostasis through impeding neutrophil extracellular trap formation. FEBS Lett. 2013, 587, 2241–2246. [Google Scholar] [CrossRef]
- Monnier, L.; Mas, E.; Ginet, C.; Michel, F.; Villon, L.; Cristol, J.P.; Colette, C. Activation of oxidative stress by acute glucose fluctuations compared with sustained chronic hyperglycemia in patients with type 2 diabetes. JAMA 2006, 295, 1681–1687. [Google Scholar] [CrossRef]
- Xu, F.; Zhao, L.H.; Su, J.B.; Chen, T.; Wang, X.Q.; Chen, J.F.; Wu, G.; Jin, Y.; Wang, X.H. The relationship between glycemic variability and diabetic peripheral neuropathy in type 2 diabetes with well-controlled HbA1c. Diabetol. Metab. Syndr. 2014, 6, 139. [Google Scholar] [CrossRef]
- Greb, J.E.; Goldminz, A.M.; Elder, J.T.; Lebwohl, M.G.; Gladman, D.D.; Wu, J.J.; Mehta, N.N.; Finlay, A.Y.; Gottlieb, A.B. Psoriasis. Nat. Rev. Dis. Primers 2016, 2, 16082. [Google Scholar] [CrossRef]
- Sticherling, M. Psoriasis and autoimmunity. Autoimmun. Rev. 2016, 15, 1167–1170. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.B.; Lebwohl, M.G. Psoriasis: Which therapy for which patient: Psoriasis comorbidities and preferred systemic agents. J. Am. Acad. Dermatol. 2019, 80, 27–40. [Google Scholar] [CrossRef]
- Hau, C.S.; Kanda, N.; Tada, Y.; Shibata, S.; Uozaki, H.; Fukusato, T.; Sato, S.; Watanabe, S. Lipocalin-2 exacerbates psoriasiform skin inflammation by augmenting T-helper 17 response. J. Dermatol. 2016, 43, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Shao, S.; Fang, H.; Dang, E.; Xue, K.; Zhang, J.; Li, B.; Qiao, H.; Cao, T.; Zhuang, Y.; Shen, S.; et al. Neutrophil Extracellular Traps Promote Inflammatory Responses in Psoriasis via Activating Epidermal TLR4/IL-36R Crosstalk. Front. Immunol. 2019, 10, 746. [Google Scholar] [CrossRef]
- Lin, A.M.; Rubin, C.J.; Khandpur, R.; Wang, J.Y.; Riblett, M.; Yalavarthi, S.; Villanueva, E.C.; Shah, P.; Kaplan, M.J.; Bruce, A.T. Mast cells and neutrophils release IL-17 through extracellular trap formation in psoriasis. J. Immunol. 2011, 187, 490–500. [Google Scholar] [CrossRef]
- Schon, M.P.; Erpenbeck, L. The Interleukin-23/Interleukin-17 Axis Links Adaptive and Innate Immunity in Psoriasis. Front. Immunol. 2018, 9, 1323. [Google Scholar] [CrossRef] [PubMed]
- Reich, K.; Papp, K.A.; Matheson, R.T.; Tu, J.H.; Bissonnette, R.; Bourcier, M.; Gratton, D.; Kunynetz, R.A.; Poulin, Y.; Rosoph, L.A.; et al. Evidence that a neutrophil-keratinocyte crosstalk is an early target of IL-17A inhibition in psoriasis. Exp. Dermatol. 2015, 24, 529–535. [Google Scholar] [CrossRef]
- von Stebut, E.; Reich, K.; Thaci, D.; Koenig, W.; Pinter, A.; Korber, A.; Rassaf, T.; Waisman, A.; Mani, V.; Yates, D.; et al. Impact of Secukinumab on Endothelial Dysfunction and Other Cardiovascular Disease Parameters in Psoriasis Patients over 52 Weeks. J. Investig. Dermatol. 2019, 139, 1054–1062. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Pathological Condition | NETs Inducers | Vascular Manifestation | References |
|---|---|---|---|
| Bacterial infection | Methicillin-resistant S. aureus, α-enolase, lipopolysaccharides, early secretory antigen-6 protein | Endocarditis, acute myocardial infection, myeloid cells recruitment to atherosclerotic lesions, microvascular thrombosis, thrombosis of injured heart valves, enhanced atherosclerotic lesion size | [7,11,12,13,14,15,16,17,18] |
| Viral infection | High levels of intracellular reactive oxygen species | Immunothrombosis, microvascular thrombi in the lung, kidney, and heart and organ damage | [19,20,21,22] |
| Atherosclerosis | Cholesterol crystal, interleukin (IL) IL-1β and IL-6, IL-8, oxLDL, high levels of anti-ApoA-1, metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) | Endothelial damage in SMC-rich plaques, plaque macrophage inflammation, increased plaque vulnerability, enhanced lesion size and carotid artery thrombosis | [23,24,25,26,27,28,29,30,31,32] |
| Thrombosis | High mobility group box 1 (HMGB1), hemodynamic force | Occlusion of microvessels and bigger vessels, enhanced infarct size | [33,34,35,36,37] |
| Autoimmune diseases | low-density granulocytes (LDGs), neutrophil antimicrobial peptide LL37 and HNP | Proatherogenic NETs-derived lipoprotein oxidation, endothelial damage, macrophage inflammation and atherosclerosis | [31,38,39,40,41,42] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thakur, M.; Evans, B.; Schindewolf, M.; Baumgartner, I.; Döring, Y. Neutrophil Extracellular Traps Affecting Cardiovascular Health in Infectious and Inflammatory Diseases. Cells 2021, 10, 1689. https://doi.org/10.3390/cells10071689
Thakur M, Evans B, Schindewolf M, Baumgartner I, Döring Y. Neutrophil Extracellular Traps Affecting Cardiovascular Health in Infectious and Inflammatory Diseases. Cells. 2021; 10(7):1689. https://doi.org/10.3390/cells10071689
Chicago/Turabian StyleThakur, Manovriti, Bryce Evans, Marc Schindewolf, Iris Baumgartner, and Yvonne Döring. 2021. "Neutrophil Extracellular Traps Affecting Cardiovascular Health in Infectious and Inflammatory Diseases" Cells 10, no. 7: 1689. https://doi.org/10.3390/cells10071689
APA StyleThakur, M., Evans, B., Schindewolf, M., Baumgartner, I., & Döring, Y. (2021). Neutrophil Extracellular Traps Affecting Cardiovascular Health in Infectious and Inflammatory Diseases. Cells, 10(7), 1689. https://doi.org/10.3390/cells10071689