
Current Knowledge of Endolysosomal and Autophagy Defects in Hereditary Spastic Paraplegia


Abstract
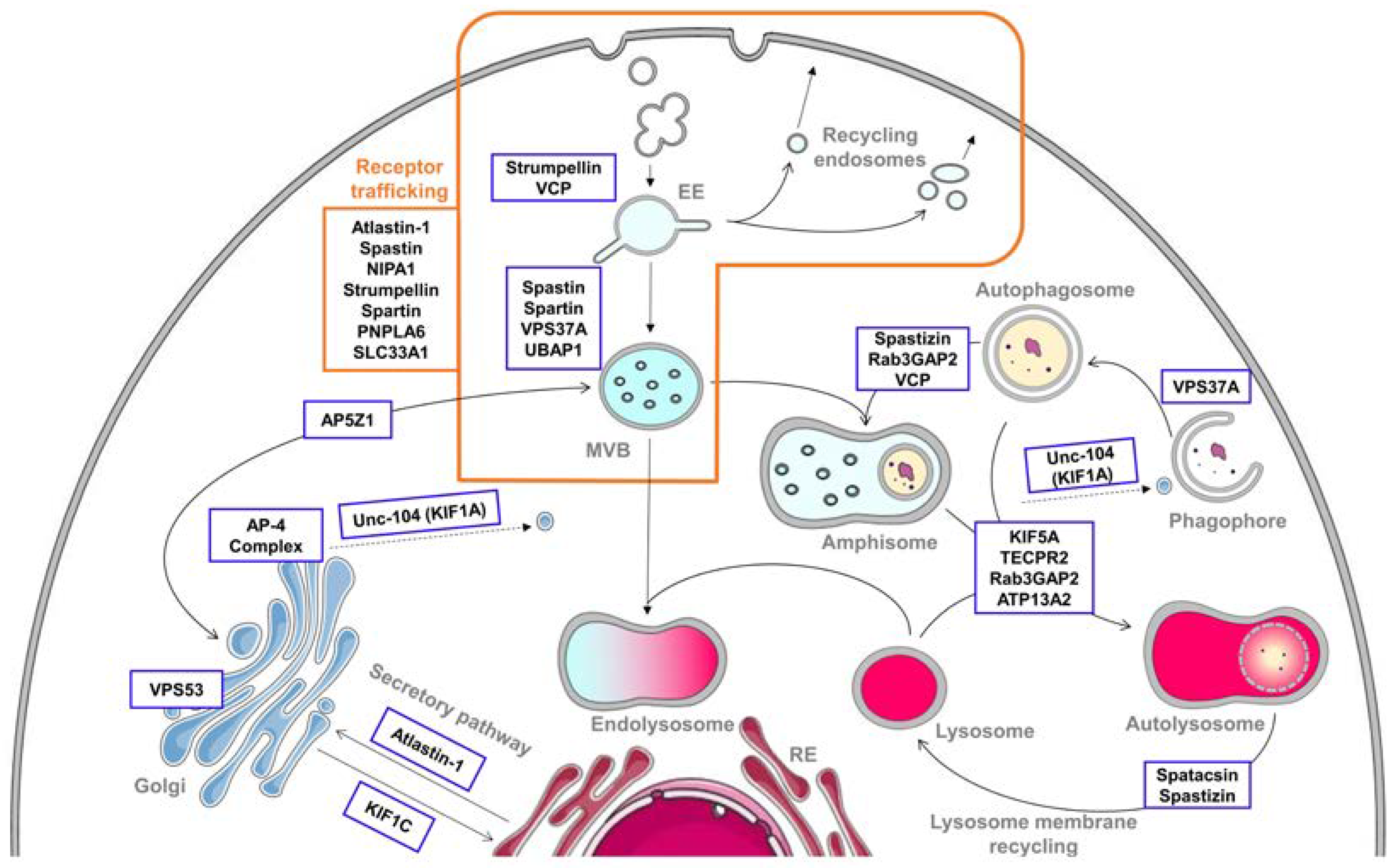
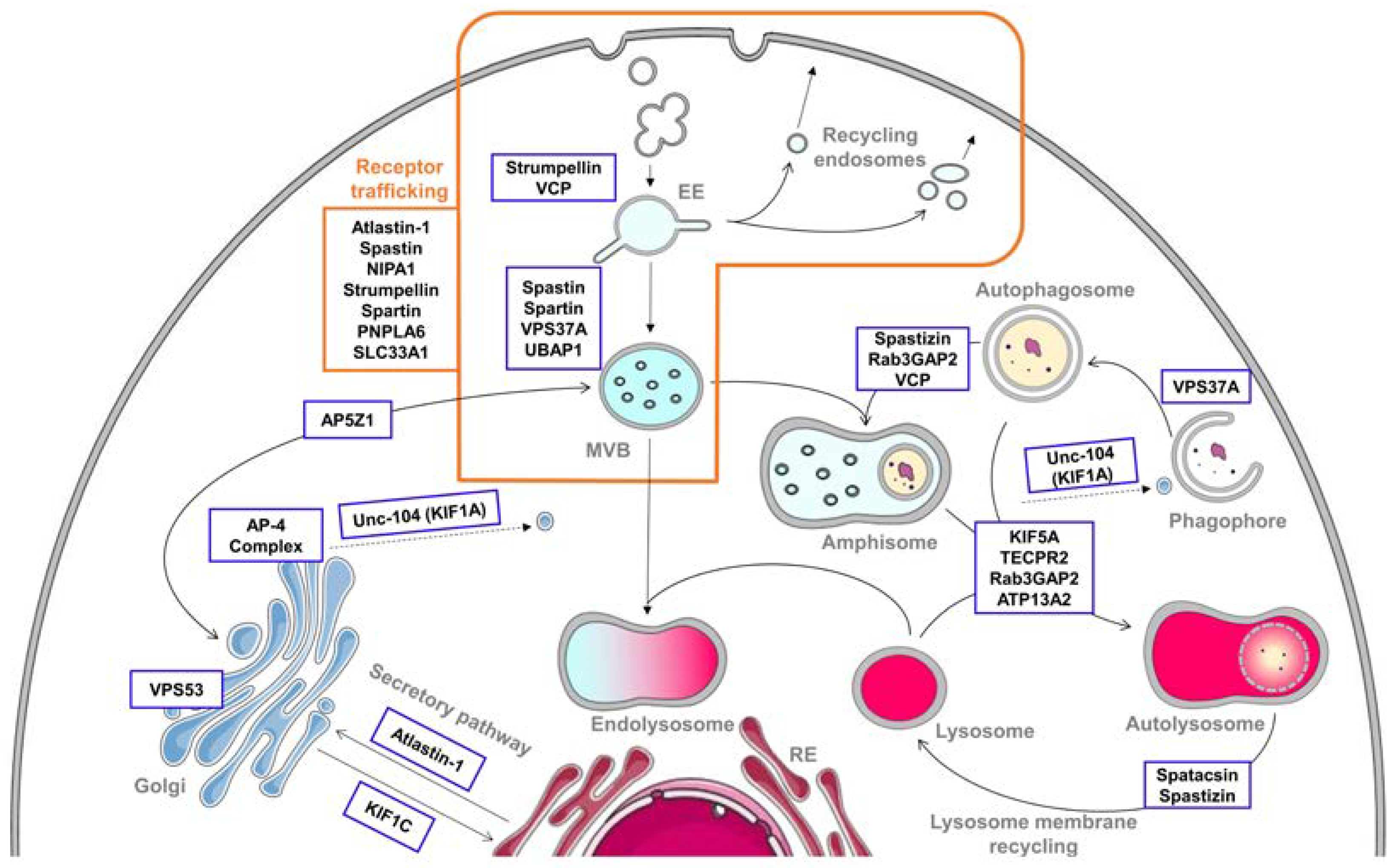
:1. Introduction
2. Alteration of the Endolysosomal Pathway in HSP
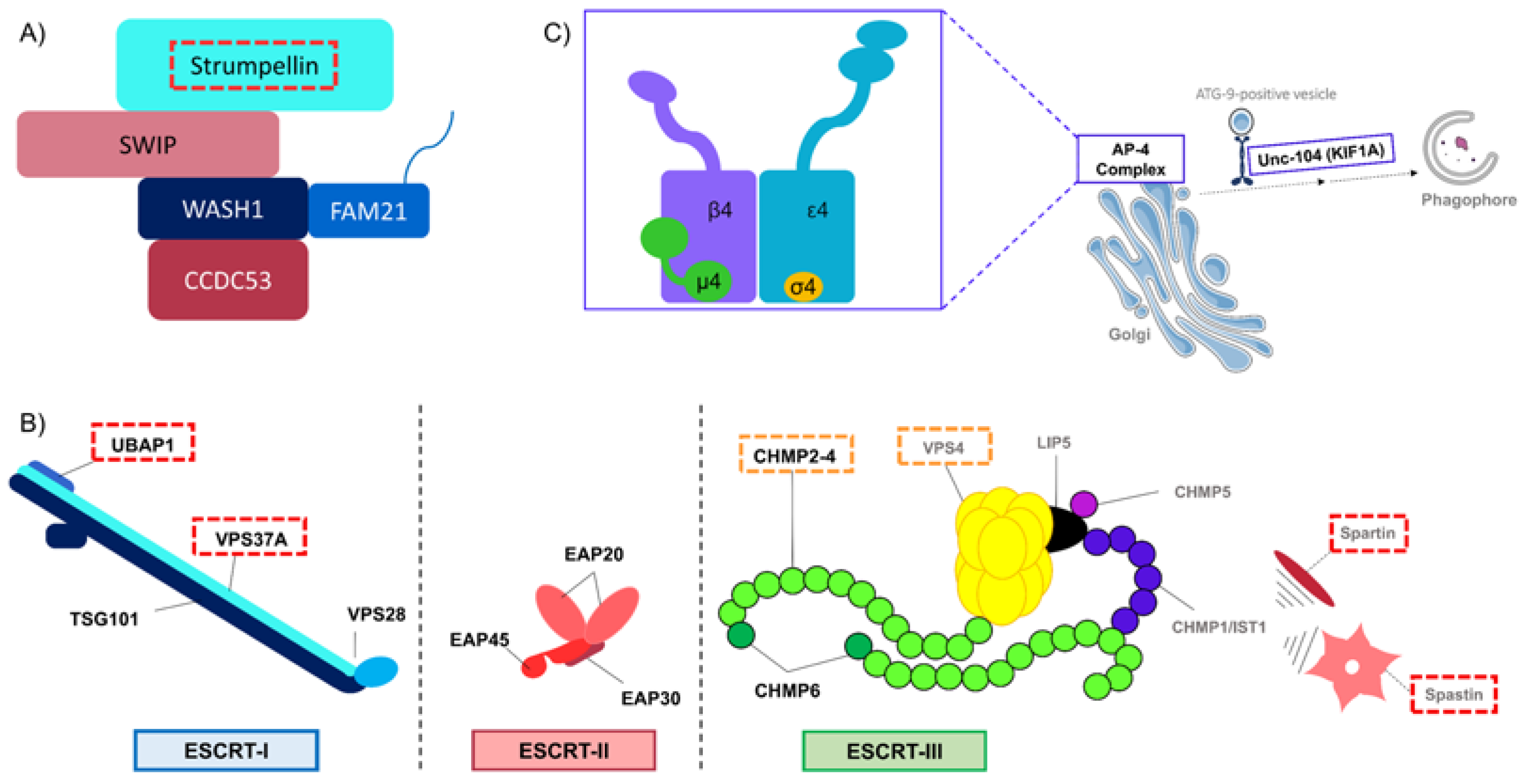
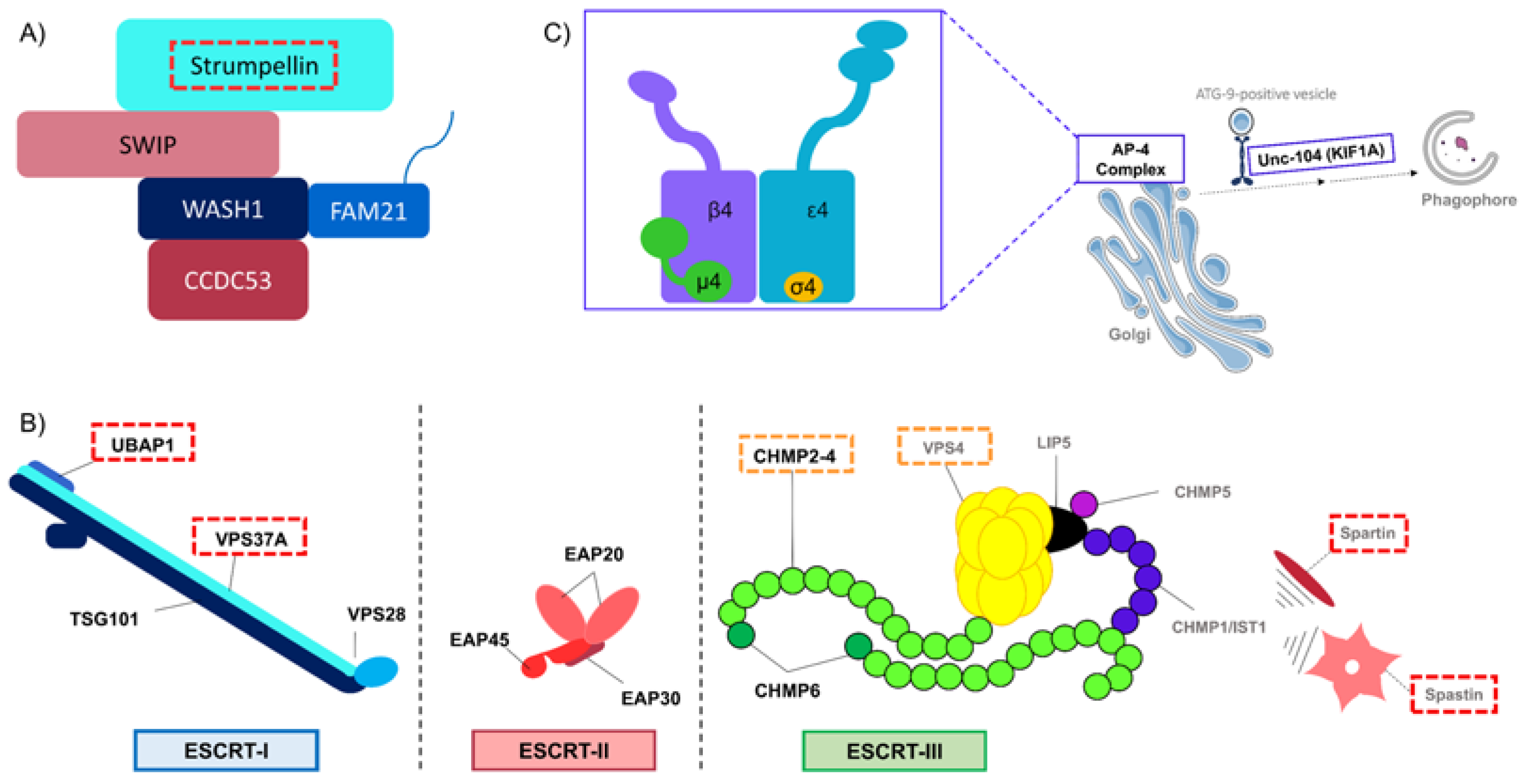
2.1. Endocytosis and Endosomes’ Dynamic

{kind=link}
{kind=link}
| SPG | Gene | Protein | Relevant Section in the Text | Contribution in the Pathway | Cellular Consequences of Mutation/Depletion | References |
|---|---|---|---|---|---|---|
| SPG3A | ATL1 | Atlastin-1 | 2.2 Receptor trafficking 2.3 Secretory pathway | Regulation of BMP signaling ER-Golgi trafficking | Overactivation of BMP signaling Impairment of ER-Golgi trafficking and Golgi morphogenesis | [26,27,28,29] |
| SPG4 | SPAST | Spastin | 2.1 Endocytosis and endosomes’ dynamic 2.2 Receptor trafficking | Regulation of ESCRT-III | Perturbation of endosomal tubulation Overactivation of BMP signaling | [30,31,32,33,34] |
| SPG6 | NIPA1 | NIPA1 | 2.2 Receptor trafficking | Regulation of BMP signaling | Overactivation of BMP signaling | [28,35,36] |
| SPG8 | KIAA0196/ WASHC5 | Strumpellin/ WASHC5 | 2.1 Endocytosis and endosomes’ dynamic 2.2 Receptor trafficking | Member of WASH complex | Perturbation of clathrin-independent pathway Impairment of endosomal tubulation | [23,24,25] |
| SPG10 | KIF5 | KIF5A | 3.2 Autophagosome—lysosome fusion | Kinesin, motor protein | Impairment of the axonal transport and autophagic flux | [37] |
| SPG11 | SPG11 | Spatacsin | 3.3 Lysosome membrane recycling | Recruitment of Dynamin Interacts with spastizin and AP-5 | Autophagy defects due to reduction of autolysosome tubulation Accumulation of autophagic compartments Defective lysosomal clearance of gangliosides | [16,38,39,40,41] |
| SPG15 | ZFYVE26 | Spastizin | 3.3 Lysosome membrane recycling 3.4 Crossroads between endocytic and autophagic pathways | Interacts with spatacsin and AP-5 Interaction with Rab5A and Rab11 | Autophagy defects due to reduction of autolysosome tubulation Accumulation of autophagic compartments Altered maturation of autophagosomes | [38,39,40,42,43] |
| SPG20 | SPART | Spartin | 2.1 Endocytosis and endosomes’ dynamic 2.2 Receptor trafficking | Regulation of ESCRT-III | Perturbation of endosomal trafficking Overactivation of BMP signaling | [44,45,46,47,48,49] |
| SPG30 | KIF1A | KIF1A/Unc-104 | 3.1 Autophagosome biogenesis | Kinesin, motor protein | Impaired transport of ATG-9-positive vesicles leading to defects in autophagosome biogenesis | [50] |
| SPG39 | PNPLA6 | PNPLA6 | 2.2 Receptor trafficking | Regulation of BMP signaling | Overactivation of BMP signaling | [51] |
| SPG42 | SLC33A1 | SLC33A1 | 2.2 Receptor trafficking | Regulation of BMP signaling | Overactivation of BMP signaling | [52] |
| SPG47 | AP4B1 | AP4B1 | 2.3 Secretory pathway 3.1 Autophagosome biogenesis 3.3 Lysosome membrane recycling | Subunit of AP-4 complex | Impairment of ATG9A’s sorting and thus autophagosome biogenesis | [53,54,55] |
| SPG48 | AP5Z1 | AP5Z1 | 3.3 Lysosome membrane recycling 3.4 Crossroads between endocytic and autophagic pathways | AP-5 subunit spatacsin and spastizin interactor | Reduction of autolysosome tubulation Impaired endolysosomal system due to accumulation of endolysosomes Impairment of CIMPR trafficking towards TGN | [56,57,58,59] |
| SPG49 | TECPR2 | TECPR2 | 3.2 Autophagosome—lysosome fusion | Interactor of HOPS and ATG8 family members | Accumulation of autophagosomes due to impaired autophagosome—lysosome fusion | [60,61] |
| SPG50 | AP4M1 | AP4M1 | 2.3 Secretory pathway 3.1 Autophagosome biogenesis 3.3 Lysosome membrane recycling | Subunit of AP-4 complex | Impairment of ATG9A’s sorting and thus autophagosome biogenesis | [53,54,55] |
| SPG51 | AP4E1 | AP4E1 | 2.3 Secretory pathway 3.1 Autophagosome biogenesis 3.3 Lysosome membrane recycling | Subunit of AP-4 complex | Impairment of ATG9A’s sorting and thus autophagosome biogenesis | [53,54,55] |
| SPG52 | AP4S1 | AP4S1 | 2.3 Secretory pathway 3.1 Autophagosome biogenesis 3.3 Lysosome membrane recycling | Subunit of AP-4 complex | Impairment of ATG9A’s sorting and thus autophagosome biogenesis | [53,54,55] |
| SPG53 | VPS37A | VPS37A | 2.1 Endocytosis and endosomes’ dynamic 3.1 Autophagosome biogenesis | Subunit of ESCRT-I | Perturbation of endosomal sorting Altered capacity to recruit ESCRT-I subunits at the PAS leading to impaired autophagosome closure | [62,63] |
| SPG58 | KIF1C | KIF1C | 2.3 Secretory pathway | Kinesin, motor protein | Impairment of Golgi-ER transport | [64,65,66] |
| SPG69 | RAB3GAP2 | Rab3GAP2 | 3.4 Crossroads between endocytic and autophagic pathways | Subunit of Rab3GAP complex | Autophagy defects | [67,68] |
| SPG78 | ATP13A2/ PARK9 | ATP13A2 | 3.2 Autophagosome—lysosome fusion | Still unclear | Autophagy defects due to accumulation of autophagic compartments | [69,70] |
| SPG80 | UBAP1 | UBAP1 | 2.1 Endocytosis and endosomes’ dynamic | Subunit of ESCRT-I | Perturbation of endosomal sorting | [71,72] |
| \ | VCP | VCP | 2.1 Endocytosis and endosomes’ dynamic 3.4 Crossroads between endocytic and autophagic pathways | Interaction with strumpellin Autophagosome maturation | Perturbation of strumpellin localization and function Autophagy defects | [73,74,75,76] |
| \ | VPS53 | VPS53 | 2.3 Secretory pathway | Subunit of GARP complex | Still unclear | [77] |
2.2. Receptor Trafficking Impairment
2.3. Secretory Pathway
3. Autophagy Defects in HSP
3.1. Autophagosome Biogenesis
3.2. Autophagosome–Lysosome Fusion
3.3. Lysosome Membrane Recycling
3.4. Crossroads between Endocytic and Autophagic Pathways
4. Lessons and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ALR | Autophagic lysosome reformation |
| ALS | Amyotrophic lateral sclerosis |
| AP | Adaptor protein |
| ATG | Autophagy-related |
| BMP | Bone morphogenic protein |
| BMPRI | Type I BMP receptors |
| BMPRII | Type II BMP receptors |
| CAV1 | Caveolin1 |
| CEDNIK | Cerebral dysgenesis, neuropathy, ichthyosis, and keratoderma |
| CGN | Cis-Golgi network |
| CHMP | Charged multivesicular body proteins |
| CIMPR | Cation-independent mannose 6-phosphate receptor |
| CMT | Charcot–Marie–Tooth |
| EE | Early endosomes |
| EGFR | Epidermal growth factor receptors |
| ER | Endoplasmic reticulum |
| ESCRT | Endosomal sorting complexes required for transport |
| FTD | Frontotemporal dementia |
| GARP | Golgi-associated retrograde proteins |
| HOPS | Homotypic fusion and vacuole protein sorting |
| HSAN | Hereditary sensory and autonomic neuropathy |
| HSP | Hereditary spastic paraplegia |
| IBMPFD | Inclusion body myopathy with Paget disease and frontotemporal dementia |
| ILV | Intraluminal vesicles |
| LC3 | Light chain 3 |
| LIR | LC3-interacting region |
| M6PR | Mannose-6-phosphate receptor |
| MEF | Mouse embryonic fibroblast |
| MIT | Microtubule interacting and trafficking |
| MVB | Multivesicular body |
| PAS | Phagophore assembly site |
| SNAP29 | Synaptosomal-associated protein 29 |
| SNARE | Soluble N-ethylmaleimide-sensitive factor attachment protein receptors |
| SPG | Spastic Paraplegia Gene |
| SPICT | Spichthyin |
| STX17 | Syntaxin 17 |
| TECPR2 | Tectonin beta-propeller repeat containing 2 |
| TGFβ | Transforming growth factor beta |
| TGN | Trans-Golgi network |
| VAMP8 | Vesicle-associated membrane protein 8 |
| VCP | Valosin-containing protein |
| WASH | Wiskott–Aldrich syndrome protein and SCAR homolog |
References
- Cullen, P.J.; Steinberg, F. To degrade or not to degrade: Mechanisms and significance of endocytic recycling. Nat. Rev. Mol. Cell Biol. 2018, 19, 679–696. [Google Scholar] [CrossRef] [PubMed]
- Doherty, G.J.; McMahon, H.T. Mechanisms of endocytosis. Annu. Rev. Biochem. 2009, 78, 857–902. [Google Scholar] [CrossRef] [Green Version]
- Scott, C.C.; Vacca, F.; Gruenberg, J. Endosome maturation, transport and functions. Semin. Cell Dev. Biol. 2014, 31, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Saimani, U.; Kim, K. Traffic from the endosome towards trans-golgi network. Eur. J. Cell Biol. 2017, 96, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Filimonenko, M.; Stuffers, S.; Raiborg, C.; Yamamoto, A.; Malerød, L.; Fisher, E.M.C.; Isaacs, A.; Brech, A.; Stenmark, H.; Simonsen, A. Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J. Cell Biol. 2007, 179, 485–500. [Google Scholar] [CrossRef]
- Xie, Z.; Klionsky, D.J. Autophagosome formation: Core machinery and adaptations. Nat. Cell Biol. 2007, 9, 1102–1109. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S. Regulation of autophagy by MTOR-dependent and MTOR-independent pathways: Autophagy dysfunction in neurodegenerative diseases and therapeutic application of autophagy enhancers. Biochem. Soc. Trans. 2013, 41, 1103–1130. [Google Scholar] [CrossRef] [Green Version]
- Wartosch, L.; Günesdogan, U.; Graham, S.C.; Luzio, J.P. Recruitment of VPS33A to HOPS by VPS16 is required for lysosome fusion with endosomes and autophagosomes. Traffic 2015, 16, 727–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreij, A.M.A.; Fon, E.A.; McPherson, P.S. Endocytic membrane trafficking and neurodegenerative disease. Cell. Mol. Life Sci. 2016, 73, 1529–1545. [Google Scholar] [CrossRef] [PubMed]
- Walusinski, O. A historical approach to hereditary spastic paraplegia. Rev. Neurol. 2020, 176, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Boutry, M.; Morais, S.; Stevanin, G. Update on the genetics of spastic paraplegias. Curr. Neurol. Neurosci. Rep. 2019, 19, 18. [Google Scholar] [CrossRef]
- Hedera, P. Hereditary Spastic Paraplegia Overview. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Mirzaa, G., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Novarino, G.; Fenstermaker, A.G.; Zaki, M.S.; Hofree, M.; Silhavy, J.L.; Heiberg, A.D.; Abdellateef, M.; Rosti, B.; Scott, E.; Mansour, L.; et al. Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science 2014, 343, 506–511. [Google Scholar] [CrossRef] [Green Version]
- Fra, A.M.; Masserini, M.; Palestini, P.; Sonnino, S.; Simons, K. A photo-reactive derivative of ganglioside GM1 specifically cross-links VIP21-caveolin on the cell surface. FEBS Lett. 1995, 375, 11–14. [Google Scholar] [CrossRef] [Green Version]
- Stevanin, G.; Santorelli, F.M.; Azzedine, H.; Coutinho, P.; Chomilier, J.; Denora, P.S.; Martin, E.; Ouvrard-Hernandez, A.-M.; Tessa, A.; Bouslam, N.; et al. Mutations in SPG11, encoding spatacsin, are a major cause of spastic paraplegia with thin corpus callosum. Nat. Genet. 2007, 39, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Boutry, M.; Branchu, J.; Lustremant, C.; Pujol, C.; Pernelle, J.; Matusiak, R.; Seyer, A.; Poirel, M.; Chu-Van, E.; Pierga, A.; et al. Inhibition of lysosome membrane recycling causes accumulation of gangliosides that contribute to neurodegeneration. Cell Rep. 2018, 23, 3813–3826. [Google Scholar] [CrossRef] [PubMed]
- Boukhris, A.; Schule, R.; Loureiro, J.L.; Lourenço, C.M.; Mundwiller, E.; Gonzalez, M.A.; Charles, P.; Gauthier, J.; Rekik, I.; Acosta Lebrigio, R.F.; et al. Alteration of ganglioside biosynthesis responsible for complex hereditary spastic paraplegia. Am. J. Hum. Genet. 2013, 93, 118–123. [Google Scholar] [CrossRef] [Green Version]
- Valdmanis, P.N.; Meijer, I.A.; Reynolds, A.; Lei, A.; MacLeod, P.; Schlesinger, D.; Zatz, M.; Reid, E.; Dion, P.A.; Drapeau, P.; et al. Mutations in the KIAA0196 gene at the SPG8 locus cause hereditary spastic paraplegia. Am. J. Hum. Genet. 2007, 80, 152–161. [Google Scholar] [CrossRef] [Green Version]
- Jia, D.; Gomez, T.S.; Metlagel, Z.; Umetani, J.; Otwinowski, Z.; Rosen, M.K.; Billadeau, D.D. WASH and WAVE actin regulators of the Wiskott–Aldrich Syndrome Protein (WASP) family are controlled by analogous structurally related complexes. Proc. Natl. Acad. Sci. USA 2010, 107, 10442–10447. [Google Scholar] [CrossRef] [Green Version]
- McNally, K.E.; Cullen, P.J. Endosomal retrieval of cargo: Retromer is not alone. Trends Cell Biol. 2018, 28, 807–822. [Google Scholar] [CrossRef] [Green Version]
- Suetsugu, S.; Gautreau, A. Synergistic BAR-NPF interactions in actin-driven membrane remodeling. Trends Cell Biol. 2012, 22, 141–150. [Google Scholar] [CrossRef]
- Gautreau, A.; Oguievetskaia, K.; Ungermann, C. Function and regulation of the endosomal fusion and fission machineries. Cold Spring Harb. Perspect. Biol. 2014, 6, a016832. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Rijal, R.; Karow, M.; Stumpf, M.; Hahn, O.; Park, L.; Insall, R.; Schröder, R.; Hofmann, A.; Clemen, C.S.; et al. Expression of N471D strumpellin leads to defects in the endolysosomal system. Dis. Model. Mech. 2018, 11, dmm033449. [Google Scholar] [CrossRef] [Green Version]
- Freeman, C.; Seaman, M.N.J.; Reid, E. The hereditary spastic paraplegia protein strumpellin: Characterisation in neurons and of the effect of disease mutations on WASH complex assembly and function. Biochim. Biophys. Acta 2013, 1832, 160–173. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Park, H.; Zhu, P.-P.; Jung, S.-Y.; Blackstone, C.; Chang, J. Hereditary spastic paraplegia SPG8 mutations impair CAV1-dependent, integrin-mediated cell adhesion. Sci. Signal. 2020, 13, eaau7500. [Google Scholar] [CrossRef] [PubMed]
- Fassier, C.; Hutt, J.A.; Scholpp, S.; Lumsden, A.; Giros, B.; Nothias, F.; Schneider-Maunoury, S.; Houart, C.; Hazan, J. Zebrafish atlastin controls motility and spinal motor axon architecture via inhibition of the BMP pathway. Nat. Neurosci. 2010, 13, 1380–1387. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Hedera, P. Hereditary spastic paraplegia-causing mutations in atlastin-1 interfere with BMPRII trafficking. Mol. Cell. Neurosci. 2013, 52, 87–96. [Google Scholar] [CrossRef] [Green Version]
- Botzolakis, E.J.; Zhao, J.; Gurba, K.N.; Macdonald, R.L.; Hedera, P. The effect of HSP-causing mutations in SPG3A and NIPA1 on the assembly, trafficking, and interaction between atlastin-1 and NIPA1. Mol. Cell. Neurosci. 2011, 46, 122–135. [Google Scholar] [CrossRef] [Green Version]
- Namekawa, M.; Muriel, M.-P.; Janer, A.; Latouche, M.; Dauphin, A.; Debeir, T.; Martin, E.; Duyckaerts, C.; Prigent, A.; Depienne, C.; et al. Mutations in the SPG3A gene encoding the GTPase atlastin interfere with vesicle trafficking in the ER/Golgi interface and golgi morphogenesis. Mol. Cell. Neurosci. 2007, 35, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Reid, E.; Connell, J.; Edwards, T.L.; Duley, S.; Brown, S.E.; Sanderson, C.M. The hereditary spastic paraplegia protein spastin interacts with the ESCRT-III complex-associated endosomal protein CHMP1B. Hum. Mol. Genet. 2005, 14, 19–38. [Google Scholar] [CrossRef] [PubMed]
- Allison, R.; Edgar, J.R.; Reid, E. Spastin MIT domain disease-associated mutations disrupt lysosomal function. Front. Neurosci. 2019, 13, 1179. [Google Scholar] [CrossRef] [Green Version]
- Allison, R.; Lumb, J.H.; Fassier, C.; Connell, J.W.; Ten Martin, D.; Seaman, M.N.J.; Hazan, J.; Reid, E. An ESCRT–spastin interaction promotes fission of recycling tubules from the endosome. J. Cell Biol. 2013, 202, 527–543. [Google Scholar] [CrossRef] [Green Version]
- Connell, J.W.; Allison, R.J.; Rodger, C.E.; Pearson, G.; Zlamalova, E.; Reid, E. ESCRT-III-associated proteins and spastin inhibit protrudin-dependent polarised membrane traffic. Cell. Mol. Life Sci. 2020, 77, 2641–2658. [Google Scholar] [CrossRef] [Green Version]
- Jardin, N.; Giudicelli, F.; Ten Martín, D.; Vitrac, A.; De Gois, S.; Allison, R.; Houart, C.; Reid, E.; Hazan, J.; Fassier, C. BMP- and neuropilin 1-mediated motor axon navigation relies on spastin alternative translation. Dev. Camb. Engl. 2018, 145. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Shaw, W.R.; Tsang, H.T.H.; Reid, E.; O’Kane, C.J. Drosophila Spichthyin Inhibits BMP Signaling and Regulates Synaptic Growth and Axonal Microtubules. Nat. Neurosci. 2007, 10, 177–185. [Google Scholar] [CrossRef] [Green Version]
- Tsang, H.T.H.; Edwards, T.L.; Wang, X.; Connell, J.W.; Davies, R.J.; Durrington, H.J.; O’Kane, C.J.; Luzio, J.P.; Reid, E. The hereditary spastic paraplegia proteins NIPA1, spastin and spartin are inhibitors of mammalian BMP signalling. Hum. Mol. Genet. 2009, 18, 3805–3821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Pi, H.; Xi, Y.; Wang, L.; Tian, L.; Chen, M.; Xie, J.; Deng, P.; Zhang, T.; Zhou, C.; et al. KIF5A-dependent axonal transport deficiency disrupts autophagic flux in trimethyltin chloride-induced neurotoxicity. Autophagy 2020, 903–924. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Lee, S.; Blackstone, C. Spastic Paraplegia proteins spastizin and spatacsin mediate autophagic lysosome reformation. J. Clin. Invest. 2014, 124, 5249–5262. [Google Scholar] [CrossRef] [Green Version]
- Renvoisé, B.; Chang, J.; Singh, R.; Yonekawa, S.; FitzGibbon, E.J.; Mankodi, A.; Vanderver, A.; Schindler, A.B.; Toro, C.; Gahl, W.A.; et al. Lysosomal abnormalities in hereditary spastic paraplegia types SPG 15 and SPG 11. Ann. Clin. Transl. Neurol. 2014, 1, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Khundadze, M.; Ribaudo, F.; Hussain, A.; Stahlberg, H.; Brocke-Ahmadinejad, N.; Franzka, P.; Varga, R.-E.; Zarkovic, M.; Pungsrinont, T.; Kokal, M.; et al. Mouse models for hereditary spastic paraplegia uncover a role of PI4K2A in autophagic lysosome reformation. Autophagy 2021. [Google Scholar] [CrossRef] [PubMed]
- Varga, R.-E.; Khundadze, M.; Damme, M.; Nietzsche, S.; Hoffmann, B.; Stauber, T.; Koch, N.; Hennings, J.C.; Franzka, P.; Huebner, A.K.; et al. In vivo evidence for lysosome depletion and impaired autophagic clearance in hereditary spastic paraplegia type spg11. PLOS Genet. 2015, 11, e1005454. [Google Scholar] [CrossRef] [PubMed]
- Vantaggiato, C.; Crimella, C.; Airoldi, G.; Polishchuk, R.; Bonato, S.; Brighina, E.; Scarlato, M.; Musumeci, O.; Toscano, A.; Martinuzzi, A.; et al. Defective autophagy in spastizin mutated patients with hereditary spastic paraparesis type 15. Brain 2013, 136, 3119–3139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vantaggiato, C.; Panzeri, E.; Castelli, M.; Citterio, A.; Arnoldi, A.; Santorelli, F.M.; Liguori, R.; Scarlato, M.; Musumeci, O.; Toscano, A.; et al. ZFYVE26/SPASTIZIN and SPG11/SPATACSIN mutations in hereditary spastic paraplegia types AR-SPG15 and AR-SPG11 have different effects on autophagy and endocytosis. Autophagy 2019, 15, 34–57. [Google Scholar] [CrossRef]
- Patel, H.; Cross, H.; Proukakis, C.; Hershberger, R.; Bork, P.; Ciccarelli, F.D.; Patton, M.A.; McKusick, V.A.; Crosby, A.H. SPG20 is mutated in troyer syndrome, an hereditary spastic paraplegia. Nat. Genet. 2002, 31, 347–348. [Google Scholar] [CrossRef] [PubMed]
- Robay, D.; Patel, H.; Simpson, M.A.; Brown, N.A.; Crosby, A.H. Endogenous spartin, mutated in hereditary spastic paraplegia, has a complex subcellular localization suggesting diverse roles in neurons. Exp. Cell Res. 2006, 312, 2764–2777. [Google Scholar] [CrossRef]
- Ciccarelli, F.D.; Proukakis, C.; Patel, H.; Cross, H.; Azam, S.; Patton, M.A.; Bork, P.; Crosby, A.H. The identification of a conserved domain in both spartin and spastin, mutated in hereditary spastic paraplegia. Genomics 2003, 81, 437–441. [Google Scholar] [CrossRef]
- Renvoisé, B.; Stadler, J.; Singh, R.; Bakowska, J.C.; Blackstone, C. Spg20−/−mice reveal multimodal functions for troyer syndrome protein spartin in lipid droplet maintenance, cytokinesis and BMP signaling. Hum. Mol. Genet. 2012, 21, 3604–3618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakowska, J.C.; Jupille, H.; Fatheddin, P.; Puertollano, R.; Blackstone, C. Troyer syndrome protein spartin is mono-ubiquitinated and functions in EGF RECEPTOR TRAFfiCKing D. Mol. Biol. Cell 2007, 18, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nahm, M.; Lee, M.-J.; Parkinson, W.; Lee, M.; Kim, H.; Kim, Y.-J.; Kim, S.; Cho, Y.S.; Min, B.-M.; Bae, Y.C.; et al. Spartin regulates synaptic growth and neuronal survival by inhibiting BMP-mediated microtubule stabilization. Neuron 2013, 77, 680–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stavoe, A.K.H.; Hill, S.E.; Hall, D.H.; Colón-Ramos, D.A. KIF1A/UNC-104 transports ATG-9 to regulate neurodevelopment and autophagy at synapses. Dev. Cell 2016, 38, 171–185. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Wang, M.; Mao, F.; Shao, M.; Zhao, B.; Song, Z.; Shao, C.; Gong, Y. Knockdown of Pnpla6 protein results in motor neuron defects in zebrafish. Dis. Model. Mech. 2013, 6, 404–413. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Jiang, B.; Ma, J.; Lin, P.; Zhang, Y.; Shao, C.; Sun, W.; Gong, Y. S113R Mutation in SLC33A1 leads to neurodegeneration and augmented BMP signaling in a mouse model. Dis. Model. Mech. 2017, 10, 53–62. [Google Scholar] [CrossRef] [Green Version]
- Behne, R.; Teinert, J.; Wimmer, M.; D’Amore, A.; Davies, A.K.; Scarrott, J.M.; Eberhardt, K.; Brechmann, B.; Chen, I.P.-F.; Buttermore, E.D.; et al. Adaptor protein complex 4 deficiency: A paradigm of childhood-onset hereditary spastic paraplegia caused by defective protein trafficking. Hum. Mol. Genet. 2020, 29, 320–334. [Google Scholar] [CrossRef]
- Mattera, R.; Park, S.Y.; De Pace, R.; Guardia, C.M.; Bonifacino, J.S. AP-4 mediates export of ATG9A from the Trans -golgi network to promote autophagosome formation. Proc. Natl. Acad. Sci. USA 2017, 114, E10697–E10706. [Google Scholar] [CrossRef] [Green Version]
- Ivankovic, D.; Drew, J.; Lesept, F.; White, I.J.; López Doménech, G.; Tooze, S.A.; Kittler, J.T. Axonal autophagosome maturation defect through failure of ATG9A sorting underpins pathology in AP-4 deficiency syndrome. Autophagy 2020, 16, 391–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khundadze, M.; Ribaudo, F.; Hussain, A.; Rosentreter, J.; Nietzsche, S.; Thelen, M.; Winter, D.; Hoffmann, B.; Afzal, M.A.; Hermann, T.; et al. A mouse model for SPG48 reveals a block of autophagic flux upon disruption of adaptor protein complex five. Neurobiol. Dis. 2019, 127, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Hirst, J.; Hesketh, G.G.; Gingras, A.-C.; Robinson, M.S. Rag GTPases and phosphatidylinositol 3-phosphate mediate recruitment of the AP-5/SPG11/SPG15 complex. J. Cell Biol. 2021, 220, e202002075. [Google Scholar] [CrossRef] [PubMed]
- Hirst, J.; Edgar, J.R.; Esteves, T.; Darios, F.; Madeo, M.; Chang, J.; Roda, R.H.; Dürr, A.; Anheim, M.; Gellera, C.; et al. Loss of AP-5 results in accumulation of aberrant endolysosomes: Defining a new type of lysosomal storage disease. Hum. Mol. Genet. 2015, 24, 4984–4996. [Google Scholar] [CrossRef] [Green Version]
- Hirst, J.; Itzhak, D.N.; Antrobus, R.; Borner, G.H.H.; Robinson, M.S. Role of the AP-5 adaptor protein complex in late endosome-to-golgi retrieval. PLOS Biol. 2018, 16, e2004411. [Google Scholar] [CrossRef] [Green Version]
- Fraiberg, M.; Tamim-Yecheskel, B.-C.; Kokabi, K.; Subic, N.; Heimer, G.; Eck, F.; Nalbach, K.; Behrends, C.; Ben-Zeev, B.; Shatz, O.; et al. Lysosomal targeting of autophagosomes by the TECPR domain of TECPR2. Autophagy 2020. [Google Scholar] [CrossRef]
- Tamim-Yecheskel, B.-C.; Fraiberg, M.; Kokabi, K.; Freud, S.; Shatz, O.; Marvaldi, L.; Subic, N.; Brenner, O.; Tsoory, M.; Eilam-Altstadter, R.; et al. A Tecpr2 knockout mouse exhibits age-dependent neuroaxonal dystrophy associated with autophagosome accumulation. Autophagy 2020. [Google Scholar] [CrossRef]
- Zivony-Elboum, Y.; Westbroek, W.; Kfir, N.; Savitzki, D.; Shoval, Y.; Bloom, A.; Rod, R.; Khayat, M.; Gross, B.; Samri, W.; et al. A Founder mutation in Vps37A causes autosomal recessive complex hereditary spastic paraparesis. J. Med. Genet. 2012, 49, 462–472. [Google Scholar] [CrossRef]
- Takahashi, Y.; Liang, X.; Hattori, T.; Tang, Z.; He, H.; Chen, H.; Liu, X.; Abraham, T.; Imamura-Kawasawa, Y.; Buchkovich, N.J.; et al. VPS37A directs ESCRT recruitment for phagophore closure. J. Cell Biol. 2019, 218, 3336–3354. [Google Scholar] [CrossRef] [Green Version]
- Marchionni, E.; Méneret, A.; Keren, B.; Melki, J.; Denier, C.; Durr, A.; Apartis, E.; Boespflug-Tanguy, O.; Mochel, F. KIF1C variants are associated with hypomyelination, ataxia, tremor, and dystonia in fraternal twins. Tremor Hyperkinetic Mov. 2019, 9. [Google Scholar] [CrossRef]
- Duchesne, A.; Vaiman, A.; Frah, M.; Floriot, S.; Legoueix-Rodriguez, S.; Desmazières, A.; Fritz, S.; Beauvallet, C.; Albaric, O.; Venot, E.; et al. Progressive ataxia of charolais cattle highlights a role of KIF1C in sustainable myelination. PLOS Genet. 2018, 14, e1007550. [Google Scholar] [CrossRef] [PubMed]
- Dorner, C.; Ciossek, T.; Müller, S.; Møller, N.P.H.; Ullrich, A.; Lammers, R. Characterization of KIF1C, a new kinesin-like protein involved in vesicle transport from the golgi apparatus to the endoplasmic reticulum *. J. Biol. Chem. 1998, 273, 20267–20275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spang, N.; Feldmann, A.; Huesmann, H.; Bekbulat, F.; Schmitt, V.; Hiebel, C.; Koziollek-Drechsler, I.; Clement, A.M.; Moosmann, B.; Jung, J.; et al. RAB3GAP1 and RAB3GAP2 modulate basal and rapamycin-induced autophagy. Autophagy 2014, 10, 2297–2309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takáts, S.; Lévay, L.; Boda, A.; Tóth, S.; Simon-Vecsei, Z.; Rubics, A.; Varga, Á.; Lippai, M.; Lőrincz, P.; Glatz, G.; et al. The warburg micro syndrome-associated Rab3GAP-Rab18 module promotes autolysosome maturation through the Vps34 complex, I. FEBS J. 2021, 288, 190–211. [Google Scholar] [CrossRef]
- Estrada-Cuzcano, A.; Martin, S.; Chamova, T.; Synofzik, M.; Timmann, D.; Holemans, T.; Andreeva, A.; Reichbauer, J.; De Rycke, R.; Chang, D.-I.; et al. Loss-of-function mutations in the ATP13A2/ PARK9 gene cause complicated hereditary spastic paraplegia (SPG78). Brain 2017, 140, 287–305. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Tan, J.; Chen, T.; Han, H.; Tian, R.; Tan, Y.; Wu, Y.; Cui, J.; Chen, F.; Li, J.; et al. ATP13A2 facilitates HDAC6 recruitment to lysosome to promote autophagosome–lysosome fusion. J. Cell Biol. 2019, 218, 267–284. [Google Scholar] [CrossRef] [PubMed]
- Nan, H.; Ichinose, Y.; Tanaka, M.; Koh, K.; Ishiura, H.; Mitsui, J.; Mizukami, H.; Morimoto, M.; Hamada, S.; Ohtsuka, T.; et al. UBAP1 mutations cause juvenile-onset hereditary spastic paraplegias (SPG80) and impair UBAP1 targeting to endosomes. J. Hum. Genet. 2019, 64, 1055–1065. [Google Scholar] [CrossRef]
- Lin, X.; Su, H.-Z.; Dong, E.-L.; Lin, X.-H.; Zhao, M.; Yang, C.; Wang, C.; Wang, J.; Chen, Y.-J.; Yu, H.; et al. Stop-gain mutations in UBAP1 cause pure autosomal-dominant spastic paraplegia. Brain J. Neurol. 2019, 142, 2238–2252. [Google Scholar] [CrossRef] [Green Version]
- de Bot, S.T.; Schelhaas, H.J.; Kamsteeg, E.-J.; van de Warrenburg, B.P.C. Hereditary spastic paraplegia caused by a mutation in the VCP gene. Brain 2012, 135, e223. [Google Scholar] [CrossRef]
- Nakamura, T.; Kawarabayashi, T.; Koh, K.; Takiyama, Y.; Ikeda, Y.; Shoji, M. Spastic paraplegia with paget’s disease of bone due to a VCP gene mutation. Intern. Med. Tokyo Jpn. 2021, 60, 141–144. [Google Scholar] [CrossRef]
- Clemen, C.S.; Tangavelou, K.; Strucksberg, K.-H.; Just, S.; Gaertner, L.; Regus-Leidig, H.; Stumpf, M.; Reimann, J.; Coras, R.; Morgan, R.O.; et al. Strumpellin is a novel valosin-containing protein binding partner linking hereditary spastic paraplegia to protein aggregation diseases. Brain J. Neurol. 2010, 133, 2920–2941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tresse, E.; Salomons, F.A.; Vesa, J.; Bott, L.C.; Kimonis, V.; Yao, T.-P.; Dantuma, N.P.; Taylor, J.P. VCP/P97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy 2010, 6, 217–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hausman-Kedem, M.; Ben-Shachar, S.; Menascu, S.; Geva, K.; Sagie, L.; Fattal-Valevski, A. VPS53 gene is associated with a new phenotype of complicated hereditary spastic paraparesis. Neurogenetics 2019, 20, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, M.A.; Feely, S.M.; Speziani, F.; Strickland, A.V.; Danzi, M.; Bacon, C.; Lee, Y.; Chou, T.-F.; Blanton, S.H.; Weihl, C.C.; et al. A novel mutation in VCP causes charcot–marie–tooth type 2 disease. Brain 2014, 137, 2897–2902. [Google Scholar] [CrossRef] [PubMed]
- Siddique, N.; Siddique, T. Amyotrophic Lateral Sclerosis Overview. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2001. [Google Scholar]
- Kakizuka, A. Roles of VCP in human neurodegenerative disorders. Biochem. Soc. Trans. 2008, 36, 105–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raiborg, C.; Stenmark, H. The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature 2009, 458, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Christ, L.; Raiborg, C.; Wenzel, E.M.; Campsteijn, C.; Stenmark, H. Cellular functions and molecular mechanisms of the ESCRT membrane-scission machinery. Trends Biochem. Sci. 2017, 42, 42–56. [Google Scholar] [CrossRef]
- Remec Pavlin, M.; Hurley, J.H. The ESCRTs–converging on mechanism. J. Cell Sci. 2020, 133. [Google Scholar] [CrossRef]
- Guo, E.Z.; Xu, Z. Distinct Mechanisms of recognizing endosomal sorting complex required for transport III (ESCRT-III) protein IST1 by different Microtubule Interacting and Trafficking (MIT) domains. J. Biol. Chem. 2015, 290, 8396–8408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henne, W.M.; Buchkovich, N.J.; Emr, S.D. The ESCRT pathway. Dev. Cell 2011, 21, 77–91. [Google Scholar] [CrossRef] [Green Version]
- Hanson, P.I.; Shim, S.; Merrill, S.A. Cell biology of the ESCRT machinery. Curr. Opin. Cell Biol. 2009, 21, 568–574. [Google Scholar] [CrossRef]
- Adell, M.A.Y.; Vogel, G.F.; Pakdel, M.; Müller, M.; Lindner, H.; Hess, M.W.; Teis, D. Coordinated binding of Vps4 to ESCRT-III drives membrane neck constriction during MVB vesicle formation. J. Cell Biol. 2014, 205, 33–49. [Google Scholar] [CrossRef]
- Skibinski, G.; Parkinson, N.J.; Brown, J.M.; Chakrabarti, L.; Lloyd, S.L.; Hummerich, H.; Nielsen, J.E.; Hodges, J.R.; Spillantini, M.G.; Thusgaard, T.; et al. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat. Genet. 2005, 37, 806–808. [Google Scholar] [CrossRef]
- Parkinson, N.; Ince, P.G.; Smith, M.O.; Highley, R.; Skibinski, G.; Andersen, P.M.; Morrison, K.E.; Pall, H.S.; Hardiman, O.; Collinge, J.; et al. ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B). Neurology 2006, 67, 1074–1077. [Google Scholar] [CrossRef]
- Cox, L.E.; Ferraiuolo, L.; Goodall, E.F.; Heath, P.R.; Higginbottom, A.; Mortiboys, H.; Hollinger, H.C.; Hartley, J.A.; Brockington, A.; Burness, C.E.; et al. Mutations in CHMP2B in lower motor neuron predominant Amyotrophic Lateral Sclerosis (ALS). PLoS ONE 2010, 5, e9872. [Google Scholar] [CrossRef] [PubMed]
- Ugbode, C.; West, R.J.H. Lessons learned from CHMP2B, implications for frontotemporal dementia and amyotrophic lateral sclerosis. Neurobiol. Dis. 2021, 147, 105144. [Google Scholar] [CrossRef]
- Rodger, C.; Flex, E.; Allison, R.J.; Sanchis-Juan, A.; Hasenahuer, M.A.; Cecchetti, S.; French, C.E.; Edgar, J.R.; Carpentieri, G.; Ciolfi, A.; et al. De Novo VPS4A mutations cause multisystem disease with abnormal neurodevelopment. Am. J. Hum. Genet. 2020, 107, 1129–1148. [Google Scholar] [CrossRef] [PubMed]
- Allison, R.; Edgar, J.R.; Pearson, G.; Rizo, T.; Newton, T.; Günther, S.; Berner, F.; Hague, J.; Connell, J.W.; Winkler, J.; et al. Defects in ER–endosome contacts impact lysosome function in hereditary spastic paraplegia. J. Cell Biol. 2017, 216, 1337–1355. [Google Scholar] [CrossRef]
- Çobanoğlu, G.; Ozansoy, M.; Başak, A.N. Are alsin and spartin novel interaction partners? Biochem. Biophys. Res. Commun. 2012, 427, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Huotari, J.; Helenius, A. Endosome maturation: Endosome maturation. EMBO J. 2011, 30, 3481–3500. [Google Scholar] [CrossRef] [PubMed]
- Mao, F.; Li, Z.; Zhao, B.; Lin, P.; Liu, P.; Zhai, M.; Liu, Q.; Shao, C.; Sun, W.; Gong, Y. Identification and functional analysis of a SLC33A1: C.339T>G (p.Ser113Arg) variant in the original SPG42 family. Hum. Mutat. 2015, 36, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Lowery, J.W.; Rosen, V. The BMP pathway and its inhibitors in the skeleton. Physiol. Rev. 2018, 98, 2431–2452. [Google Scholar] [CrossRef] [Green Version]
- Gámez, B.; Rodriguez-Carballo, E.; Ventura, F. BMP signaling in telencephalic neural cell specification and maturation. Front. Cell. Neurosci. 2013, 7, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, J.; Zou, H. BMP signaling in axon regeneration. Curr. Opin. Neurobiol. 2014, 0, 127–134. [Google Scholar] [CrossRef] [Green Version]
- Kurth, I. Hereditary Sensory and Autonomic Neuropathy Type II. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2010. [Google Scholar]
- Makaraci, P.; Kim, K. Trans-golgi network-bound cargo traffic. Eur. J. Cell Biol. 2018, 97, 137–149. [Google Scholar] [CrossRef]
- Siniossoglou, S.; Pelham, H.R.B. Vps51p Links the VFT complex to the SNARE Tlg1p. J. Biol. Chem. 2002, 277, 48318–48324. [Google Scholar] [CrossRef] [Green Version]
- Schmitt-John, T. VPS54 and the wobbler mouse. Front. Neurosci. 2015, 9, 381. [Google Scholar] [CrossRef] [Green Version]
- Gershlick, D.C.; Ishida, M.; Jones, J.R.; Bellomo, A.; Bonifacino, J.S.; Everman, D.B. A neurodevelopmental disorder caused by mutations in the VPS51 subunit of the GARP and EARP complexes. Hum. Mol. Genet. 2019, 28, 1548–1560. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Sirkis, D.W.; Schekman, R. Protein sorting at the trans-golgi network. Annu. Rev. Cell Dev. Biol. 2014, 30, 169–206. [Google Scholar] [CrossRef]
- Gadbery, J.E.; Abraham, A.; Needle, C.D.; Moth, C.; Sheehan, J.; Capra, J.A.; Jackson, L.P. Integrating structural and evolutionary data to interpret variation and pathogenicity in adapter protein complex 4. Protein Sci. Publ. Protein Soc. 2020, 29, 1535–1549. [Google Scholar] [CrossRef]
- Ebrahimi-Fakhari, D.; Cheng, C.; Dies, K.; Diplock, A.; Pier, D.B.; Ryan, C.S.; Lanpher, B.C.; Hirst, J.; Chung, W.K.; Sahin, M.; et al. Clinical and genetic characterization of AP4B1 -associated SPG47. Am. J. Med. Genet. A 2018, 176, 311–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abou Jamra, R.; Philippe, O.; Raas-Rothschild, A.; Eck, S.H.; Graf, E.; Buchert, R.; Borck, G.; Ekici, A.; Brockschmidt, F.F.; Nöthen, M.M.; et al. Adaptor protein complex 4 deficiency causes severe autosomal-recessive intellectual disability, progressive spastic paraplegia, shy character, and short stature. Am. J. Hum. Genet. 2011, 88, 788–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, X.-F.; Bousfiha, A.; Rouissi, A.; Itan, Y.; Abhyankar, A.; Bryant, V.; Okada, S.; Ailal, F.; Bustamante, J.; Casanova, J.-L.; et al. A Novel homozygous p.R1105X mutation of the AP4E1 gene in twins with hereditary spastic paraplegia and mycobacterial disease. PLoS ONE 2013, 8, e58286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tessa, A.; Battini, R.; Rubegni, A.; Storti, E.; Marini, C.; Galatolo, D.; Pasquariello, R.; Santorelli, F.M. Identification of mutations in AP4S1/ SPG52 through next generation sequencing in three families. Eur. J. Neurol. 2016, 23, 1580–1587. [Google Scholar] [CrossRef]
- Bettencourt, C.; Salpietro, V.; Efthymiou, S.; Chelban, V.; Hughes, D.; Pittman, A.M.; Federoff, M.; Bourinaris, T.; Spilioti, M.; Deretzi, G.; et al. Genotype-phenotype correlations and expansion of the molecular spectrum of AP4M1-related hereditary spastic paraplegia. Orphanet J. Rare Dis. 2017, 12. [Google Scholar] [CrossRef] [Green Version]
- Raiborg, C.; Wenzel, E.M.; Stenmark, H. ER–endosome contact sites: Molecular compositions and functions. EMBO J. 2015, 34, 1848–1858. [Google Scholar] [CrossRef] [Green Version]
- Blackstone, C. Cellular pathways of hereditary spastic paraplegia. Annu. Rev. Neurosci. 2012, 35, 25–47. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.A.; Blackstone, C. ER Morphology and endo-lysosomal crosstalk: Functions and disease implications. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158544. [Google Scholar] [CrossRef]
- Blackstone, C. Converging cellular themes for the hereditary spastic paraplegias. Curr. Opin. Neurobiol. 2018, 51, 139–146. [Google Scholar] [CrossRef]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Overhoff, M.; De Bruyckere, E.; Kononenko, N.L. Mechanisms of neuronal survival safeguarded by endocytosis and autophagy. J. Neurochem. 2020, jnc.15194. [Google Scholar] [CrossRef]
- Ravussin, A.; Brech, A.; Tooze, S.A.; Stenmark, H. The phosphatidylinositol 3-phosphate-binding protein SNX4 controls ATG9A recycling and autophagy. J. Cell Sci. 2021, 134, jcs250670. [Google Scholar] [CrossRef]
- Tamura, H.; Shibata, M.; Koike, M.; Sasaki, M.; Uchiyama, Y. Atg9A protein, an autophagy-related membrane protein, is localized in the neurons of mouse brains. J. Histochem. Cytochem. 2010, 58, 443–453. [Google Scholar] [CrossRef] [Green Version]
- Pennings, M.; Schouten, M.I.; van Gaalen, J.; Meijer, R.P.P.; de Bot, S.T.; Kriek, M.; Saris, C.G.J.; van den Berg, L.H.; van Es, M.A.; Zuidgeest, D.M.H.; et al. KIF1A variants are a frequent cause of autosomal dominant hereditary spastic paraplegia. Eur. J. Hum. Genet. 2020, 28, 40–49. [Google Scholar] [CrossRef]
- Klebe, S.; Lossos, A.; Azzedine, H.; Mundwiller, E.; Sheffer, R.; Gaussen, M.; Marelli, C.; Nawara, M.; Carpentier, W.; Meyer, V.; et al. KIF1A missense mutations in SPG30, an autosomal recessive spastic paraplegia: Distinct phenotypes according to the nature of the mutations. Eur. J. Hum. Genet. EJHG 2012, 20, 645–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabrych, D.R.; Lau, V.Z.; Niwa, S.; Silverman, M.A. Going too far is the same as falling short†: Kinesin-3 family members in hereditary spastic paraplegia. Front. Cell. Neurosci. 2019, 13, 419. [Google Scholar] [CrossRef] [PubMed]
- Lőrincz, P.; Juhász, G. Autophagosome-Lysosome Fusion. J. Mol. Biol. 2020, 432, 2462–2482. [Google Scholar] [CrossRef] [PubMed]
- Guardia, C.M.; Farías, G.G.; Jia, R.; Pu, J.; Bonifacino, J.S. BORC functions upstream of kinesins 1 and 3 to coordinate regional movement of lysosomes along different microtubule tracks. Cell Rep. 2016, 17, 1950–1961. [Google Scholar] [CrossRef] [Green Version]
- Kanai, Y.; Okada, Y.; Tanaka, Y.; Harada, A.; Terada, S.; Hirokawa, N. KIF5C, a novel neuronal kinesin enriched in motor neurons. J. Neurosci. 2000, 20, 6374–6384. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, C.M.P.; Groth-Pedersen, L.; Høyer-Hansen, M.; Kirkegaard, T.; Andersen, J.S. Depletion of kinesin 5B affects lysosomal distribution and stability and induces peri-nuclear accumulation of autophagosomes in cancer cells. PLoS ONE 2009, 4, e4424. [Google Scholar] [CrossRef] [Green Version]
- Du, W.; Su, Q.P.; Chen, Y.; Zhu, Y.; Jiang, D.; Rong, Y.; Zhang, S.; Zhang, Y.; Ren, H.; Zhang, C.; et al. Kinesin 1 drives autolysosome tubulation. Dev. Cell 2016, 37, 326–336. [Google Scholar] [CrossRef]
- Reid, E.; Kloos, M.; Ashley-Koch, A.; Hughes, L.; Bevan, S.; Svenson, I.K.; Graham, F.L.; Gaskell, P.C.; Dearlove, A.; Pericak-Vance, M.A.; et al. A Kinesin Heavy Chain (KIF5A) mutation in hereditary spastic paraplegia (SPG10). Am. J. Hum. Genet. 2002, 71, 1189–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crimella, C.; Baschirotto, C.; Arnoldi, A.; Tonelli, A.; Tenderini, E.; Airoldi, G.; Martinuzzi, A.; Trabacca, A.; Losito, L.; Scarlato, M.; et al. Mutations in the motor and stalk domains of KIF5A in spastic paraplegia type 10 and in axonal charcot-marie-tooth type 2. Clin. Genet. 2012, 82, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, A.; Kenna, K.P.; Renton, A.E.; Ticozzi, N.; Faghri, F.; Chia, R.; Dominov, J.A.; Kenna, B.J.; Nalls, M.A.; Keagle, P.; et al. Genome-wide analyses identify KIF5A as a novel ALS gene. Neuron 2018, 97, 1268–1283.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, J.; Liu, X.; Tang, L.; Zhao, C.; He, J.; Fan, D. Whole-exome sequencing identified novel KIF5A mutations in Chinese patients with amyotrophic lateral sclerosis and charcot-marie-tooth type 2. J. Neurol. Neurosurg. Psychiatry 2020, 91, 326–328. [Google Scholar] [CrossRef]
- Wang, L.; Brown, A. A hereditary spastic paraplegia mutation in kinesin-1A/KIF5A disrupts neurofilament transport. Mol. Neurodegener. 2010, 5, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karle, K.N.; Möckel, D.; Reid, E.; Schöls, L. Axonal transport deficit in a KIF5A–/–mouse model. Neurogenetics 2012, 13, 169–179. [Google Scholar] [CrossRef] [Green Version]
- Campbell, P.D.; Shen, K.; Sapio, M.R.; Glenn, T.D.; Talbot, W.S.; Marlow, F.L. Unique function of kinesin Kif5A in localization of mitochondria in axons. J. Neurosci. 2014, 34, 14717–14732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamal, A.; Stokin, G.B.; Yang, Z.; Xia, C.-H.; Goldstein, L.S.B. Axonal transport of amyloid precursor protein is mediated by direct binding to the kinesin light chain subunit of kinesin-I. Neuron 2000, 28, 449–459. [Google Scholar] [CrossRef] [Green Version]
- Jia, R.; Guardia, C.M.; Pu, J.; Chen, Y.; Bonifacino, J.S. BORC coordinates encounter and fusion of lysosomes with autophagosomes. Autophagy 2017, 13, 1648–1663. [Google Scholar] [CrossRef] [Green Version]
- Itakura, E.; Kishi-Itakura, C.; Mizushima, N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 2012, 151, 1256–1269. [Google Scholar] [CrossRef] [Green Version]
- Edvardson, S.; Gerhard, F.; Jalas, C.; Lachmann, J.; Golan, D.; Saada, A.; Shaag, A.; Ungermann, C.; Elpeleg, O. Hypomyelination and developmental delay associated with VPS11 mutation in ashkenazi-jewish patients. J. Med. Genet. 2015, 52, 749–753. [Google Scholar] [CrossRef]
- Hörtnagel, K.; Krägeloh-Mann, I.; Bornemann, A.; Döcker, M.; Biskup, S.; Mayrhofer, H.; Battke, F.; du Bois, G.; Harzer, K. The second report of a new hypomyelinating disease due to a defect in the VPS11 gene discloses a massive lysosomal involvement. J. Inherit. Metab. Dis. 2016, 39, 849–857. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Lachance, V.; Schaffner, A.; Li, X.; Fedick, A.; Kaye, L.E.; Liao, J.; Rosenfeld, J.; Yachelevich, N.; Chu, M.-L.; et al. A founder mutation in VPS11 causes an autosomal recessive leukoencephalopathy linked to autophagic defects. PLOS Genet. 2016, 12, e1005848. [Google Scholar] [CrossRef] [Green Version]
- Van der Beek, J.; Jonker, C.; van der Welle, R.; Liv, N.; Klumperman, J. CORVET, CHEVI and HOPS–multisubunit tethers of the endo-lysosomal system in health and disease. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monfrini, E.; Zech, M.; Steel, D.; Kurian, M.A.; Winkelmann, J.; Di Fonzo, A. HOPS-associated neurological disorders (HOPSANDs): Linking endolysosomal dysfunction to the pathogenesis of dystonia. Brain 2021. [Google Scholar] [CrossRef]
- Cai, X.; Chen, X.; Wu, S.; Liu, W.; Zhang, X.; Zhang, D.; He, S.; Wang, B.; Zhang, M.; Zhang, Y.; et al. Homozygous mutation of VPS16 gene is responsible for an autosomal recessive adolescent-onset primary dystonia. Sci. Rep. 2016, 6, 25834. [Google Scholar] [CrossRef] [Green Version]
- Sanderson, L.E.; Lanko, K.; Alsagob, M.; Almass, R.; Al-Ahmadi, N.; Najafi, M.; Al-Muhaizea, M.A.; Alzaidan, H.; AlDhalaan, H.; Perenthaler, E.; et al. Bi-allelic variants in HOPS complex subunit VPS41 cause cerebellar ataxia and abnormal membrane trafficking. Brain 2021, 144, 769–780. [Google Scholar] [CrossRef]
- Steel, D.; Zech, M.; Zhao, C.; Barwick, K.E.S.; Burke, D.; Demailly, D.; Kumar, K.R.; Zorzi, G.; Nardocci, N.; Kaiyrzhanov, R.; et al. Loss-of-function variants in HOPS complex genes VPS16 and VPS41 cause early onset dystonia associated with lysosomal abnormalities. Ann. Neurol. 2020, 88, 867–877. [Google Scholar] [CrossRef]
- Van der Welle, R.E.N.; Jobling, R.; Burns, C.; Sanza, P.; van der Beek, J.A.; Fasano, A.; Chen, L.; Zwartkruis, F.J.; Zwakenberg, S.; Griffin, E.F.; et al. Neurodegenerative VPS41 variants inhibit HOPS function and MTORC1-dependent TFEB/TFE3 regulation. EMBO Mol. Med. 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Fuchs-Telem, D.; Stewart, H.; Rapaport, D.; Nousbeck, J.; Gat, A.; Gini, M.; Lugassy, Y.; Emmert, S.; Eckl, K.; Hennies, H.C.; et al. CEDNIK syndrome results from loss-of-function mutations in SNAP29. Br. J. Dermatol. 2011, 164, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Sprecher, E.; Ishida-Yamamoto, A.; Mizrahi-Koren, M.; Rapaport, D.; Goldsher, D.; Indelman, M.; Topaz, O.; Chefetz, I.; Keren, H.; O’Brien, T.J.; et al. A mutation in SNAP29, coding for a SNARE protein involved in intracellular trafficking, causes a novel neurocutaneous syndrome characterized by cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma. Am. J. Hum. Genet. 2005, 77, 242–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poojary, S.; Shah, K.S.; Bhalala, K.B.; Hegde, A.U. CEDNIK syndrome in an indian patient with a novel mutation of the SNAP29 gene. Pediatr. Dermatol. 2019, 36, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Stadel, D.; Millarte, V.; Tillmann, K.D.; Huber, J.; Tamin-Yecheskel, B.-C.; Akutsu, M.; Demishtein, A.; Ben-Zeev, B.; Anikster, Y.; Perez, F.; et al. TECPR2 cooperates with LC3C to regulate COPII-dependent ER export. Mol. Cell 2015, 60, 89–104. [Google Scholar] [CrossRef] [Green Version]
- Heimer, G.; Oz-Levi, D.; Eyal, E.; Edvardson, S.; Nissenkorn, A.; Ruzzo, E.K.; Szeinberg, A.; Maayan, C.; Mai-Zahav, M.; Efrati, O.; et al. TECPR2 mutations cause a new subtype of familial dysautonomia like hereditary sensory autonomic neuropathy with intellectual disability. Eur. J. Paediatr. Neurol. 2016, 20, 69–79. [Google Scholar] [CrossRef]
- Oz-Levi, D.; Ben-Zeev, B.; Ruzzo, E.K.; Hitomi, Y.; Gelman, A.; Pelak, K.; Anikster, Y.; Reznik-Wolf, H.; Bar-Joseph, I.; Olender, T.; et al. Mutation in TECPR2 reveals a role for autophagy in hereditary spastic paraparesis. Am. J. Hum. Genet. 2012, 91, 1065–1072. [Google Scholar] [CrossRef] [Green Version]
- Van Veen, S.; Sørensen, D.M.; Holemans, T.; Holen, H.W.; Palmgren, M.G.; Vangheluwe, P. Cellular function and pathological role of ATP13A2 and related P-type transport ATPases in Parkinson’s disease and other neurological disorders. Front. Mol. Neurosci. 2014, 7. [Google Scholar] [CrossRef] [Green Version]
- Odake, Y.; Koh, K.; Takiyama, Y.; Ishiura, H.; Tsuji, S.; Yamada, M.; Yoshita, M. Identification of a novel mutation in ATP13A2 associated with a complicated form of hereditary spastic paraplegia. Neurol. Genet. 2020, 6. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-S.; Blair, N.F.; Sue, C.M. The role of ATP13A2 in Parkinson’s disease: Clinical phenotypes and molecular mechanisms: ATP13A2 in Parkinson’s disease. Mov. Disord. 2015, 30, 770–779. [Google Scholar] [CrossRef] [PubMed]
- Sridhar, S.; Patel, B.; Aphkhazava, D.; Macian, F.; Santambrogio, L.; Shields, D.; Cuervo, A.M. The Lipid Kinase PI4KIIIβ preserves lysosomal identity. EMBO J. 2013, 32, 324–339. [Google Scholar] [CrossRef]
- Chen, Y.; Yu, L. Autophagic lysosome reformation. Exp. Cell Res. 2013, 319, 142–146. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yu, L. Recent progress in autophagic lysosome reformation. Traffic 2017, 18, 358–361. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.; Liu, M.; Ma, L.; Du, W.; Zhang, H.; Tian, Y.; Cao, Z.; Li, Y.; Ren, H.; Zhang, C.; et al. Clathrin and phosphatidylinositol-4,5-bisphosphate regulate autophagic lysosome reformation. Nat. Cell Biol. 2012, 14, 924–934. [Google Scholar] [CrossRef]
- Denora, P.S.; Smets, K.; Zolfanelli, F.; Ceuterick-de Groote, C.; Casali, C.; Deconinck, T.; Sieben, A.; Gonzales, M.; Zuchner, S.; Darios, F.; et al. Motor Neuron degeneration in spastic paraplegia 11 mimics amyotrophic lateral sclerosis lesions. Brain 2016. [Google Scholar] [CrossRef]
- Orlacchio, A.; Babalini, C.; Borreca, A.; Patrono, C.; Massa, R.; Basaran, S.; Munhoz, R.P.; Rogaeva, E.A.; St George-Hyslop, P.H.; Bernardi, G.; et al. SPATACSIN mutations cause autosomal recessive juvenile amyotrophic lateral sclerosis. Brain 2010, 133, 591–598. [Google Scholar] [CrossRef] [Green Version]
- Montecchiani, C.; Pedace, L.; Lo Giudice, T.; Casella, A.; Mearini, M.; Gaudiello, F.; Pedroso, J.L.; Terracciano, C.; Caltagirone, C.; Massa, R.; et al. ALS5/SPG11/ KIAA1840 mutations cause autosomal recessive axonal charcot–marie–tooth disease. Brain 2016, 139, 73–85. [Google Scholar] [CrossRef] [Green Version]
- Hanein, S.; Martin, E.; Boukhris, A.; Byrne, P.; Goizet, C.; Hamri, A.; Benomar, A.; Lossos, A.; Denora, P.; Fernandez, J.; et al. Identification of the SPG15 gene, encoding spastizin, as a frequent cause of complicated autosomal-recessive spastic paraplegia, including kjellin syndrome. Am. J. Hum. Genet. 2008, 82, 992–1002. [Google Scholar] [CrossRef] [Green Version]
- Słabicki, M.; Theis, M.; Krastev, D.B.; Samsonov, S.; Mundwiller, E.; Junqueira, M.; Paszkowski-Rogacz, M.; Teyra, J.; Heninger, A.-K.; Poser, I.; et al. A genome-scale DNA repair RNAi screen identifies SPG48 as a novel gene associated with hereditary spastic paraplegia. PLoS Biol. 2010, 8, e1000408. [Google Scholar] [CrossRef] [PubMed]
- Murmu, R.P.; Martin, E.; Rastetter, A.; Esteves, T.; Muriel, M.-P.; El Hachimi, K.H.; Denora, P.S.; Dauphin, A.; Fernandez, J.C.; Duyckaerts, C.; et al. Cellular distribution and subcellular localization of spatacsin and spastizin, two proteins involved in hereditary spastic paraplegia. Mol. Cell. Neurosci. 2011, 47, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Hirst, J.; Borner, G.H.H.; Edgar, J.; Hein, M.Y.; Mann, M.; Buchholz, F.; Antrobus, R.; Robinson, M.S. Interaction between AP-5 and the hereditary spastic paraplegia proteins SPG11 and SPG15. Mol. Biol. Cell 2013, 24, 2558–2569. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Brangulí, F.; Mishra, H.K.; Prots, I.; Havlicek, S.; Kohl, Z.; Saul, D.; Rummel, C.; Dorca-Arevalo, J.; Regensburger, M.; Graef, D.; et al. Dysfunction of spatacsin leads to axonal pathology in SPG11-linked hereditary spastic paraplegia. Hum. Mol. Genet. 2014, 23, 4859–4874. [Google Scholar] [CrossRef]
- Pozner, T.; Regensburger, M.; Engelhorn, T.; Winkler, J.; Winner, B. Janus-faced spatacsin (SPG11): Involvement in neurodevelopment and multisystem neurodegeneration. Brain 2020, 143, 2369–2379. [Google Scholar] [CrossRef] [PubMed]
- Khundadze, M.; Kollmann, K.; Koch, N.; Biskup, C.; Nietzsche, S.; Zimmer, G.; Hennings, J.C.; Huebner, A.K.; Symmank, J.; Jahic, A.; et al. A hereditary spastic paraplegia mouse model supports a role of ZFYVE26/SPASTIZIN for the endolysosomal system. PLoS Genet. 2013, 9, e1003988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Branchu, J.; Boutry, M.; Sourd, L.; Depp, M.; Leone, C.; Corriger, A.; Vallucci, M.; Esteves, T.; Matusiak, R.; Dumont, M.; et al. Loss of spatacsin function alters lysosomal lipid clearance leading to upper and lower motor neuron degeneration. Neurobiol. Dis. 2017, 102, 21–37. [Google Scholar] [CrossRef]
- Stavoe, A.K.H.; Holzbaur, E.L.F. Autophagy in neurons. Annu. Rev. Cell Dev. Biol. 2019, 35, 477–500. [Google Scholar] [CrossRef]
- Tooze, S.A.; Abada, A.; Elazar, Z. Endocytosis and autophagy: Exploitation or cooperation? Cold Spring Harb. Perspect. Biol. 2014, 6. [Google Scholar] [CrossRef]
- Fader, C.M.; Colombo, M.I. Autophagy and multivesicular bodies: Two closely related partners. Cell Death Differ. 2009, 16, 70–78. [Google Scholar] [CrossRef] [Green Version]
- Ravikumar, B.; Imarisio, S.; Sarkar, S.; O’Kane, C.J.; Rubinsztein, D.C. Rab5 modulates mutant huntingtin aggregation/toxicity in cell and fly models via macroautophagy. J. Cell Sci. 2008, 121, 1649–1660. [Google Scholar] [CrossRef] [Green Version]
- Szatmári, Z.; Kis, V.; Lippai, M.; Hegedűs, K.; Faragó, T.; Lőrincz, P.; Tanaka, T.; Juhász, G.; Sass, M. Rab11 facilitates cross-talk between autophagy and endosomal pathway through regulation of hook localization. Mol. Biol. Cell 2014, 25, 522–531. [Google Scholar] [CrossRef]
- De Souza, P.V.S.; de Rezende Pinto, W.B.V.; de Rezende Batistella, G.N.; Bortholin, T.; Oliveira, A.S.B. Hereditary spastic paraplegia: Clinical and genetic hallmarks. Cerebellum 2017, 16, 525–551. [Google Scholar] [CrossRef] [PubMed]
- Handley, M.T.; Morris-Rosendahl, D.J.; Brown, S.; Macdonald, F.; Hardy, C.; Bem, D.; Carpanini, S.M.; Borck, G.; Martorell, L.; Izzi, C.; et al. Mutation spectrum in RAB3GAP1, RAB3GAP2, and RAB18 and genotype–phenotype correlations in warburg micro syndrome and martsolf syndrome. Hum. Mutat. 2013, 34, 686–696. [Google Scholar] [CrossRef] [PubMed]
- Handley, M.T.; Aligianis, I.A. RAB3GAP1, RAB3GAP2 and RAB18: Disease genes in micro and martsolf syndromes. Biochem. Soc. Trans. 2012, 40, 1394–1397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, B.R.; Maddison, D.C.; Smith, G.A.; Peters, O.M. Autophagic and endo-lysosomal dysfunction in neurodegenerative disease. Mol. Brain 2019, 12, 100. [Google Scholar] [CrossRef] [PubMed]
- Whyte, L.S.; Lau, A.A.; Hemsley, K.M.; Hopwood, J.J.; Sargeant, T.J. Endo-lysosomal and autophagic dysfunction: A driving factor in Alzheimer’s disease? J. Neurochem. 2017, 140, 703–717. [Google Scholar] [CrossRef] [Green Version]
- Sjödin, S.; Brinkmalm, G.; Öhrfelt, A.; Parnetti, L.; Paciotti, S.; Hansson, O.; Hardy, J.; Blennow, K.; Zetterberg, H.; Brinkmalm, A. Endo-lysosomal proteins and ubiquitin CSF concentrations in Alzheimer’s and Parkinson’s disease. Alzheimers Res. Ther. 2019, 11, 1–16. [Google Scholar] [CrossRef]
- Miranda, A.M.; Di Paolo, G. Endolysosomal dysfunction and exosome secretion: Implications for neurodegenerative disorders. Cell Stress 2018, 2, 115–118. [Google Scholar] [CrossRef]
- Vidyadhara, D.J.; Lee, J.E.; Chandra, S.S. Role of the endolysosomal system in Parkinson’s disease. J. Neurochem. 2019, 150, 487–506. [Google Scholar] [CrossRef] [Green Version]
- Giovedì, S.; Ravanelli, M.M.; Parisi, B.; Bettegazzi, B.; Guarnieri, F.C. Dysfunctional autophagy and endolysosomal system in neurodegenerative diseases: Relevance and therapeutic options. Front. Cell. Neurosci. 2020, 14. [Google Scholar] [CrossRef] [PubMed]
- Yarwood, R.; Hellicar, J.; Woodman, P.G.; Lowe, M. Membrane trafficking in health and disease. Dis. Model. Mech. 2020, 13. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Kang, J.-H.; Lee, S. Autophagy in neurodegenerative diseases: A hunter for aggregates. Int. J. Mol. Sci. 2020, 21, 3369. [Google Scholar] [CrossRef] [PubMed]
- Otomo, A.; Pan, L.; Hadano, S. Dysregulation of the autophagy-endolysosomal system in amyotrophic lateral sclerosis and related motor neuron diseases. Neurol. Res. Int. 2012, 2012. [Google Scholar] [CrossRef]
- Rudolf, R.; Khan, M.M.; Wild, F.; Hashemolhosseini, S. The impact of autophagy on peripheral synapses in health and disease. Front. Biosci. Landmark Ed. 2016, 21, 1474–1487. [Google Scholar] [CrossRef] [Green Version]
- Tarrade, A.; Fassier, C.; Courageot, S.; Charvin, D.; Vitte, J.; Peris, L.; Thorel, A.; Mouisel, E.; Fonknechten, N.; Roblot, N.; et al. A mutation of spastin is responsible for swellings and impairment of transport in a region of axon characterized by changes in microtubule composition. Hum. Mol. Genet. 2006, 15, 3544–3558. [Google Scholar] [CrossRef] [Green Version]
- De Pace, R.; Skirzewski, M.; Damme, M.; Mattera, R.; Mercurio, J.; Foster, A.M.; Cuitino, L.; Jarnik, M.; Hoffmann, V.; Morris, H.D.; et al. Altered distribution of ATG9A and accumulation of axonal aggregates in neurons from a mouse model of AP-4 deficiency syndrome. PLOS Genet. 2018, 14, e1007363. [Google Scholar] [CrossRef] [Green Version]
- Denton, K.R.; Xu, C.; Shah, H.; Li, X.-J. Modeling axonal defects in hereditary spastic paraplegia with human pluripotent stem cells. Front. Biol. 2016, 11, 339–354. [Google Scholar] [CrossRef] [Green Version]
- Butler, R.; Wood, J.D.; Landers, J.A.; Cunliffe, V.T. Genetic and chemical modulation of spastin-dependent axon outgrowth in zebrafish embryos indicates a role for impaired microtubule dynamics in hereditary spastic paraplegia. Dis. Model. Mech. 2010, 3, 743–751. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Hedera, P. Strumpellin and spartin, hereditary spastic paraplegia proteins, are binding partners. J. Exp. Neurosci. 2015, 9, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Sharma, J.; di Ronza, A.; Lotfi, P.; Sardiello, M. Lysosomes and brain health. Annu. Rev. Neurosci. 2018, 41, 255–276. [Google Scholar] [CrossRef]
- Darios, F.; Stevanin, G. Impairment of lysosome function and autophagy in rare neurodegenerative diseases. J. Mol. Biol. 2020, 432, 2714–2734. [Google Scholar] [CrossRef]
- Pérez-Brangulí, F.; Buchsbaum, I.Y.; Pozner, T.; Regensburger, M.; Fan, W.; Schray, A.; Börstler, T.; Mishra, H.; Gräf, D.; Kohl, Z.; et al. Human SPG11 cerebral organoids reveal cortical neurogenesis impairment. Hum. Mol. Genet. 2019, 28, 961–971. [Google Scholar] [CrossRef] [Green Version]
- Chiba, K.; Takahashi, H.; Chen, M.; Obinata, H.; Arai, S.; Hashimoto, K.; Oda, T.; McKenney, R.J.; Niwa, S. Disease-associated mutations hyperactivate KIF1A motility and anterograde axonal transport of synaptic vesicle precursors. Proc. Natl. Acad. Sci. USA 2019, 116, 18429–18434. [Google Scholar] [CrossRef] [Green Version]
- Cheon, C.K.; Lim, S.-H.; Kim, Y.-M.; Kim, D.; Lee, N.-Y.; Yoon, T.-S.; Kim, N.-S.; Kim, E.; Lee, J.-R. Autosomal dominant transmission of complicated hereditary spastic paraplegia due to a dominant negative mutation of KIF1A, SPG30 Gene. Sci. Rep. 2017, 7, 12527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Chen, Y.; Yang, M.; Xu, X.; Lin, Z.; Ma, J.; Chen, H.; Hu, Y.; Ma, Y.; Wang, X.; et al. A Rare KIF1A missense mutation enhances synaptic function and increases seizure activity. Front. Genet. 2020, 11, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breza, M.; Hirst, J.; Chelban, V.; Banneau, G.; Tissier, L.; Kol, B.; Bourinaris, T.; Ait-Said, S.; Péréon, Y.; Heinzmann, A.; et al. Expanding the spectrum of AP5Z1–related Hereditary Spastic Paraplegia (HSP-SPG48): A multi-center study on a rare disease. Mov. Dis. 2021, 36, 1034–1038. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toupenet Marchesi, L.; Leblanc, M.; Stevanin, G. Current Knowledge of Endolysosomal and Autophagy Defects in Hereditary Spastic Paraplegia. Cells 2021, 10, 1678. https://doi.org/10.3390/cells10071678
Toupenet Marchesi L, Leblanc M, Stevanin G. Current Knowledge of Endolysosomal and Autophagy Defects in Hereditary Spastic Paraplegia. Cells. 2021; 10(7):1678. https://doi.org/10.3390/cells10071678
Chicago/Turabian StyleToupenet Marchesi, Liriopé, Marion Leblanc, and Giovanni Stevanin. 2021. "Current Knowledge of Endolysosomal and Autophagy Defects in Hereditary Spastic Paraplegia" Cells 10, no. 7: 1678. https://doi.org/10.3390/cells10071678
APA StyleToupenet Marchesi, L., Leblanc, M., & Stevanin, G. (2021). Current Knowledge of Endolysosomal and Autophagy Defects in Hereditary Spastic Paraplegia. Cells, 10(7), 1678. https://doi.org/10.3390/cells10071678