
Neurotoxic Astrocytes Directly Converted from Sporadic and Familial ALS Patient Fibroblasts Reveal Signature Diversities and miR-146a Theragnostic Potential in Specific Subtypes

 , ,
, , 
Abstract
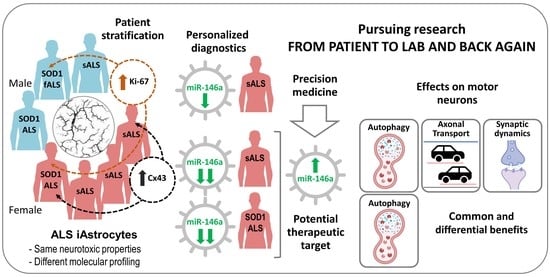
:
1. Introduction
2. Materials and Methods
2.1. Skin Fibroblast Isolation
2.2. Preparation of Retrovirus for Reprogramming
2.3. Conversion of ALS Patient Fibroblasts to iNPCs
2.4. Differentiation of iNPCs into iAstrocytes
2.5. Motor Neuron Differentiation from meSCs
2.6. Assessment of iAstrocyte Neurotoxicity on HB9/GFP-positive MNs
2.7. Immunofluorescence
2.8. Gene and miRNA Expression Profiling by RT-qPCR
2.9. Protein Analysis Using Western Blot
2.10. Isolation and Evaluation of Soluble miRNAs
2.11. Isolation and Characterization of sEVs
2.12. Upregulation of miR-146a in iAstrocytes
2.13. iAstrocytes and NSC-34 MN Co-cultures
2.14. Statistical Analysis
3. Results
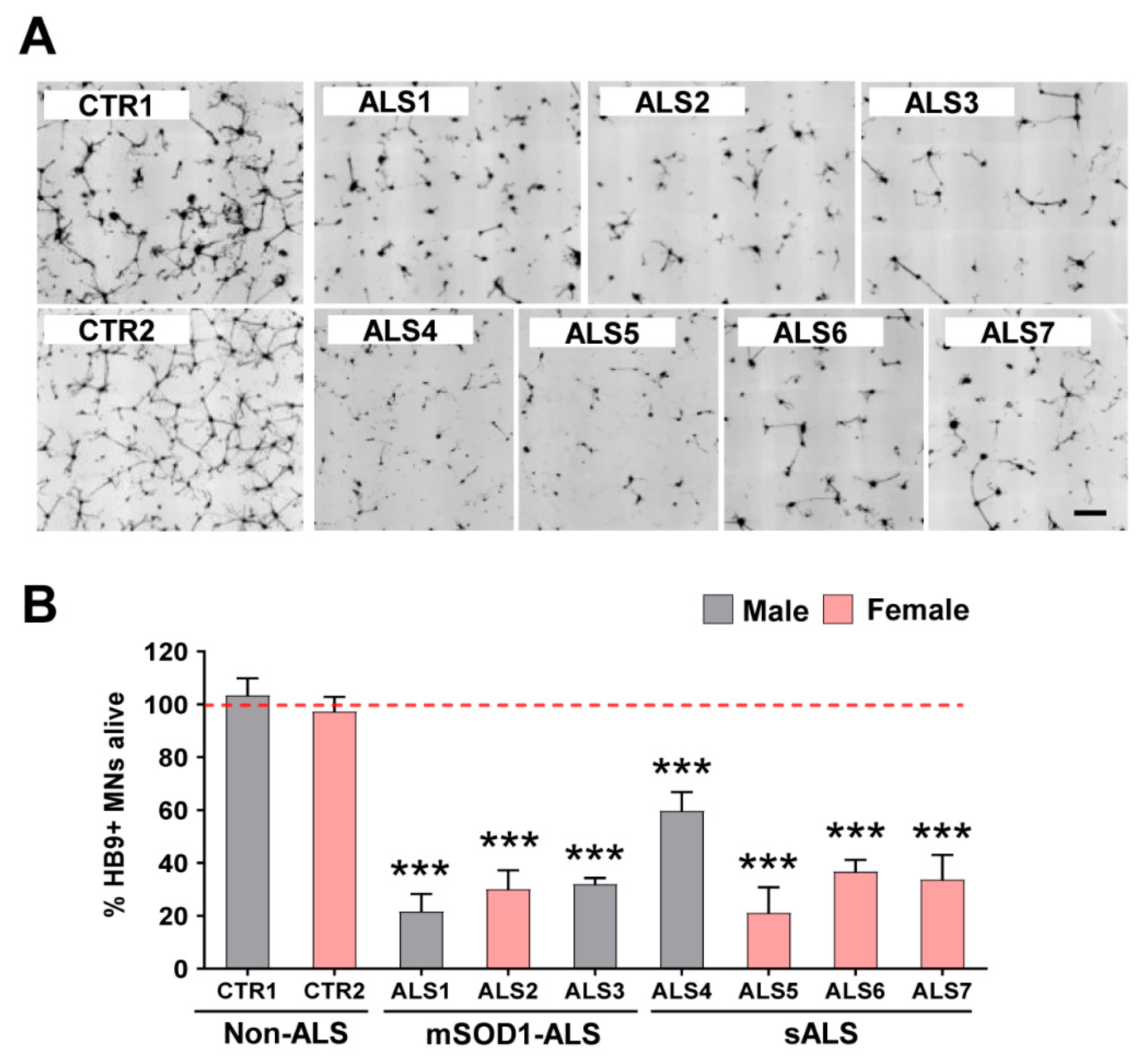
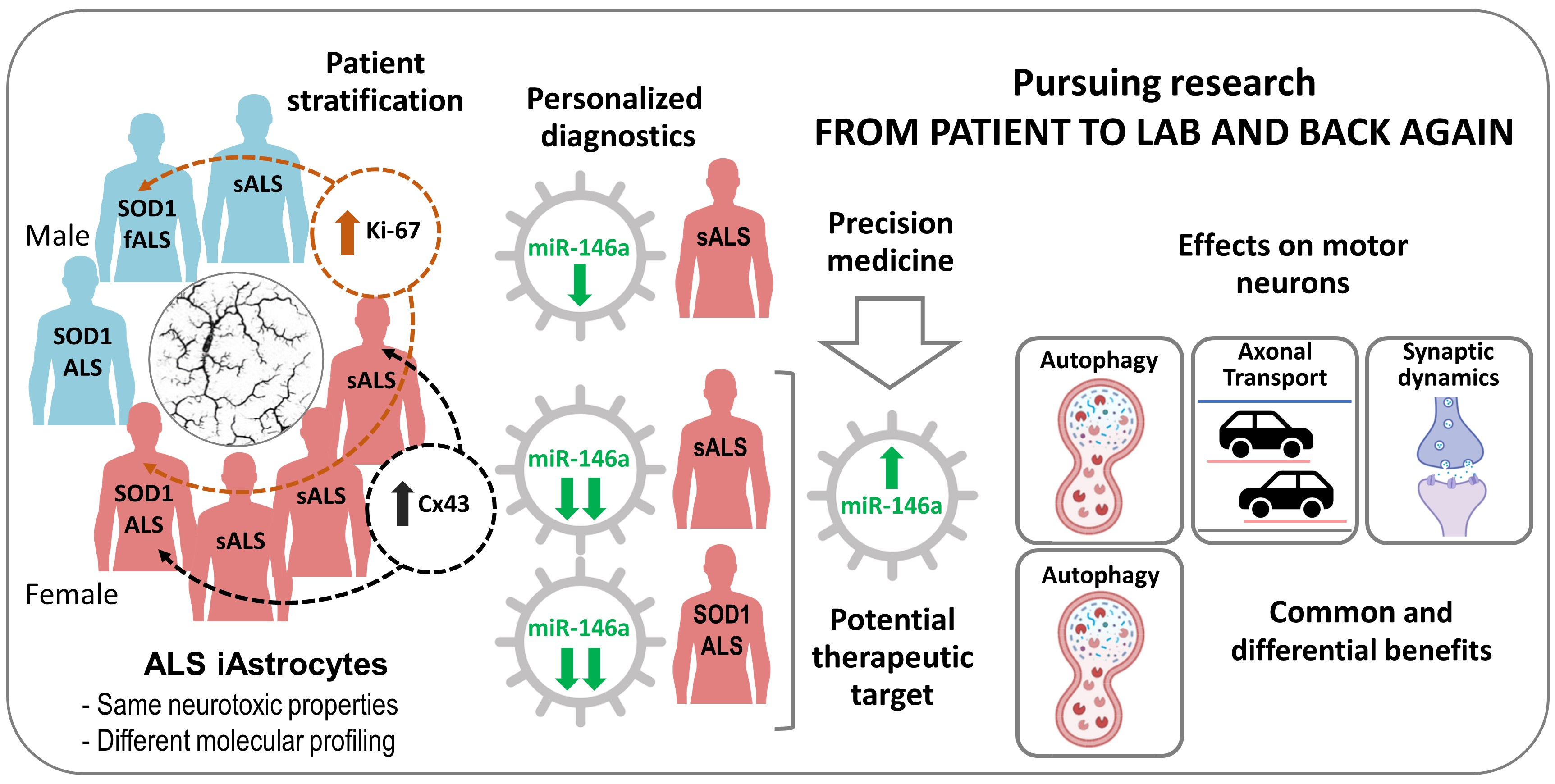
3.1. iAstrocytes Derived from Patients with ALS Show Neurotoxic Properties
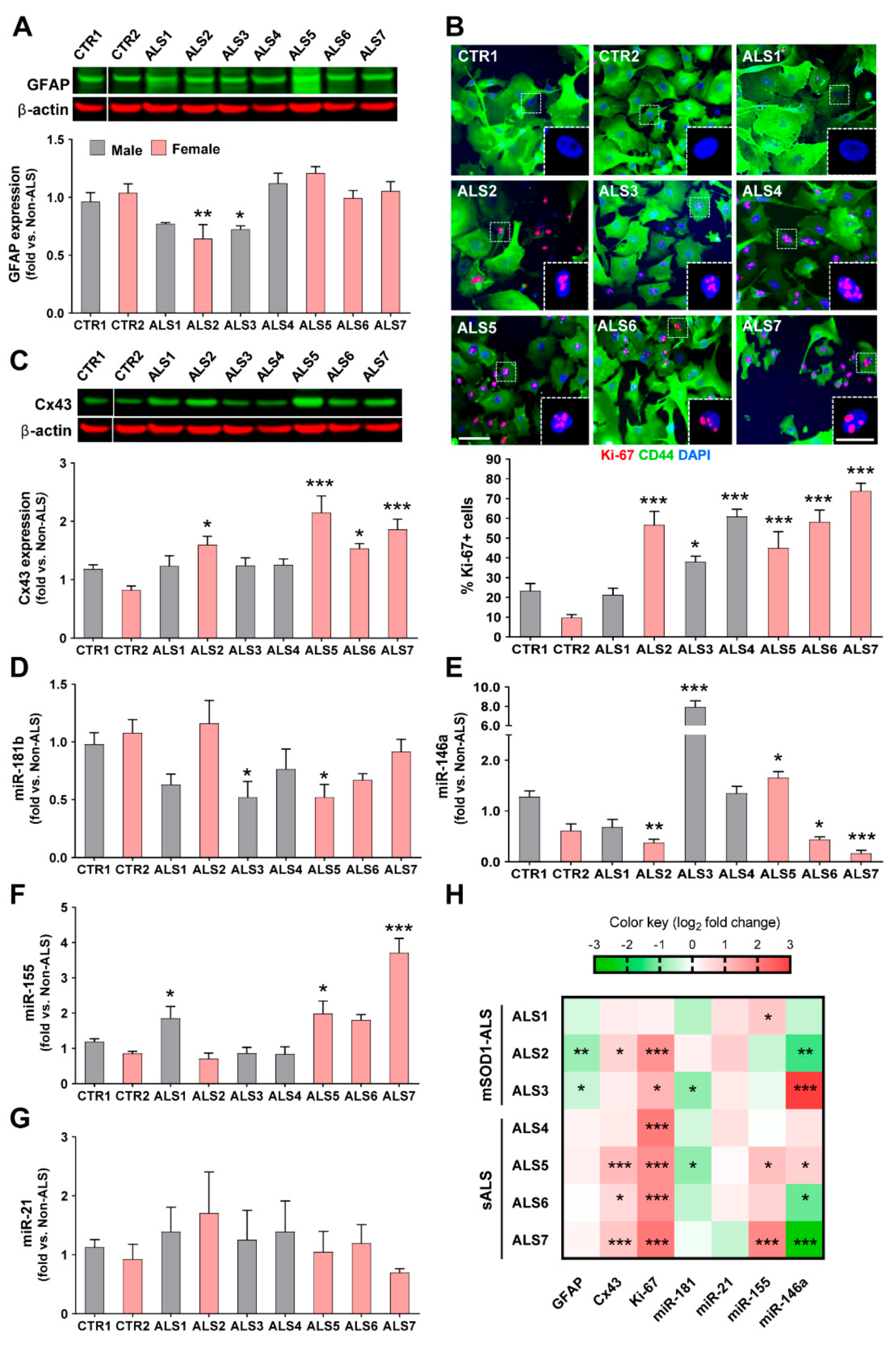
3.2. iAstrocytes from mSOD1-ALS and sALS Patients Show Distinct Cell Subtype Signatures
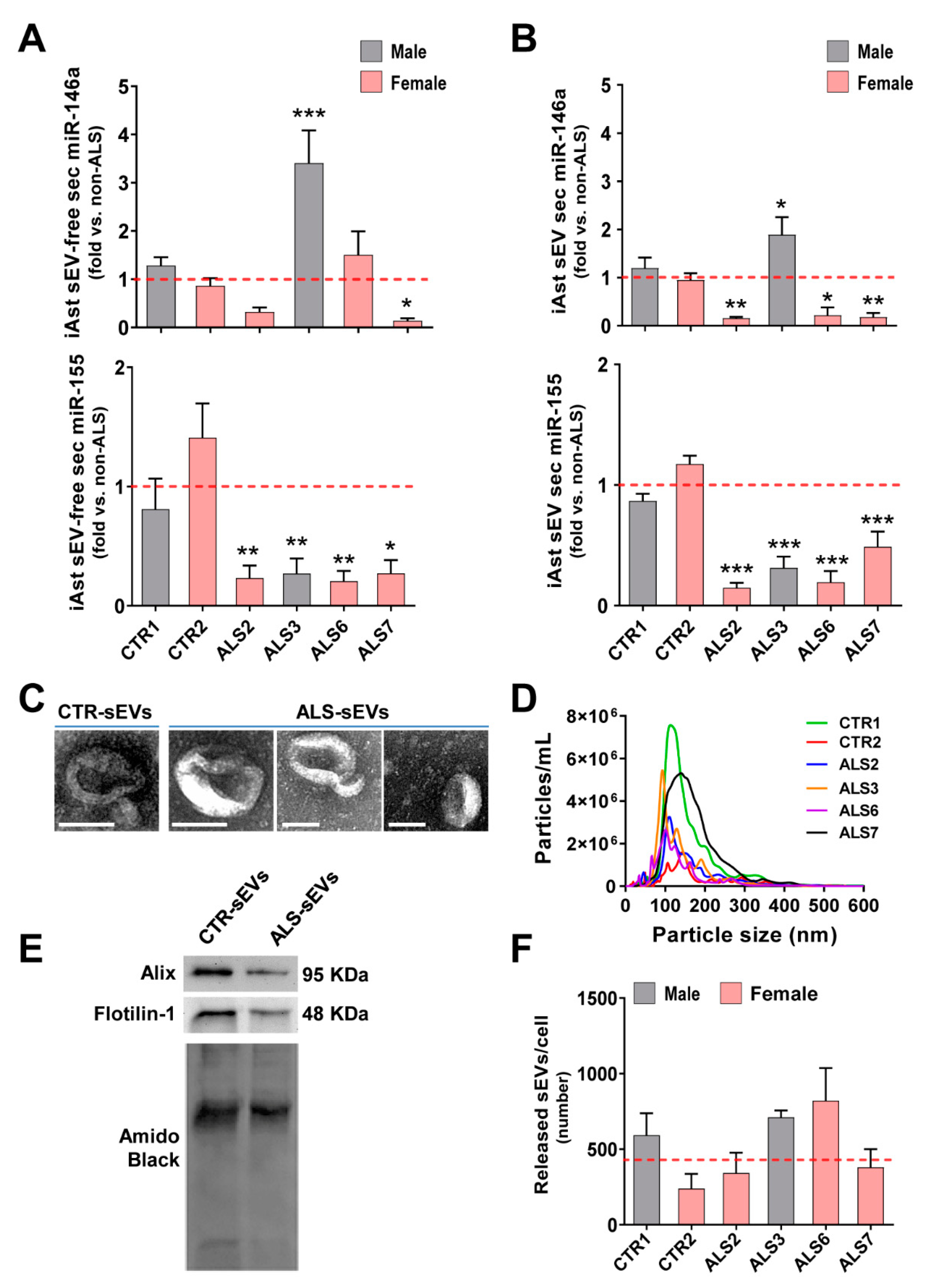
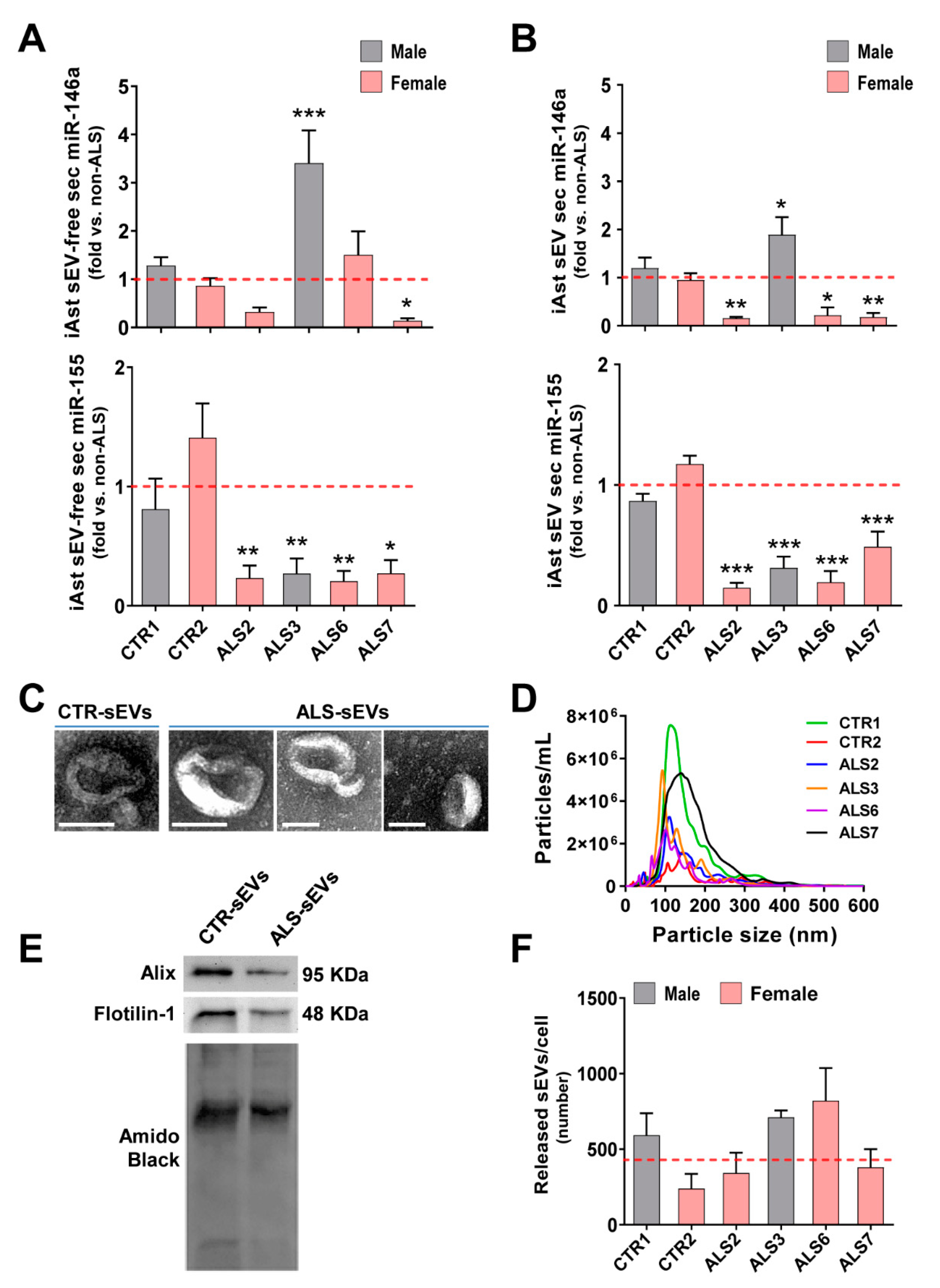
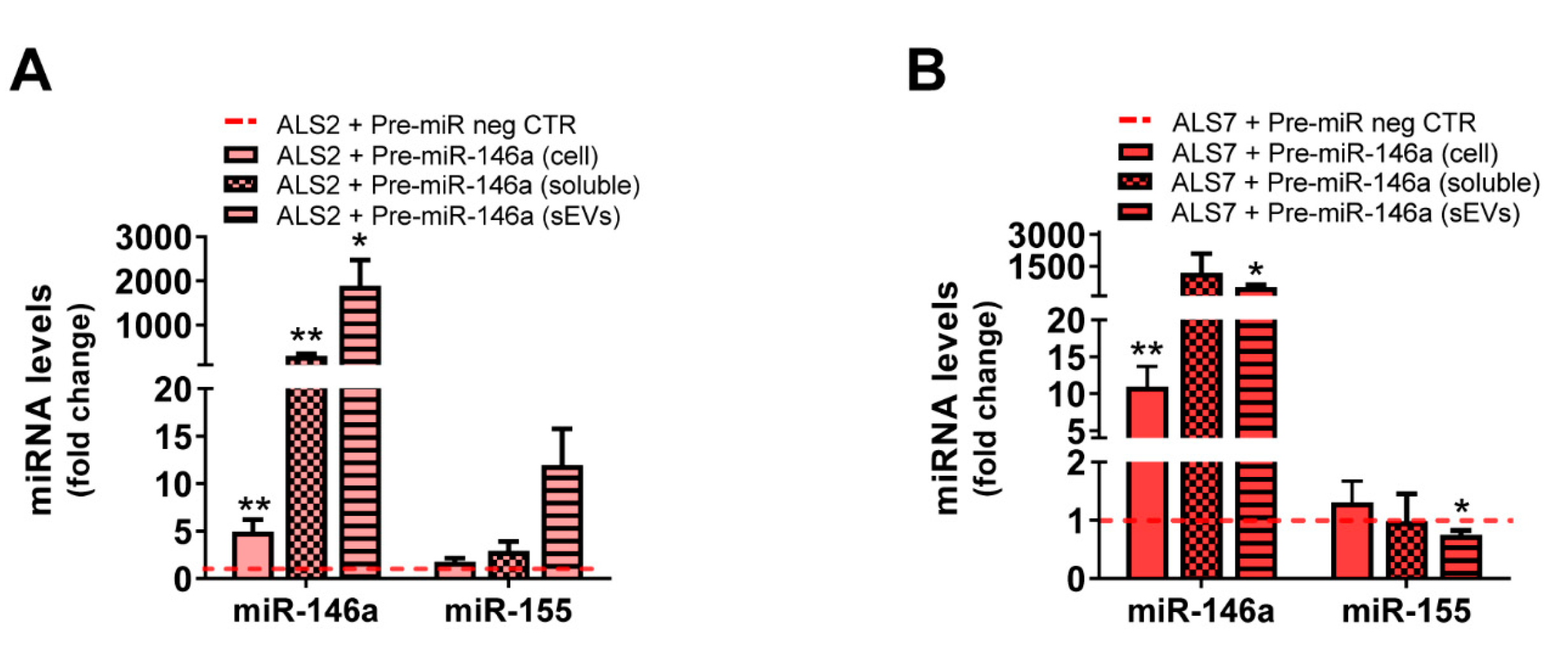
3.3. Dysregulated miR-146a Levels in ALS iAstrocytes Are Mimicked in sEV-Free Secretomes and sEV Cargoes, but the Same Is Not True for miR-155
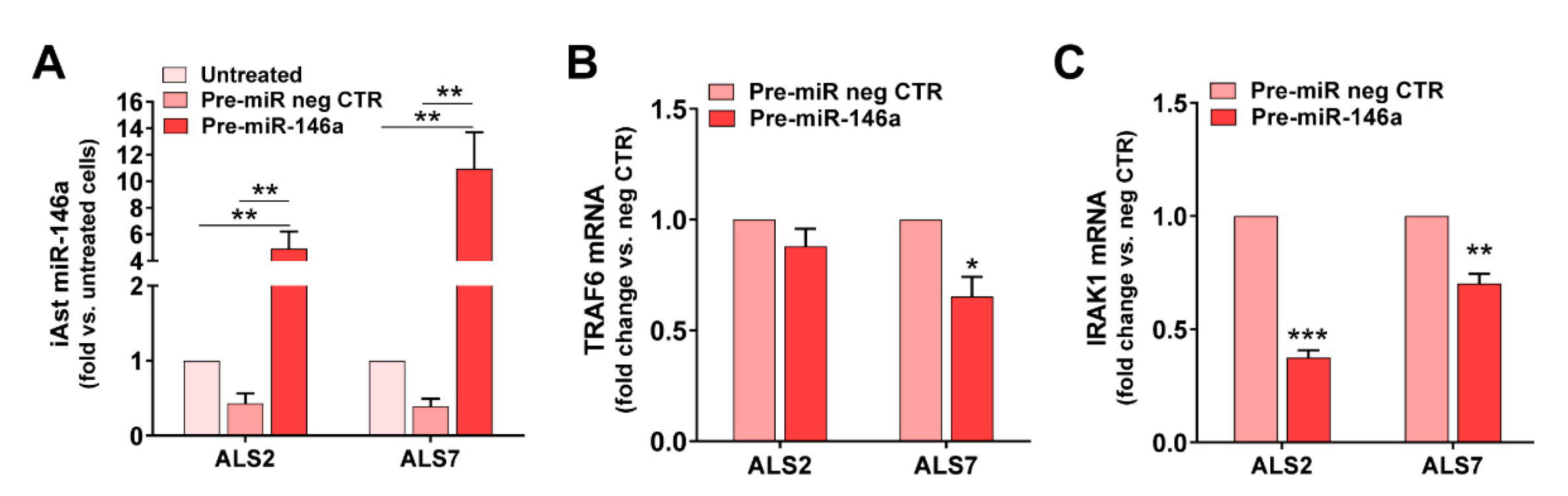
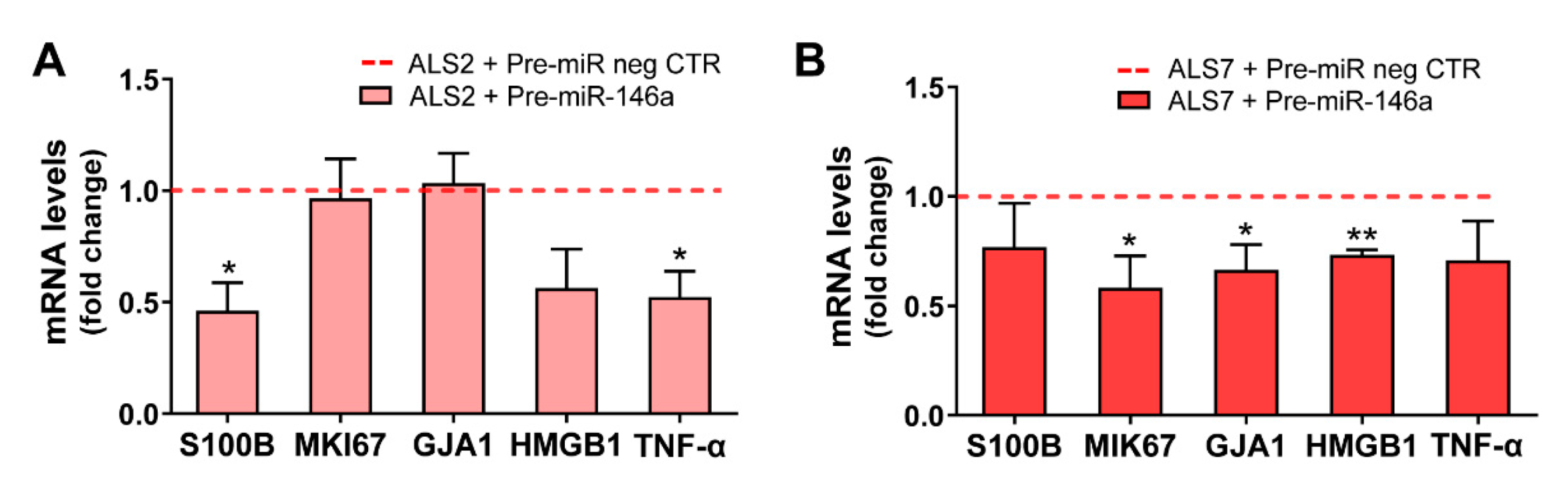
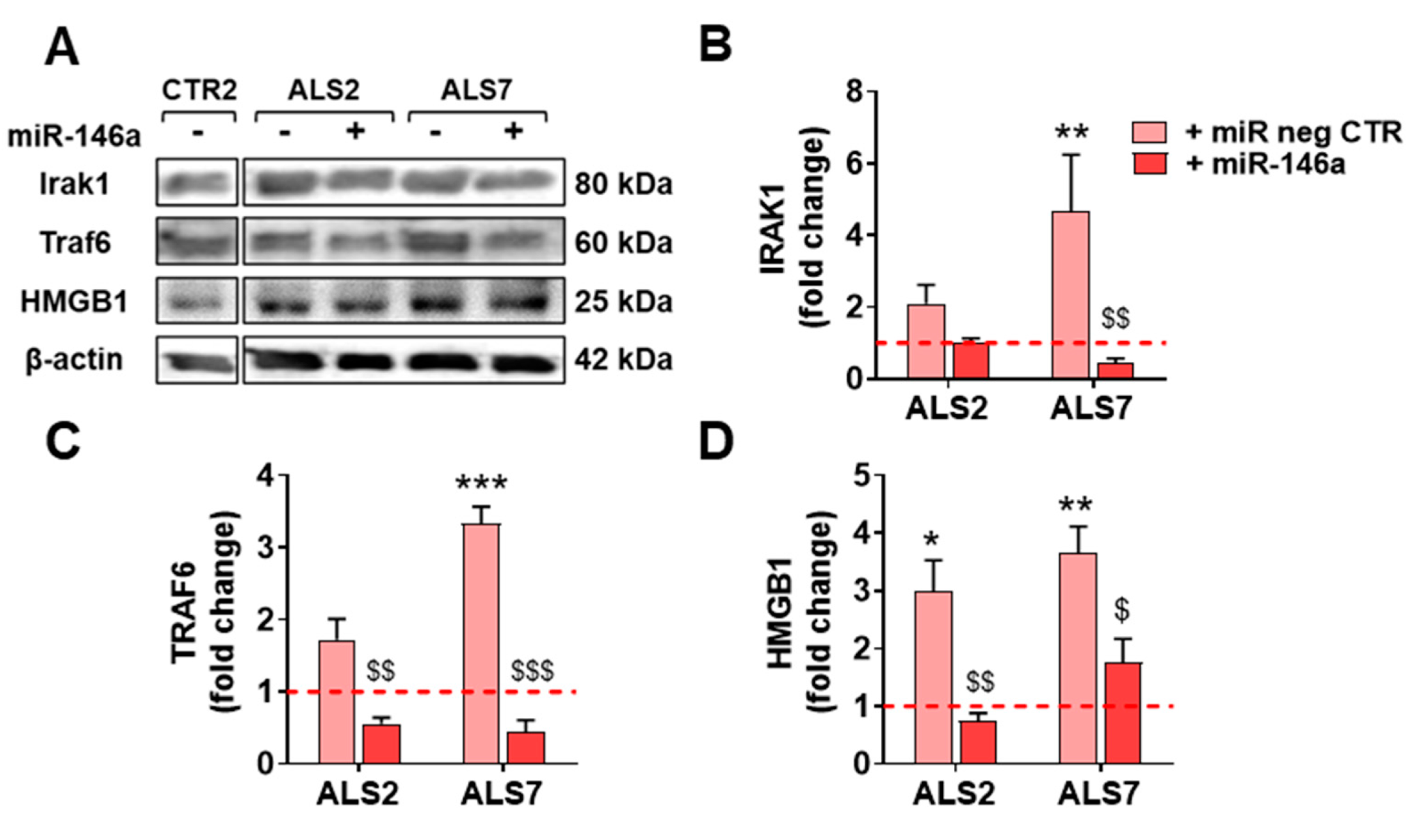
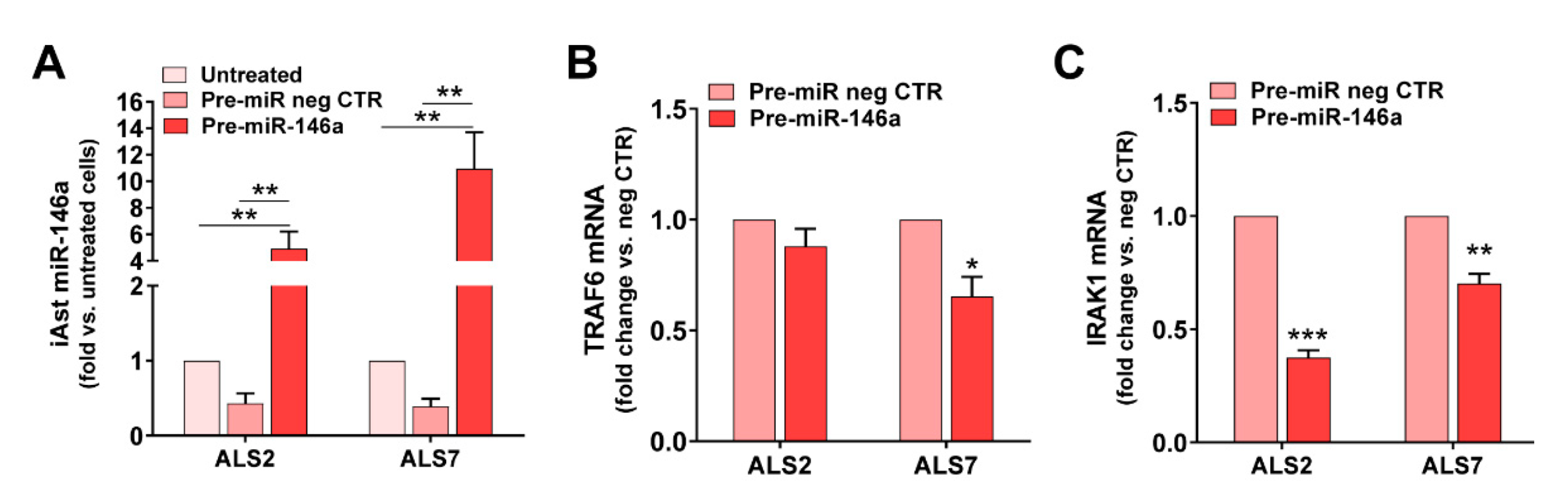
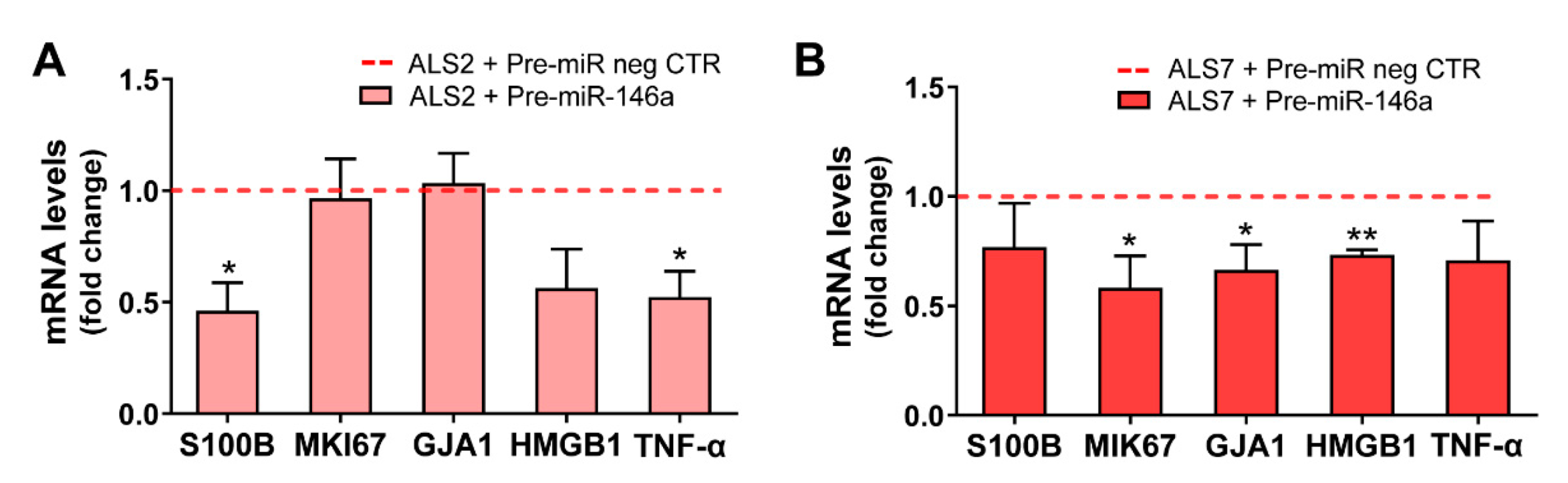
3.4. Upregulation of miR-146a in Depleted ALS2 and ALS7 iAstrocytes Differently Regulates Its Targets and Reactive Biomarkers
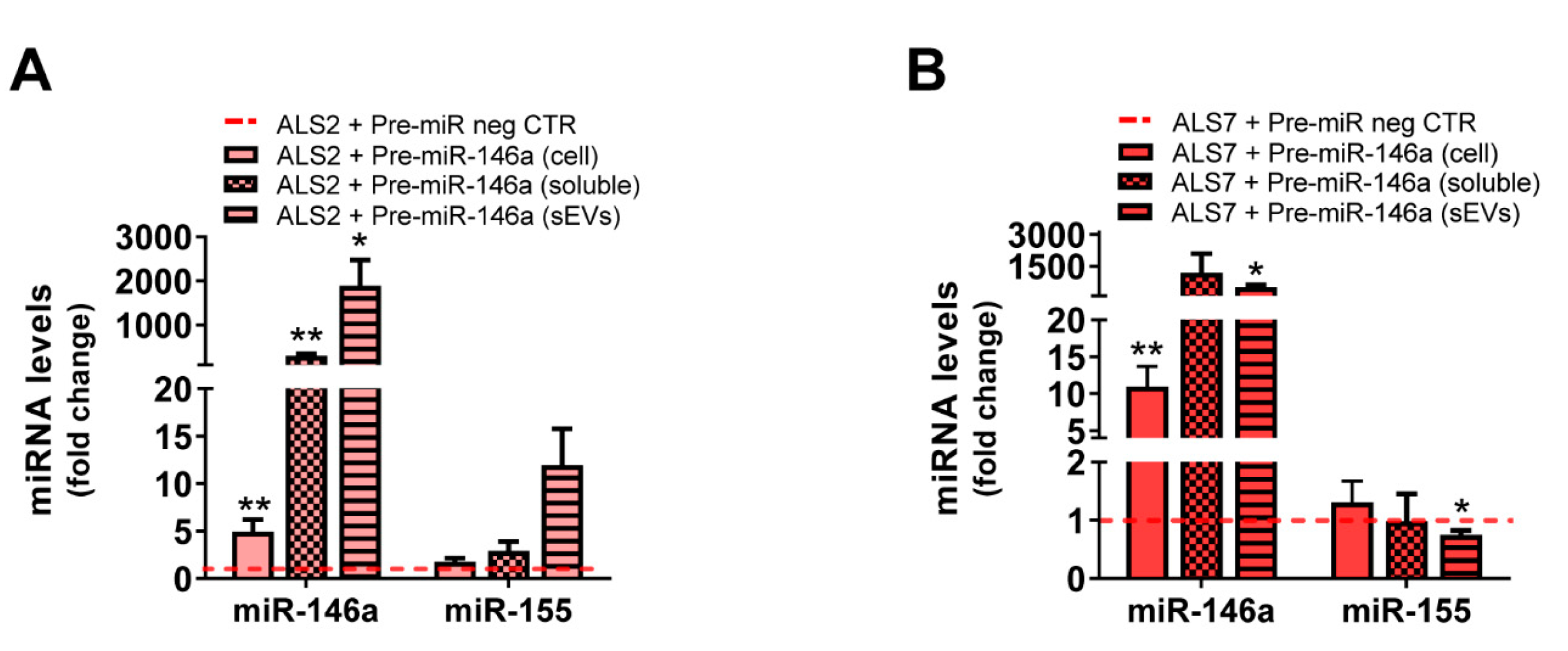
3.5. Transfection of ALS2 and ALS7 iAstrocytes with Pre-miR-146a Successfully Enhances Its Expression Levels in the Secretome, without Modifying miR-155 Paracrine Signaling
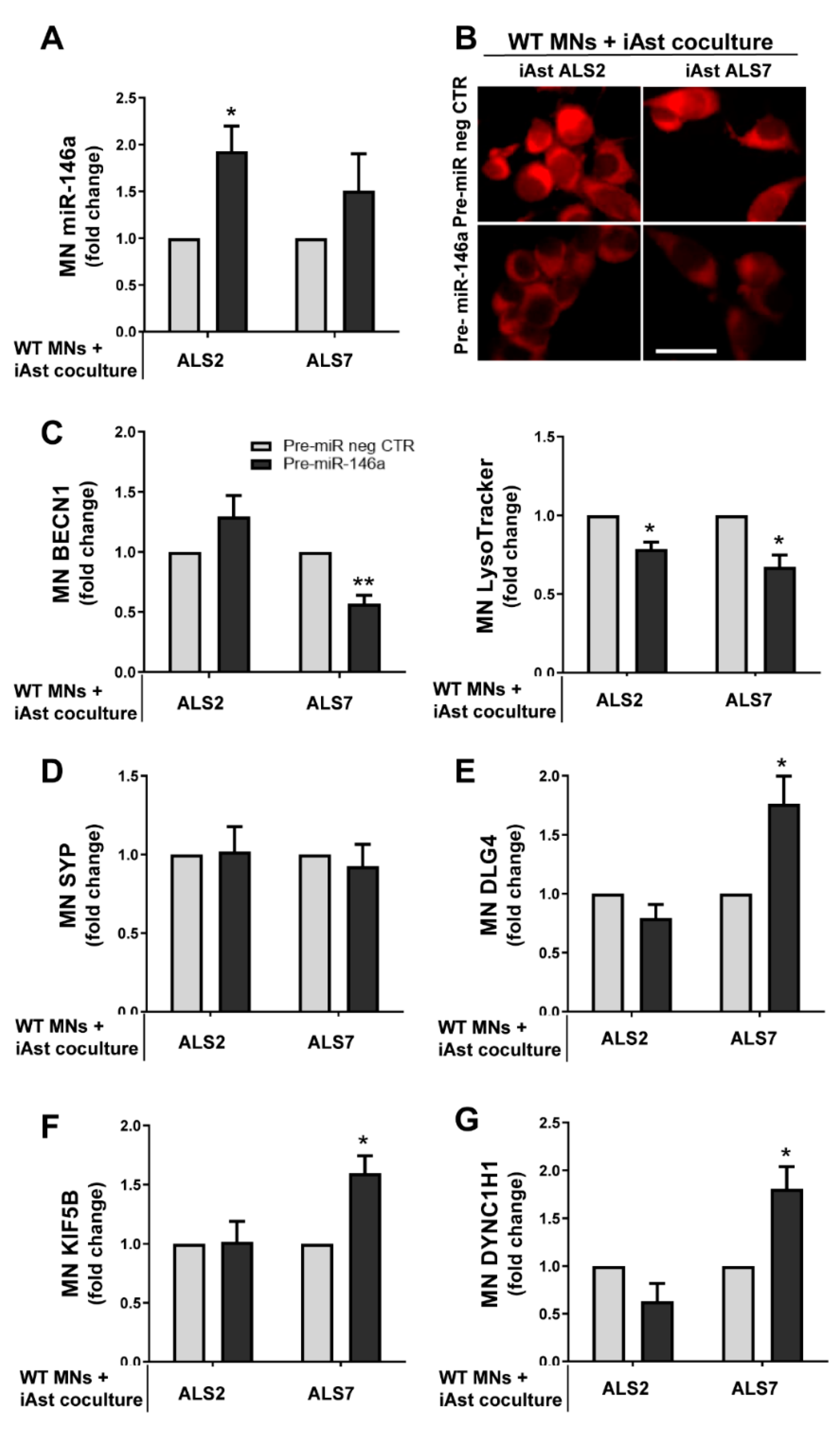
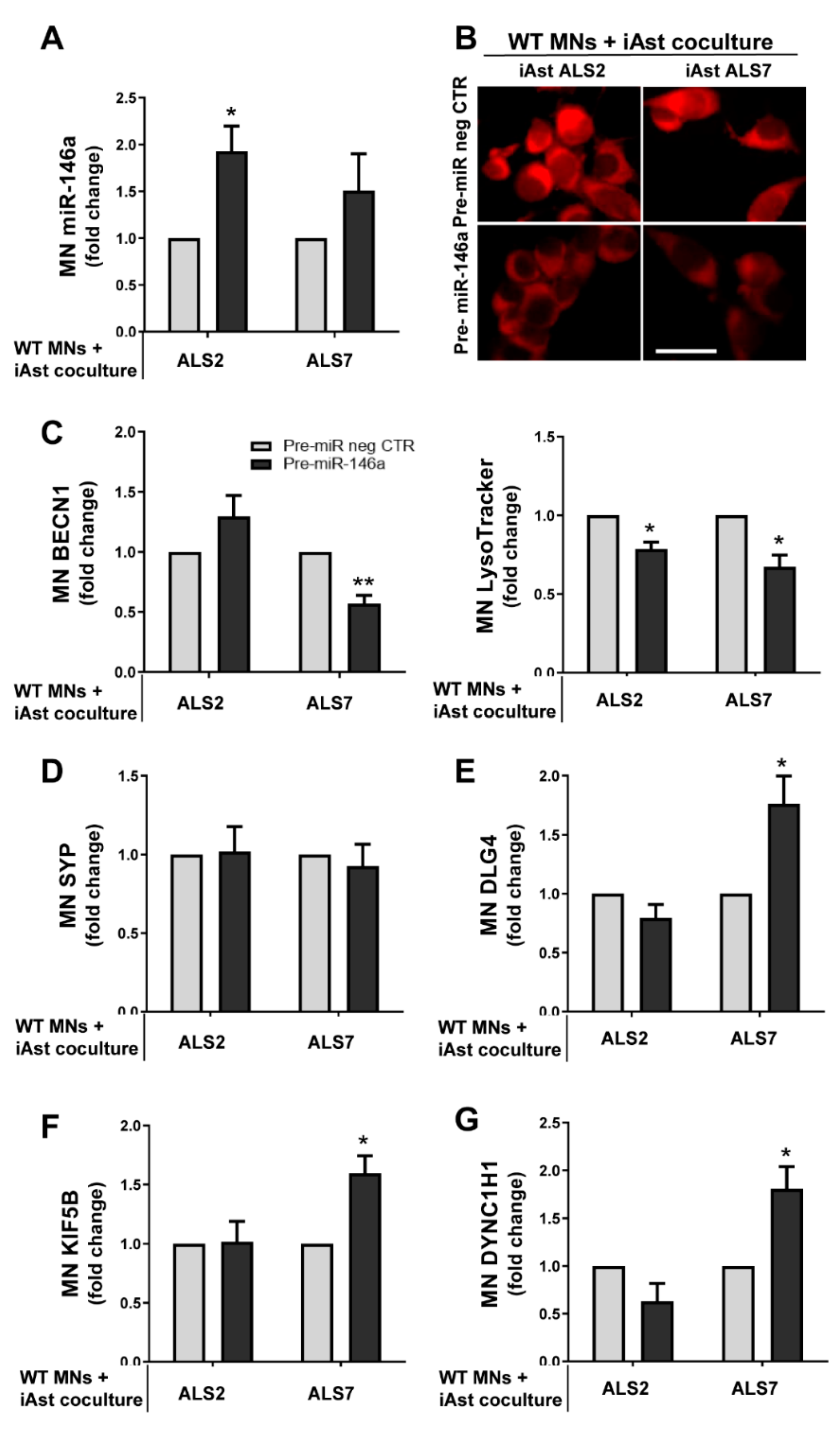
3.6. Upregulation of Depleted miR-146a in ALS2 and ALS7 iAstrocytes Diversely Contributes to the Recovery of Autophagic, Synaptic, and Axonal Transport Dynamics in NSC-34 MNs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rojas, P.; Ramirez, A.I.; Fernandez-Albarral, J.A.; Lopez-Cuenca, I.; Salobrar-Garcia, E.; Cadena, M.; Elvira-Hurtado, L.; Salazar, J.J.; de Hoz, R.; Ramirez, J.M. Amyotrophic Lateral Sclerosis: A Neurodegenerative Motor Neuron Disease with Ocular Involvement. Front. Neurosci. 2020, 14, 566858. [Google Scholar]
- Tiryaki, E.; Horak, H.A. ALS and other motor neuron diseases. Continuum (Minneap. Minn.) 2014, 20, 1185–1207. [Google Scholar]
- Swinnen, B.; Robberecht, W. The phenotypic variability of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2014, 10, 661–670. [Google Scholar]
- Katyal, N.; Govindarajan, R. Shortcomings in the Current Amyotrophic Lateral Sclerosis Trials and Potential Solutions for Improvement. Front. Neurol. 2017, 8, 521. [Google Scholar]
- Petrov, D.; Mansfield, C.; Moussy, A.; Hermine, O. ALS Clinical Trials Review: 20 Years of Failure. Are We Any Closer to Registering a New Treatment? Front. Aging Neurosci. 2017, 9, 68. [Google Scholar]
- Yamanaka, K.; Komine, O. The multi-dimensional roles of astrocytes in ALS. Neurosci. Res. 2018, 126, 31–38. [Google Scholar]
- Pehar, M.; Harlan, B.A.; Killoy, K.M.; Vargas, M.R. Role and Therapeutic Potential of Astrocytes in Amyotrophic Lateral Sclerosis. Curr. Pharm. Des. 2017, 23, 5010–5021. [Google Scholar]
- Trias, E.; Barbeito, L.; Yamanaka, K. Phenotypic heterogeneity of astrocytes in motor neuron disease. Clin. Exp. Neuroimmunol. 2018, 9, 225–234. [Google Scholar]
- Stifani, S. Taking Cellular Heterogeneity Into Consideration When Modeling Astrocyte Involvement in Amyotrophic Lateral Sclerosis Using Human Induced Pluripotent Stem Cells. Front. Cell. Neurosci. 2021, 15, 707861. [Google Scholar]
- Villarreal, A.; Vogel, T. Different Flavors of Astrocytes: Revising the Origins of Astrocyte Diversity and Epigenetic Signatures to Understand Heterogeneity after Injury. Int. J. Mol. Sci. 2021, 22, 6867. [Google Scholar] [CrossRef]
- Ajroud-Driss, S.; Siddique, T. Sporadic and hereditary amyotrophic lateral sclerosis (ALS). Biochim. Biophys. Acta 2015, 1852, 679–684. [Google Scholar]
- Al-Chalabi, A.; Hardiman, O. The epidemiology of ALS: A conspiracy of genes, environment and time. Nat. Rev. Neurol. 2013, 9, 617–628. [Google Scholar]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011, 72, 245–256. [Google Scholar]
- van Blitterswijk, M.; DeJesus-Hernandez, M.; Rademakers, R. How do C9ORF72 repeat expansions cause amyotrophic lateral sclerosis and frontotemporal dementia: Can we learn from other noncoding repeat expansion disorders? Curr. Opin. Neurol. 2012, 25, 689–700. [Google Scholar]
- Berdynski, M.; Miszta, P.; Safranow, K.; Andersen, P.M.; Morita, M.; Filipek, S.; Zekanowski, C.; Kuzma-Kozakiewicz, M. SOD1 mutations associated with amyotrophic lateral sclerosis analysis of variant severity. Sci. Rep. 2022, 12, 103. [Google Scholar]
- Chen, L.X.; Xu, H.F.; Wang, P.S.; Yang, X.X.; Wu, Z.Y.; Li, H.F. SOD1 Mutation Spectrum and Natural History of ALS Patients in a 15-Year Cohort in Southeastern China. Front. Genet. 2021, 12, 746060. [Google Scholar]
- Wei, Q.; Zhou, Q.; Chen, Y.; Ou, R.; Cao, B.; Xu, Y.; Yang, J.; Shang, H.F. Analysis of SOD1 mutations in a Chinese population with amyotrophic lateral sclerosis: A case-control study and literature review. Sci. Rep. 2017, 7, 44606. [Google Scholar]
- Haidet-Phillips, A.M.; Hester, M.E.; Miranda, C.J.; Meyer, K.; Braun, L.; Frakes, A.; Song, S.; Likhite, S.; Murtha, M.J.; Foust, K.D.; et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat. Biotechnol. 2011, 29, 824–828. [Google Scholar]
- Meyer, K.; Ferraiuolo, L.; Miranda, C.J.; Likhite, S.; McElroy, S.; Renusch, S.; Ditsworth, D.; Lagier-Tourenne, C.; Smith, R.A.; Ravits, J.; et al. Direct conversion of patient fibroblasts demonstrates non-cell autonomous toxicity of astrocytes to motor neurons in familial and sporadic ALS. Proc. Natl. Acad. Sci. USA 2014, 111, 829–832. [Google Scholar]
- Gomes, C.; Cunha, C.; Nascimento, F.; Ribeiro, J.A.; Vaz, A.R.; Brites, D. Cortical neurotoxic astrocytes with early ALS pathology and miR-146a deficit replicate gliosis markers of symptomatic SOD1G93A mouse model. Mol. Neurobiol. 2019, 56, 2137–2158. [Google Scholar]
- Diaz-Amarilla, P.; Olivera-Bravo, S.; Trias, E.; Cragnolini, A.; Martinez-Palma, L.; Cassina, P.; Beckman, J.; Barbeito, L. Phenotypically aberrant astrocytes that promote motoneuron damage in a model of inherited amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2011, 108, 18126–18131. [Google Scholar]
- Vaz, S.H.; Pinto, S.; Sebastião, A.M.; Brites, D. Astrocytes in Amyotrophic Lateral Sclerosis; Exon Publications: Brisbane, Australia, 2021; Chapter 3. [Google Scholar]
- Johann, S. Astrocytes Pathology in ALS: A Potential Therapeutic Target? Curr. Pharm. Des. 2017, 23, 5022–5036. [Google Scholar]
- Van Damme, P.; Robberecht, W.; Van Den Bosch, L. Modelling amyotrophic lateral sclerosis: Progress and possibilities. Dis. Model. Mech. 2017, 10, 537–549. [Google Scholar]
- Kelaini, S.; Cochrane, A.; Margariti, A. Direct reprogramming of adult cells: Avoiding the pluripotent state. Stem Cells Cloning 2014, 7, 19–29. [Google Scholar]
- Gatto, N.; Dos Santos Souza, C.; Shaw, A.C.; Bell, S.M.; Myszczynska, M.A.; Powers, S.; Meyer, K.; Castelli, L.M.; Karyka, E.; Mortiboys, H.; et al. Directly converted astrocytes retain the ageing features of the donor fibroblasts and elucidate the astrocytic contribution to human CNS health and disease. Aging Cell 2021, 20, e13281. [Google Scholar]
- Roybon, L.; Lamas, N.J.; Garcia, A.D.; Yang, E.J.; Sattler, R.; Lewis, V.J.; Kim, Y.A.; Kachel, C.A.; Rothstein, J.D.; Przedborski, S.; et al. Human stem cell-derived spinal cord astrocytes with defined mature or reactive phenotypes. Cell Rep. 2013, 4, 1035–1048. [Google Scholar]
- Serio, A.; Bilican, B.; Barmada, S.J.; Ando, D.M.; Zhao, C.; Siller, R.; Burr, K.; Haghi, G.; Story, D.; Nishimura, A.L.; et al. Astrocyte pathology and the absence of non-cell autonomy in an induced pluripotent stem cell model of TDP-43 proteinopathy. Proc. Natl. Acad. Sci. USA 2013, 110, 4697–4702. [Google Scholar]
- Oksanen, M.; Petersen, A.J.; Naumenko, N.; Puttonen, K.; Lehtonen, S.; Gubert Olive, M.; Shakirzyanova, A.; Leskela, S.; Sarajarvi, T.; Viitanen, M.; et al. PSEN1 mutant iPSC-derived model reveals severe astrocyte pathology in Alzheimer’s disease. Stem Cell Rep. 2017, 9, 1885–1897. [Google Scholar]
- Volonte, C.; Apolloni, S.; Parisi, C. MicroRNAs: Newcomers into the ALS picture. CNS Neurol. Disord. Drug Targets 2015, 14, 194–207. [Google Scholar]
- Sharma, S.; Lu, H.C. microRNAs in Neurodegeneration: Current Findings and Potential Impacts. J. Alzheimers Dis Parkinsonism 2018, 8, 420. [Google Scholar] [CrossRef]
- Chugh, P.; Dittmer, D.P. Potential pitfalls in microRNA profiling. Wiley Interdiscip. Rev. RNA 2012, 3, 601–616. [Google Scholar]
- Tahamtan, A.; Teymoori-Rad, M.; Nakstad, B.; Salimi, V. Anti-Inflammatory MicroRNAs and Their Potential for Inflammatory Diseases Treatment. Front. Immunol. 2018, 9, 1377. [Google Scholar]
- Quinn, S.R.; O’Neill, L.A. A trio of microRNAs that control Toll-like receptor signalling. Int. Immunol. 2011, 23, 421–425. [Google Scholar]
- Olivieri, F.; Rippo, M.R.; Procopio, A.D.; Fazioli, F. Circulating inflamma-miRs in aging and age-related diseases. Front. Genet. 2013, 4, 121. [Google Scholar]
- Cunha, C.; Santos, C.; Gomes, C.; Fernandes, A.; Correia, A.M.; Sebastião, A.M.; Vaz, A.R.; Brites, D. Downregulated glia interplay and increased miRNA-155 as promising markers to track ALS at an early stage. Mol. Neurobiol. 2018, 55, 4207–4224. [Google Scholar]
- Brites, D. Regulatory function of microRNAs in microglia. Glia 2020, 68, 1631–1642. [Google Scholar]
- Halushka, M.K.; Fromm, B.; Peterson, K.J.; McCall, M.N. Big Strides in Cellular MicroRNA Expression. Trends Genet. 2018, 34, 165–167. [Google Scholar]
- Bell, E.; Taylor, M.A. Functional roles for exosomal microRNAs in the tumour microenvironment. Comput. Struct. Biotechnol. J. 2017, 15, 8–13. [Google Scholar]
- Gomes, C.; Sequeira, C.; Barbosa, M.; Cunha, C.; Vaz, A.R.; Brites, D. Astrocyte regional diversity in ALS includes distinct aberrant phenotypes with common and causal pathological processes. Exp. Cell. Res. 2020, 395, 112209. [Google Scholar]
- Clarke, B.E.; Taha, D.M.; Tyzack, G.E.; Patani, R. Regionally encoded functional heterogeneity of astrocytes in health and disease: A perspective. Glia 2021, 69, 20–27. [Google Scholar]
- Butovsky, O.; Jedrychowski, M.P.; Cialic, R.; Krasemann, S.; Murugaiyan, G.; Fanek, Z.; Greco, D.J.; Wu, P.M.; Doykan, C.E.; Kiner, O.; et al. Targeting miR-155 restores abnormal microglia and attenuates disease in SOD1 mice. Ann. Neurol. 2015, 77, 75–99. [Google Scholar]
- Barbosa, M.; Gomes, C.; Sequeira, C.; Goncalves-Ribeiro, J.; Pina, C.C.; Carvalho, L.A.; Moreira, R.; Vaz, S.H.; Vaz, A.R.; Brites, D. Recovery of depleted miR-146a in ALS cortical astrocytes reverts cell aberrancies and prevents paracrine pathogenicity on microglia and motor neurons. Front. Cell. Dev. Biol. 2021, 9, 634355. [Google Scholar]
- Prada, I.; Gabrielli, M.; Turola, E.; Iorio, A.; D’Arrigo, G.; Parolisi, R.; De Luca, M.; Pacifici, M.; Bastoni, M.; Lombardi, M.; et al. Glia-to-neuron transfer of miRNAs via extracellular vesicles: A new mechanism underlying inflammation-induced synaptic alterations. Acta Neuropatho.l 2018, 135, 529–550. [Google Scholar]
- Pinto, S.; Cunha, C.; Barbosa, M.; Vaz, A.R.; Brites, D. Exosomes from NSC-34 cells transfected with hSOD1-G93A are enriched in miR-124 and drive alterations in microglia phenotype. Front. Neurosci. 2017, 11, 273. [Google Scholar]
- Fernandes, A.; Ribeiro, A.R.; Monteiro, M.; Garcia, G.; Vaz, A.R.; Brites, D. Secretome from SH-SY5Y APPswe cells trigger time-dependent CHME3 microglia activation phenotypes, ultimately leading to miR-21 exosome shuttling. Biochimie 2018, 155, 67–82. [Google Scholar]
- Ravnik-Glavac, M.; Glavac, D. Circulating RNAs as Potential Biomarkers in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2020, 21, 1714. [Google Scholar] [CrossRef] [Green Version]
- Dennys, C.N.; Sierra-Delgado, J.A.; Ray, S.S.; Hartlaub, A.M.; Roussel, F.S.; Rodriguez, Y.; Meyer, K. In vitro Modeling for Neurological Diseases using Direct Conversion from Fibroblasts to Neuronal Progenitor Cells and Differentiation into Astrocytes. J. Vis. Exp. 2021, 172, e62016. [Google Scholar] [CrossRef]
- Hester, M.E.; Song, S.; Miranda, C.J.; Eagle, A.; Schwartz, P.H.; Kaspar, B.K. Two factor reprogramming of human neural stem cells into pluripotency. PLoS ONE 2009, 4, e7044. [Google Scholar]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar]
- Stopford, M.J.; Allen, S.P.; Ferraiuolo, L. A High-throughput and Pathophysiologically Relevant Astrocyte-motor Neuron Co-culture Assay for Amyotrophic Lateral Sclerosis Therapeutic Discovery. Bio. Protoc. 2019, 9, e3353. [Google Scholar] [CrossRef]
- Cunha, C.; Gomes, C.; Vaz, A.R.; Brites, D. Exploring new inflammatory biomarkers and pathways during LPS-induced M1 polarization. Mediators Inflamm. 2016, 2016, 6986175. [Google Scholar]
- Caldeira, C.; Oliveira, A.F.; Cunha, C.; Vaz, A.R.; Falcão, A.S.; Fernandes, A.; Brites, D. Microglia change from a reactive to an age-like phenotype with the time in culture. Front. Cell. Neurosci. 2014, 8, 152. [Google Scholar]
- Vaz, A.R.; Cunha, C.; Gomes, C.; Schmucki, N.; Barbosa, M.; Brites, D. Glycoursodeoxycholic acid reduces matrix metalloproteinase-9 and caspase-9 activation in a cellular model of superoxide dismutase-1 neurodegeneration. Mol. Neurobiol. 2015, 51, 864–877. [Google Scholar]
- Bucchia, M.; Merwin, S.J.; Re, D.B.; Kariya, S. Limitations and Challenges in Modeling Diseases Involving Spinal Motor Neuron Degeneration in Vitro. Front. Cell. Neurosci. 2018, 12, 61. [Google Scholar]
- Ho, R.; Sances, S.; Gowing, G.; Amoroso, M.W.; O’Rourke, J.G.; Sahabian, A.; Wichterle, H.; Baloh, R.H.; Sareen, D.; Svendsen, C.N. ALS disrupts spinal motor neuron maturation and aging pathways within gene co-expression networks. Nat. Neurosci. 2016, 19, 1256–1267. [Google Scholar]
- Vargas, M.R.; Johnson, J.A. Astrogliosis in amyotrophic lateral sclerosis: Role and therapeutic potential of astrocytes. Neurotherapeutics 2010, 7, 471–481. [Google Scholar]
- Figueroa-Romero, C.; Hur, J.; Lunn, J.S.; Paez-Colasante, X.; Bender, D.E.; Yung, R.; Sakowski, S.A.; Feldman, E.L. Expression of microRNAs in human post-mortem amyotrophic lateral sclerosis spinal cords provides insight into disease mechanisms. Mol. Cell. Neurosci. 2016, 71, 34–45. [Google Scholar]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar]
- Almad, A.A.; Doreswamy, A.; Gross, S.K.; Richard, J.P.; Huo, Y.; Haughey, N.; Maragakis, N.J. Connexin 43 in astrocytes contributes to motor neuron toxicity in amyotrophic lateral sclerosis. Glia 2016, 64, 1154–1169. [Google Scholar]
- Ziff, O.J.; Clarke, B.E.; Taha, D.M.; Crerar, H.; Luscombe, N.M.; Patani, R. Meta-analysis of human and mouse ALS astrocytes reveals multi-omic signatures of inflammatory reactive states. Genome Res. 2022, 32, 71–84. [Google Scholar]
- Birger, A.; Ben-Dor, I.; Ottolenghi, M.; Turetsky, T.; Gil, Y.; Sweetat, S.; Perez, L.; Belzer, V.; Casden, N.; Steiner, D.; et al. Human iPSC-derived astrocytes from ALS patients with mutated C9ORF72 show increased oxidative stress and neurotoxicity. EBioMedicine 2019, 50, 274–289. [Google Scholar]
- Madill, M.; McDonagh, K.; Ma, J.; Vajda, A.; McLoughlin, P.; O’Brien, T.; Hardiman, O.; Shen, S. Amyotrophic lateral sclerosis patient iPSC-derived astrocytes impair autophagy via non-cell autonomous mechanisms. Mol. Brain 2017, 10, 22. [Google Scholar]
- Walgrave, H.; Zhou, L.; De Strooper, B.; Salta, E. The promise of microRNA-based therapies in Alzheimer’s disease: Challenges and perspectives. Mol. Neurodegener. 2021, 16, 76. [Google Scholar]
- Foggin, S.; Mesquita-Ribeiro, R.; Dajas-Bailador, F.; Layfield, R. Biological Significance of microRNA Biomarkers in ALS-Innocent Bystanders or Disease Culprits? Front. Neurol. 2019, 10, 578. [Google Scholar]
- Parisi, C.; Arisi, I.; D’Ambrosi, N.; Storti, A.E.; Brandi, R.; D’Onofrio, M.; Volonte, C. Dysregulated microRNAs in amyotrophic lateral sclerosis microglia modulate genes linked to neuroinflammation. Cell Death Dis. 2013, 4, e959. [Google Scholar]
- Soliman, H.M.; Ghonaim, G.A.; Gharib, S.M.; Chopra, H.; Farag, A.K.; Hassanin, M.H.; Nagah, A.; Emad-Eldin, M.; Hashem, N.E.; Yahya, G.; et al. Exosomes in Alzheimer’s Disease: From Being Pathological Players to Potential Diagnostics and Therapeutics. Int. J. Mol. Sci. 2021, 22, 10794. [Google Scholar]
- Xia, X.; Wang, Y.; Huang, Y.; Zhang, H.; Lu, H.; Zheng, J.C. Exosomal miRNAs in central nervous system diseases: Biomarkers, pathological mediators, protective factors and therapeutic agents. Prog. Neurobiol. 2019, 183, 101694. [Google Scholar]
- de Abreu, R.C.; Ramos, C.V.; Becher, C.; Lino, M.; Jesus, C.; da Costa Martins, P.A.; Martins, P.A.T.; Moreno, M.J.; Fernandes, H.; Ferreira, L. Exogenous loading of miRNAs into small extracellular vesicles. J. Extracell. Vesicles 2021, 10, e12111. [Google Scholar]
- Saeedi, S.; Israel, S.; Nagy, C.; Turecki, G. The emerging role of exosomes in mental disorders. Transl. Psychiatry 2019, 9, 122. [Google Scholar]
- Iyer, A.; Zurolo, E.; Prabowo, A.; Fluiter, K.; Spliet, W.G.; van Rijen, P.C.; Gorter, J.A.; Aronica, E. MicroRNA-146a: A key regulator of astrocyte-mediated inflammatory response. PLoS ONE 2012, 7, e44789. [Google Scholar]
- Muroi, M.; Tanamoto, K. IRAK-1-mediated negative regulation of Toll-like receptor signaling through proteasome-dependent downregulation of TRAF6. Biochim. Biophys. Acta 2012, 1823, 255–263. [Google Scholar]
- van Loo, G.; Beyaert, R. Negative regulation of NF-kappaB and its involvement in rheumatoid arthritis. Arthritis Res. Ther 2011, 13, 221. [Google Scholar]
- Sproviero, D.; La Salvia, S.; Giannini, M.; Crippa, V.; Gagliardi, S.; Bernuzzi, S.; Diamanti, L.; Ceroni, M.; Pansarasa, O.; Poletti, A.; et al. Pathological Proteins Are Transported by Extracellular Vesicles of Sporadic Amyotrophic Lateral Sclerosis Patients. Front. Neurosci. 2018, 12, 487. [Google Scholar]
- Chen, Q.Y.; Wen, T.; Wu, P.; Jia, R.; Zhang, R.; Dang, J. Exosomal Proteins and miRNAs as Mediators of Amyotrophic Lateral Sclerosis. Front. Cell Dev. Biol. 2021, 9, 718803. [Google Scholar]
- Vaz, A.R.; Vizinha, D.; Morais, H.; Colaco, A.R.; Loch-Neckel, G.; Barbosa, M.; Brites, D. Overexpression of miR-124 in motor neurons plays a key role in ALS pathological processes. Int. J. Mol. Sci. 2021, 22, 6128. [Google Scholar]
- Marques, C.R.; Pereira-Sousa, J.; Teixeira, F.G.; Sousa, R.A.; Teixeira-Castro, A.; Salgado, A.J. Mesenchymal stem cell secretome protects against alpha-synuclein-induced neurodegeneration in a Caenorhabditis elegans model of Parkinson’s disease. Cytotherapy 2021, 23, 894–901. [Google Scholar]
- Giunti, D.; Marini, C.; Parodi, B.; Usai, C.; Milanese, M.; Bonanno, G.; Kerlero de Rosbo, N.; Uccelli, A. Role of miRNAs shuttled by mesenchymal stem cell-derived small extracellular vesicles in modulating neuroinflammation. Sci. Rep. 2021, 11, 1740. [Google Scholar]
- Howe, J.R.; Bear, M.F.; Golshani, P.; Klann, E.; Lipton, S.A.; Mucke, L.; Sahin, M.; Silva, A.J. The mouse as a model for neuropsychiatric drug development. Curr. Biol. 2018, 28, R909–R914. [Google Scholar]
- Ransohoff, R.M. All (animal) models (of neurodegeneration) are wrong. Are they also useful? J. Exp. Med. 2018, 215, 2955–2958. [Google Scholar]
- Bhartiya, D. Clinical translation of stem cells for regenerative medicine. Circ. Res. 2019, 124, 840–842. [Google Scholar]
- Haake, K.; Ackermann, M.; Lachmann, N. Concise review: Towards the clinical translation of induced pluripotent stem cell-derived blood cells-ready for take-off. Stem Cells Transl. Med. 2019, 8, 332–339. [Google Scholar]
- Galieva, L.R.; James, V.; Mukhamedshina, Y.O.; Rizvanov, A.A. Therapeutic potential of extracellular vesicles for the treatment of nerve disorders. Front. Neurosci. 2019, 13, 163. [Google Scholar]
- Yang, B.; Yang, R.; Xu, B.; Fu, J.; Qu, X.; Li, L.; Dai, M.; Tan, C.; Chen, H.; Wang, X. miR-155 and miR-146a collectively regulate meningitic Escherichia coli infection-mediated neuroinflammatory responses. J. Neuroinflammation 2021, 18, 114. [Google Scholar]
- Koval, E.D.; Shaner, C.; Zhang, P.; du Maine, X.; Fischer, K.; Tay, J.; Chau, B.N.; Wu, G.F.; Miller, T.M. Method for widespread microRNA-155 inhibition prolongs survival in ALS-model mice. Hum. Mol. Genet. 2013, 22, 4127–4135. [Google Scholar]
- Waters, R.; Subham, S.; Pacelli, S.; Modaresi, S.; Chakravarti, A.R.; Paul, A. Development of microRNA-146a-enriched stem cell secretome for wound-healing applications. Mol. Pharm. 2019, 16, 4302–4312. [Google Scholar]
- Sasaki, S. Autophagy in spinal cord motor neurons in sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2011, 70, 349–359. [Google Scholar]
- Gudi, V.; Gai, L.; Herder, V.; Tejedor, L.S.; Kipp, M.; Amor, S.; Suhs, K.W.; Hansmann, F.; Beineke, A.; Baumgartner, W.; et al. Synaptophysin is a reliable marker for axonal damage. J. Neuropathol. Exp. Neurol. 2017, 76, 109–125. [Google Scholar]
- Dore, K.; Carrico, Z.; Alfonso, S.; Marino, M.; Koymans, K.; Kessels, H.W.; Malinow, R. PSD-95 protects synapses from β-amyloid. Cell. Rep. 2021, 35, 109194. [Google Scholar]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar]
- Sebastião, A.M.; Rei, N.; Ribeiro, J.A. Amyotrophic Lateral Sclerosis (ALS) and Adenosine Receptors. Front. Pharmacol. 2018, 9, 267. [Google Scholar]
- De Vos, K.J.; Hafezparast, M. Neurobiology of axonal transport defects in motor neuron diseases: Opportunities for translational research? Neurobiol. Dis. 2017, 105, 283–299. [Google Scholar]
- Ikenaka, K.; Katsuno, M.; Kawai, K.; Ishigaki, S.; Tanaka, F.; Sobue, G. Disruption of axonal transport in motor neuron diseases. Int. J. Mol. Sci. 2012, 13, 1225–1238. [Google Scholar]
- Philips, T.; Rothstein, J.D. Rodent Models of Amyotrophic Lateral Sclerosis. Curr. Protoc. Pharmacol. 2015, 69, 5–67. [Google Scholar]
- Kueffner, R.; Zach, N.; Bronfeld, M.; Norel, R.; Atassi, N.; Balagurusamy, V.; Di Camillo, B.; Chio, A.; Cudkowicz, M.; Dillenberger, D.; et al. Stratification of amyotrophic lateral sclerosis patients: A crowdsourcing approach. Sci. Rep. 2019, 9, 690. [Google Scholar]
- Devine, H.; Patani, R. The translational potential of human induced pluripotent stem cells for clinical neurology: The translational potential of hiPSCs in neurology. Cell. Biol. Toxicol. 2017, 33, 129–144. [Google Scholar]
- Traxler, L.; Edenhofer, F.; Mertens, J. Next-generation disease modeling with direct conversion: A new path to old neurons. FEBS Lett. 2019, 593, 3316–3337. [Google Scholar]
- Caiazzo, M.; Giannelli, S.; Valente, P.; Lignani, G.; Carissimo, A.; Sessa, A.; Colasante, G.; Bartolomeo, R.; Massimino, L.; Ferroni, S.; et al. Direct conversion of fibroblasts into functional astrocytes by defined transcription factors. Stem Cell Rep. 2015, 4, 25–36. [Google Scholar]
- Nagy, D.; Kato, T.; Kushner, P.D. Reactive astrocytes are widespread in the cortical gray matter of amyotrophic lateral sclerosis. J. Neurosci. Res. 1994, 38, 336–347. [Google Scholar]
- Schiffer, D.; Cordera, S.; Cavalla, P.; Migheli, A. Reactive astrogliosis of the spinal cord in amyotrophic lateral sclerosis. J. Neurol. Sci. 1996, 139, 27–33. [Google Scholar]
- Qian, K.; Huang, H.; Peterson, A.; Hu, B.; Maragakis, N.J.; Ming, G.L.; Chen, H.; Zhang, S.C. Sporadic ALS astrocytes induce neuronal degeneration in vivo. Stem Cell Rep. 2017, 8, 843–855. [Google Scholar]
- Turner, M.R.; Barnwell, J.; Al-Chalabi, A.; Eisen, A. Young-onset amyotrophic lateral sclerosis: Historical and other observations. Brain 2012, 135, 2883–2891. [Google Scholar]
- Tripathi, P.; Rodriguez-Muela, N.; Klim, J.R.; de Boer, A.S.; Agrawal, S.; Sandoe, J.; Lopes, C.S.; Ogliari, K.S.; Williams, L.A.; Shear, M.; et al. Reactive astrocytes promote ALS-like degeneration and intracellular protein aggregation in human motor neurons by disrupting autophagy through TGF-beta1. Stem Cell Rep. 2017, 9, 667–680. [Google Scholar]
- Kunze, A.; Lengacher, S.; Dirren, E.; Aebischer, P.; Magistretti, P.J.; Renaud, P. Astrocyte-neuron co-culture on microchips based on the model of SOD mutation to mimic ALS. Integr. Biol. 2013, 5, 964–975. [Google Scholar]
- Yoshii, Y.; Otomo, A.; Pan, L.; Ohtsuka, M.; Hadano, S. Loss of glial fibrillary acidic protein marginally accelerates disease progression in a SOD1(H46R) transgenic mouse model of ALS. Neurosci. Res. 2011, 70, 321–329. [Google Scholar]
- Jeffrey, J.; D’Cunha, H.; Suzuki, M. Blood Level of Glial Fibrillary Acidic Protein (GFAP) Does not Correlate With Disease Progression in a Rat Model of Familial ALS (SOD1(G93A) Transgenic). Front. Neurol. 2018, 9, 954. [Google Scholar]
- Escartin, C.; Galea, E.; Lakatos, A.; O’Callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhauser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar]
- Vicario, N.; Zappala, A.; Calabrese, G.; Gulino, R.; Parenti, C.; Gulisano, M.; Parenti, R. Connexins in the Central Nervous System: Physiological Traits and Neuroprotective Targets. Front. Physiol. 2017, 8, 1060. [Google Scholar]
- Tribulova, N.; Dupont, E.; Soukup, T.; Okruhlicova, L.; Severs, N.J. Sex differences in connexin-43 expression in left ventricles of aging rats. Physiol. Res. 2005, 54, 705–708. [Google Scholar]
- Stauffer, B.L.; Sobus, R.D.; Sucharov, C.C. Sex differences in cardiomyocyte connexin43 expression. J. Cardiovasc. Pharmacol. 2011, 58, 32–39. [Google Scholar]
- Varcianna, A.; Myszczynska, M.A.; Castelli, L.M.; O’Neill, B.; Kim, Y.; Talbot, J.; Nyberg, S.; Nyamali, I.; Heath, P.R.; Stopford, M.J.; et al. Micro-RNAs secreted through astrocyte-derived extracellular vesicles cause neuronal network degeneration in C9orf72 ALS. EBioMedicine 2019, 40, 626–635. [Google Scholar]
- Hutchison, E.R.; Kawamoto, E.M.; Taub, D.D.; Lal, A.; Abdelmohsen, K.; Zhang, Y.; Wood, W.H., 3rd; Lehrmann, E.; Camandola, S.; Becker, K.G.; et al. Evidence for miR-181 involvement in neuroinflammatory responses of astrocytes. Glia 2013, 61, 1018–1028. [Google Scholar]
- Shi, L.; Cheng, Z.; Zhang, J.; Li, R.; Zhao, P.; Fu, Z.; You, Y. Hsa-mir-181a and hsa-mir-181b function as tumor suppressors in human glioma cells. Brain Res. 2008, 1236, 185–193. [Google Scholar]
- Pegoraro, V.; Merico, A.; Angelini, C. Micro-RNAs in ALS muscle: Differences in gender, age at onset and disease duration. J. Neurol. Sci. 2017, 380, 58–63. [Google Scholar]
- Guedes, J.R.; Custodia, C.M.; Silva, R.J.; de Almeida, L.P.; Pedroso de Lima, M.C.; Cardoso, A.L. Early miR-155 upregulation contributes to neuroinflammation in Alzheimer’s disease triple transgenic mouse model. Hum. Mol. Genet. 2014, 23, 6286–6301. [Google Scholar]
- Sison, S.L.; Patitucci, T.N.; Seminary, E.R.; Villalon, E.; Lorson, C.L.; Ebert, A.D. Astrocyte-produced miR-146a as a mediator of motor neuron loss in spinal muscular atrophy. Hum. Mol. Genet. 2017, 26, 3409–3420. [Google Scholar]
- Waller, R.; Wyles, M.; Heath, P.R.; Kazoka, M.; Wollff, H.; Shaw, P.J.; Kirby, J. Small RNA Sequencing of Sporadic Amyotrophic Lateral Sclerosis Cerebrospinal Fluid Reveals Differentially Expressed miRNAs Related to Neural and Glial Activity. Front. Neurosci. 2017, 11, 731. [Google Scholar]
- Tasca, E.; Pegoraro, V.; Merico, A.; Angelini, C. Circulating microRNAs as biomarkers of muscle differentiation and atrophy in ALS. Clin. Neuropathol. 2016, 35, 22–30. [Google Scholar]
- Alexander, M.; Hu, R.; Runtsch, M.C.; Kagele, D.A.; Mosbruger, T.L.; Tolmachova, T.; Seabra, M.C.; Round, J.L.; Ward, D.M.; O’Connell, R.M. Exosome-delivered microRNAs modulate the inflammatory response to endotoxin. Nat. Commun. 2015, 6, 7321. [Google Scholar]
- Ferrara, D.; Pasetto, L.; Bonetto, V.; Basso, M. Role of Extracellular Vesicles in Amyotrophic Lateral Sclerosis. Front. Neurosci. 2018, 12, 574. [Google Scholar]
- Pashangzadeh, S.; Motallebnezhad, M.; Vafashoar, F.; Khalvandi, A.; Mojtabavi, N. Implications the Role of miR-155 in the Pathogenesis of Autoimmune Diseases. Front. Immunol. 2021, 12, 669382. [Google Scholar]
- Zilahi, E.; Tarr, T.; Papp, G.; Griger, Z.; Sipka, S.; Zeher, M. Increased microRNA-146a/b, TRAF6 gene and decreased IRAK1 gene expressions in the peripheral mononuclear cells of patients with Sjogren’s syndrome. Immunol. Lett. 2012, 141, 165–168. [Google Scholar]
- Xie, Y.F.; Shu, R.; Jiang, S.Y.; Song, Z.C.; Guo, Q.M.; Dong, J.C.; Lin, Z.K. miRNA-146 negatively regulates the production of pro-inflammatory cytokines via NF-kappaB signalling in human gingival fibroblasts. J. Inflamm. 2014, 11, 38. [Google Scholar]
- Guidotti, G.; Scarlata, C.; Brambilla, L.; Rossi, D. Tumor Necrosis Factor Alpha in Amyotrophic Lateral Sclerosis: Friend or Foe? Cells 2021, 10, 518. [Google Scholar]
- Brambilla, L.; Martorana, F.; Guidotti, G.; Rossi, D. Dysregulation of Astrocytic HMGB1 Signaling in Amyotrophic Lateral Sclerosis. Front. Neurosci. 2018, 12, 622. [Google Scholar]
- Wang, C.; Zhang, C.; Liu, L.; A, X.; Chen, B.; Li, Y.; Du, J. Macrophage-Derived mir-155-Containing Exosomes Suppress Fibroblast Proliferation and Promote Fibroblast Inflammation during Cardiac Injury. Mol. Ther. 2017, 25, 192–204. [Google Scholar]
- Nguyen, L.S.; Fregeac, J.; Bole-Feysot, C.; Cagnard, N.; Iyer, A.; Anink, J.; Aronica, E.; Alibeu, O.; Nitschke, P.; Colleaux, L. Role of miR-146a in neural stem cell differentiation and neural lineage determination: Relevance for neurodevelopmental disorders. Autism 2018, 9, 38. [Google Scholar]
- Hetz, C.; Thielen, P.; Matus, S.; Nassif, M.; Court, F.; Kiffin, R.; Martinez, G.; Cuervo, A.M.; Brown, R.H.; Glimcher, L.H. XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 2009, 23, 2294–2306. [Google Scholar]
- Nassif, M.; Valenzuela, V.; Rojas-Rivera, D.; Vidal, R.; Matus, S.; Castillo, K.; Fuentealba, Y.; Kroemer, G.; Levine, B.; Hetz, C. Pathogenic role of BECN1/Beclin 1 in the development of amyotrophic lateral sclerosis. Autophagy 2014, 10, 1256–1271. [Google Scholar]
- Casas, C.; Manzano, R.; Vaz, R.; Osta, R.; Brites, D. Synaptic Failure: Focus in an Integrative View of ALS. Brain Plast. 2016, 1, 159–175. [Google Scholar]
- Martorana, F.; Brambilla, L.; Valori, C.F.; Bergamaschi, C.; Roncoroni, C.; Aronica, E.; Volterra, A.; Bezzi, P.; Rossi, D. The BH4 domain of Bcl-X(L) rescues astrocyte degeneration in amyotrophic lateral sclerosis by modulating intracellular calcium signals. Hum. Mol. Genet. 2012, 21, 826–840. [Google Scholar]
- Rossi, D.; Brambilla, L.; Valori, C.F.; Roncoroni, C.; Crugnola, A.; Yokota, T.; Bredesen, D.E.; Volterra, A. Focal degeneration of astrocytes in amyotrophic lateral sclerosis. Cell Death Differ. 2008, 15, 1691–1700. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Diagnosis | Mutation | Age at Biopsy (yrs) | Onset Type | Symptom Onset to Biopsy | Sex |
|---|---|---|---|---|---|---|
| CTR1 | Non-ALS | - | 42 | - | - | Male |
| CTR2 | Non-ALS | - | 64 | - | - | Female |
| ALS1 | ALS | SOD1 1 | 40 | Lower limb | 3 months | Male |
| ALS2 | ALS | SOD1 2 | 63 | Lower limb | 8 months | Female |
| ALS3 | fALS | SOD1 3 | 40 | Unknown | Unknown | Male |
| ALS4 | sALS | Unknown | 29 | Bulbar | 2.7 Yrs | Male |
| ALS5 | sALS | Unknown | 62 | Distal upper extremity | 2.5 Yrs | Female |
| ALS6 | sALS | Unknown | 47 | Distal upper extremity | 1.75 Yrs | Female |
| ALS7 | sALS | Unknown | 69 | Distal upper extremity | 2.25 Yrs | Female |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomes, C.; Sequeira, C.; Likhite, S.; Dennys, C.N.; Kolb, S.J.; Shaw, P.J.; Vaz, A.R.; Kaspar, B.K.; Meyer, K.; Brites, D. Neurotoxic Astrocytes Directly Converted from Sporadic and Familial ALS Patient Fibroblasts Reveal Signature Diversities and miR-146a Theragnostic Potential in Specific Subtypes. Cells 2022, 11, 1186. https://doi.org/10.3390/cells11071186
Gomes C, Sequeira C, Likhite S, Dennys CN, Kolb SJ, Shaw PJ, Vaz AR, Kaspar BK, Meyer K, Brites D. Neurotoxic Astrocytes Directly Converted from Sporadic and Familial ALS Patient Fibroblasts Reveal Signature Diversities and miR-146a Theragnostic Potential in Specific Subtypes. Cells. 2022; 11(7):1186. https://doi.org/10.3390/cells11071186
Chicago/Turabian StyleGomes, Cátia, Catarina Sequeira, Shibi Likhite, Cassandra N. Dennys, Stephen J. Kolb, Pamela J. Shaw, Ana R. Vaz, Brian K. Kaspar, Kathrin Meyer, and Dora Brites. 2022. "Neurotoxic Astrocytes Directly Converted from Sporadic and Familial ALS Patient Fibroblasts Reveal Signature Diversities and miR-146a Theragnostic Potential in Specific Subtypes" Cells 11, no. 7: 1186. https://doi.org/10.3390/cells11071186
APA StyleGomes, C., Sequeira, C., Likhite, S., Dennys, C. N., Kolb, S. J., Shaw, P. J., Vaz, A. R., Kaspar, B. K., Meyer, K., & Brites, D. (2022). Neurotoxic Astrocytes Directly Converted from Sporadic and Familial ALS Patient Fibroblasts Reveal Signature Diversities and miR-146a Theragnostic Potential in Specific Subtypes. Cells, 11(7), 1186. https://doi.org/10.3390/cells11071186