
ER+ Breast Cancer Strongly Depends on MCL-1 and BCL-xL Anti-Apoptotic Proteins

 , , ,
, , ,  and
and 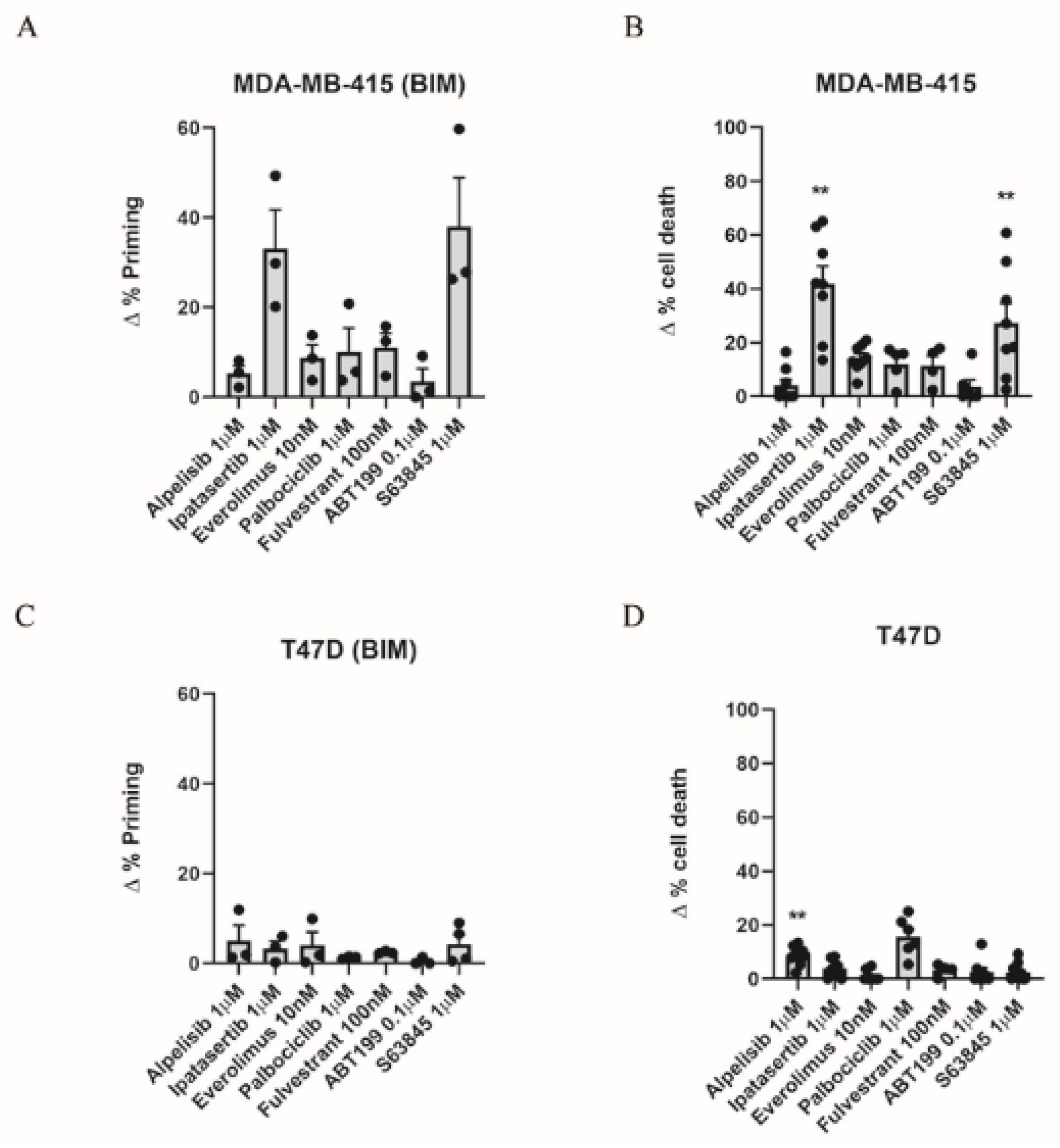{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Treatments
2.2. Dynamic BH3 Profiling
2.3. Cell Death Analysis
2.4. Protein Extraction and Quantification
2.5. Immunoprecipitation
2.6. Immunoblotting
2.7. Statistical Analysis
3. Results
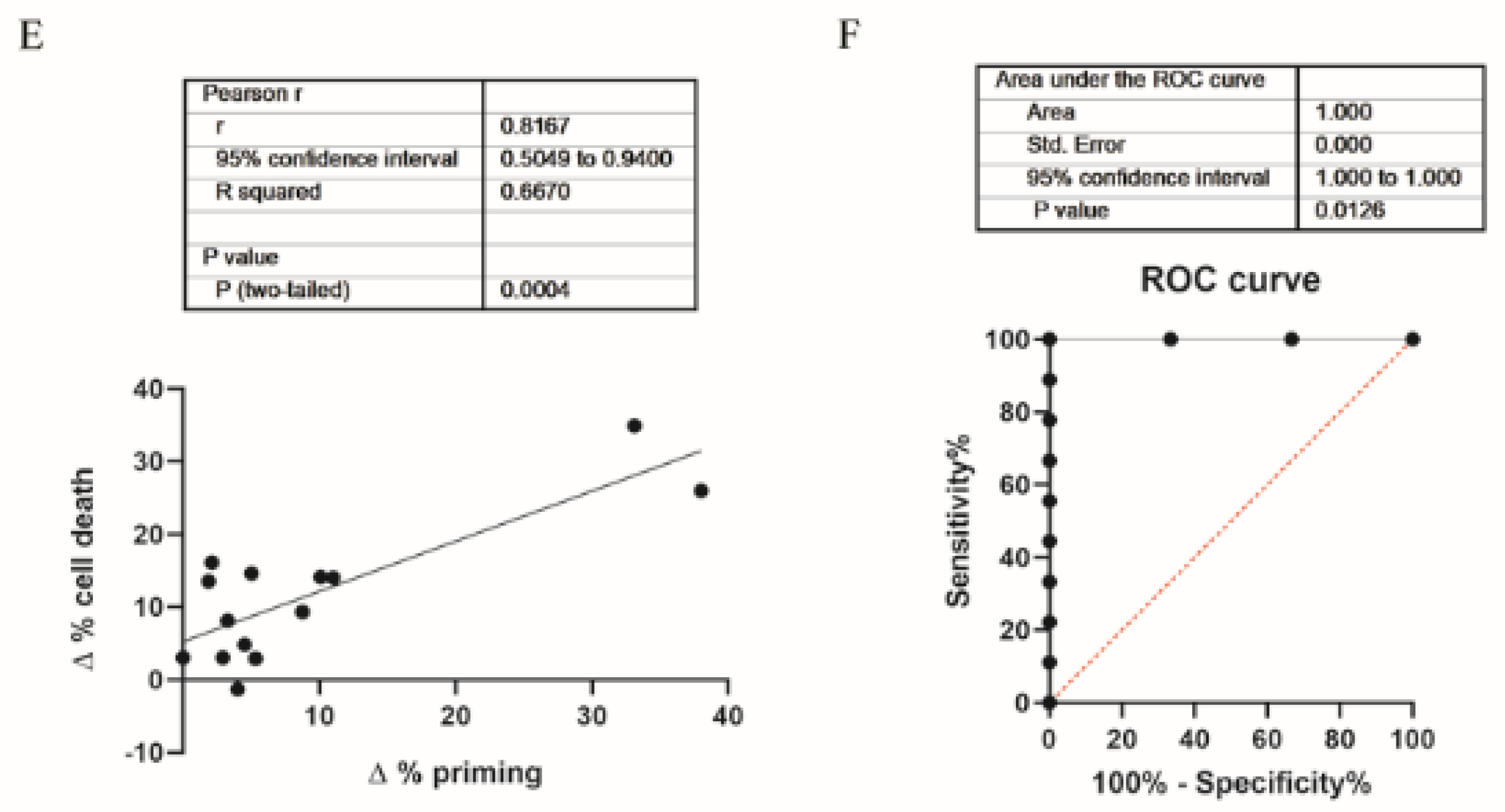
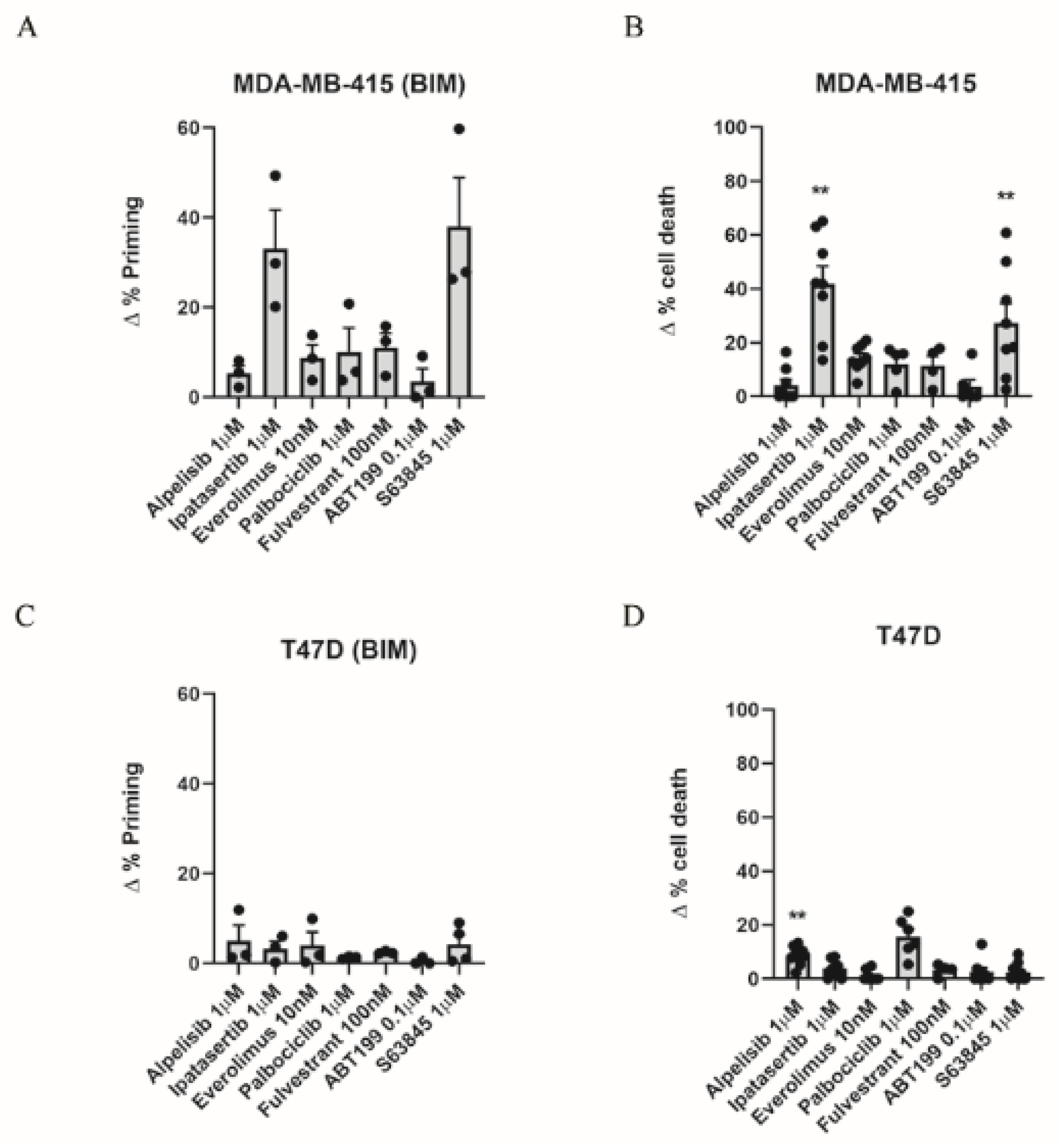
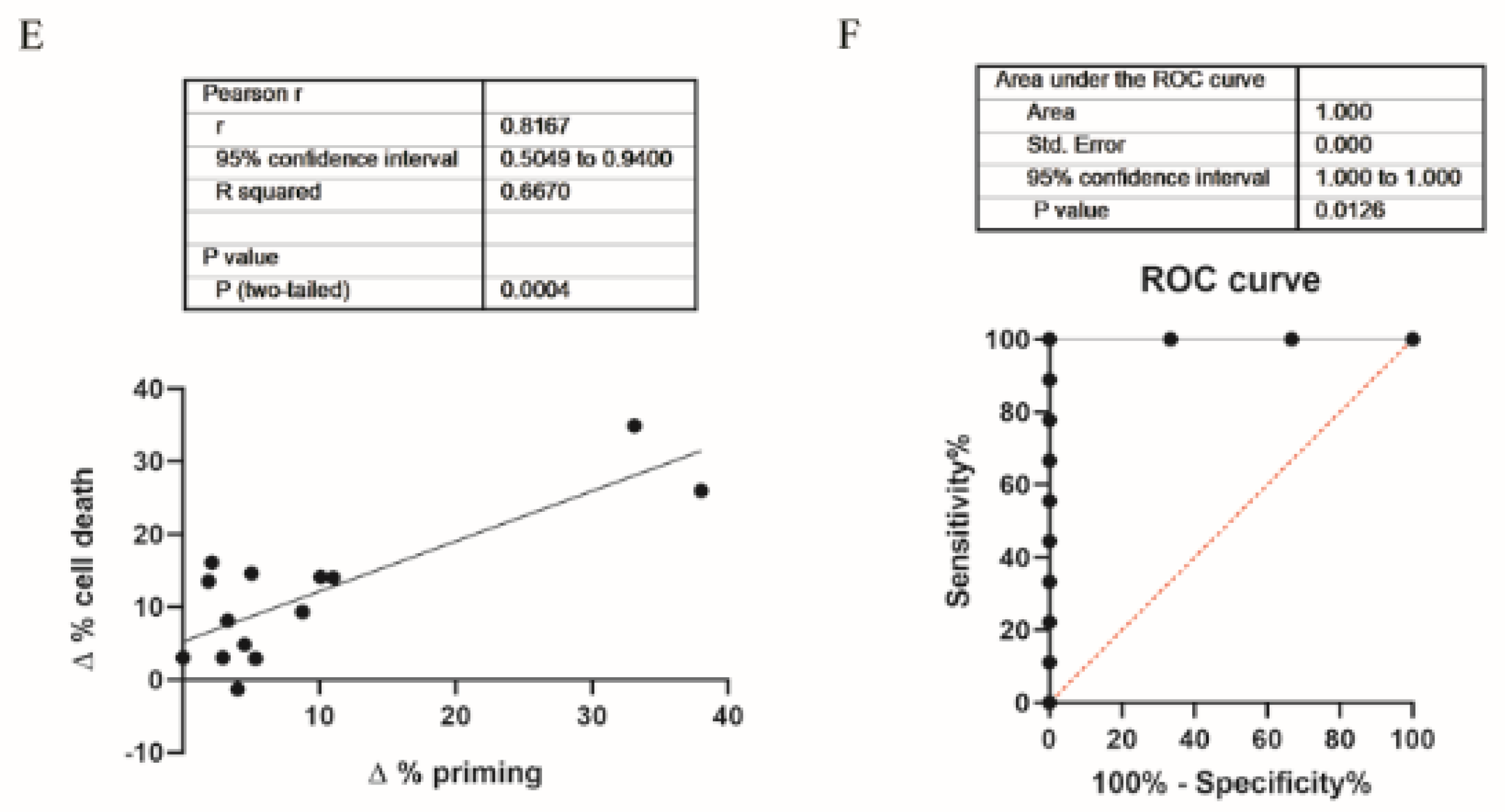
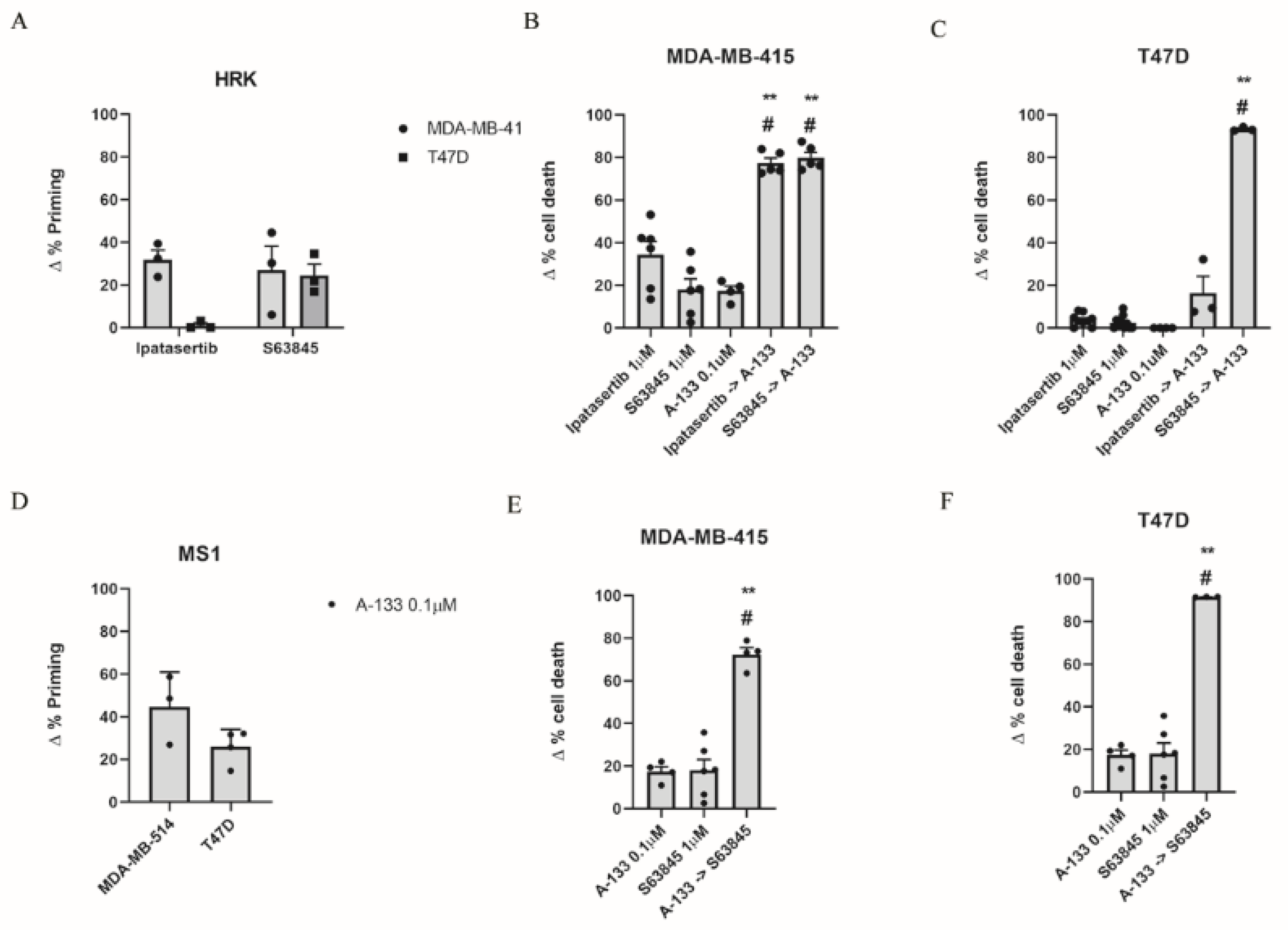
3.1. Dynamic BH3 Profiling Predicts Targeted Agents’ Effectiveness in ER+ Breast Cancer Cells
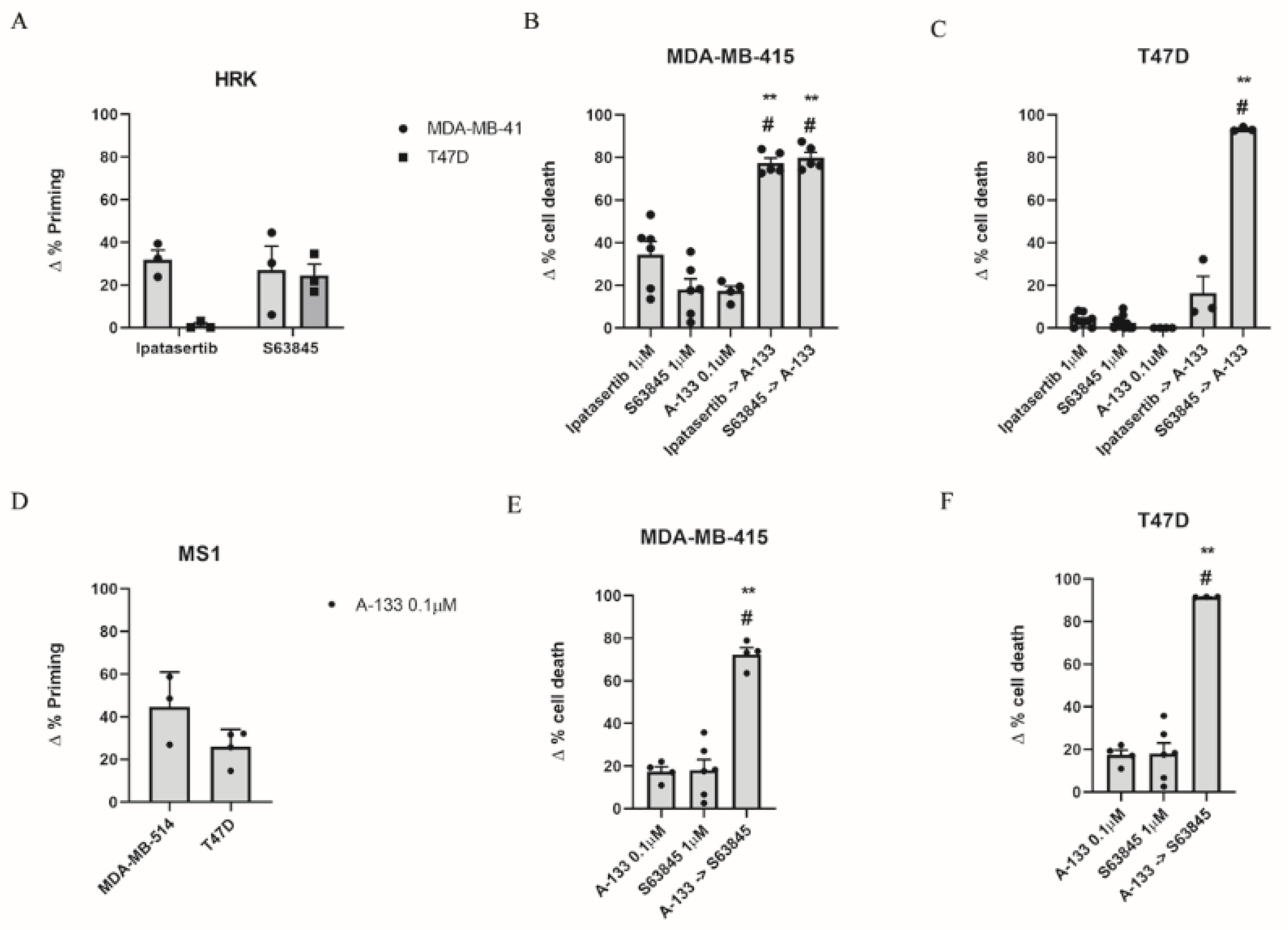
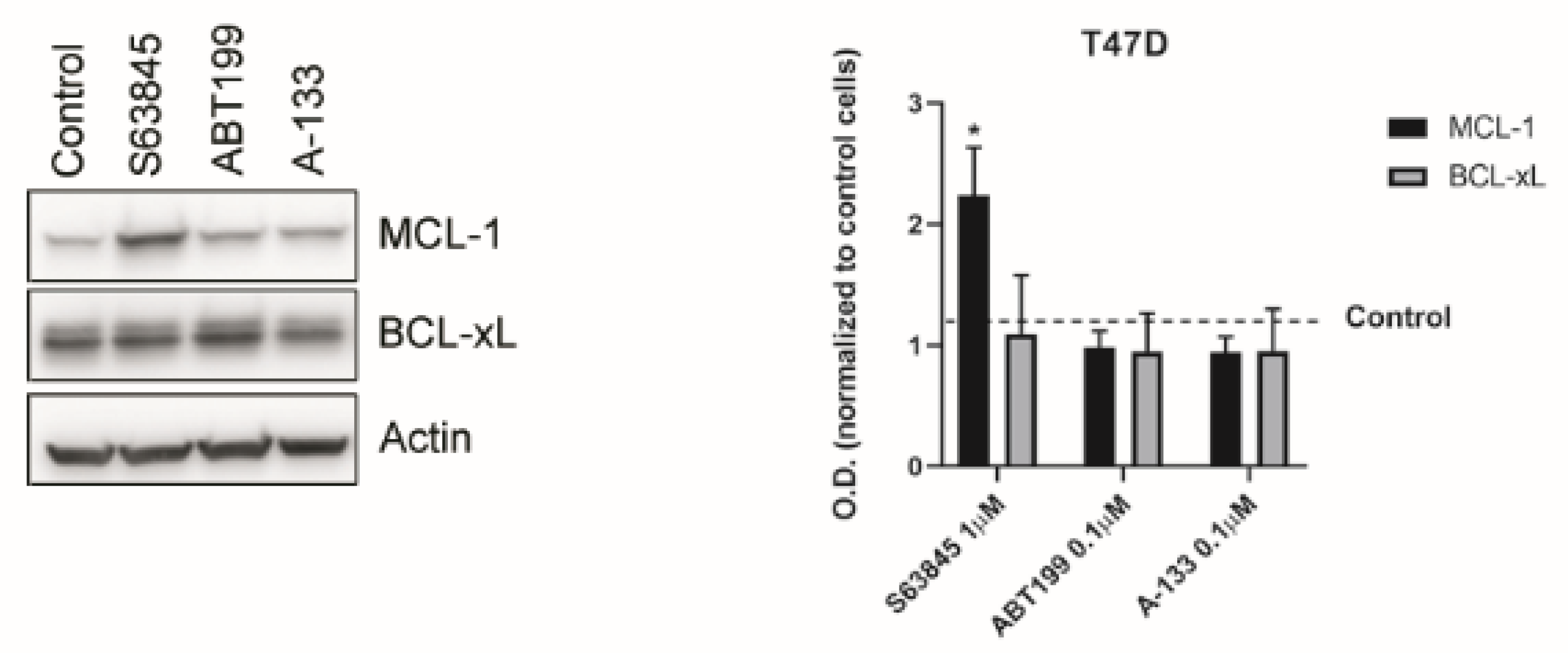
3.2. Inhibition of Anti-Apoptotic Adaptations Could Overcome Treatment-Induced Resistance
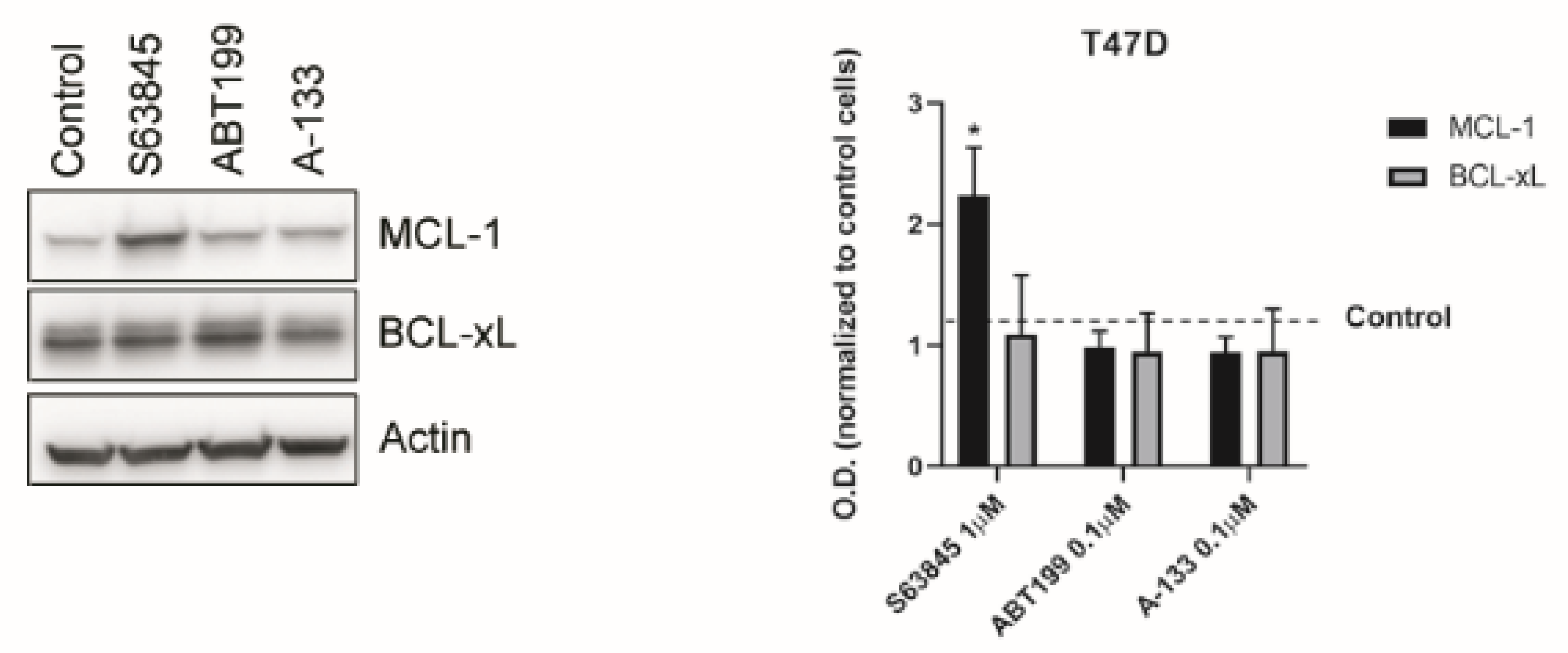
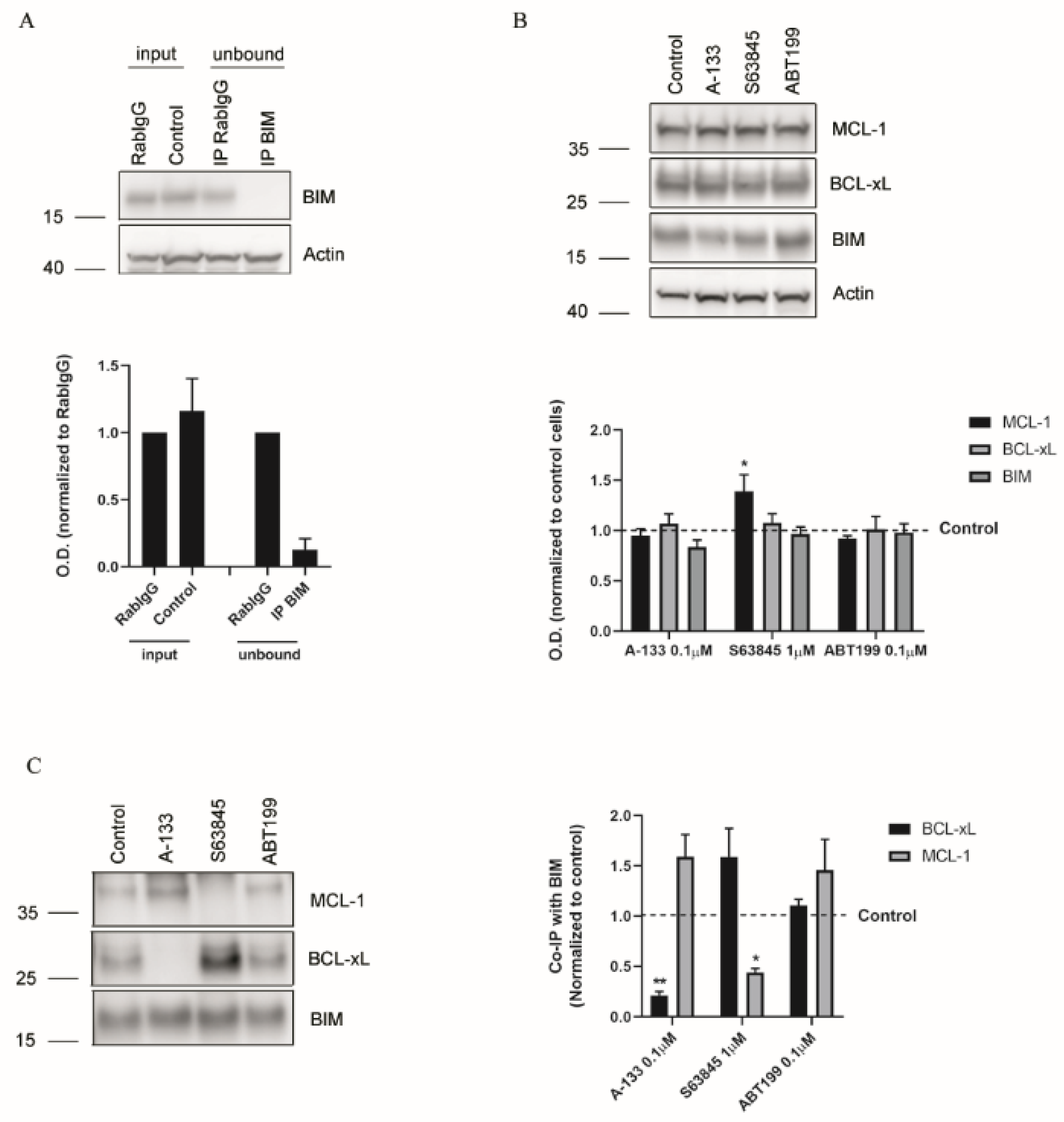
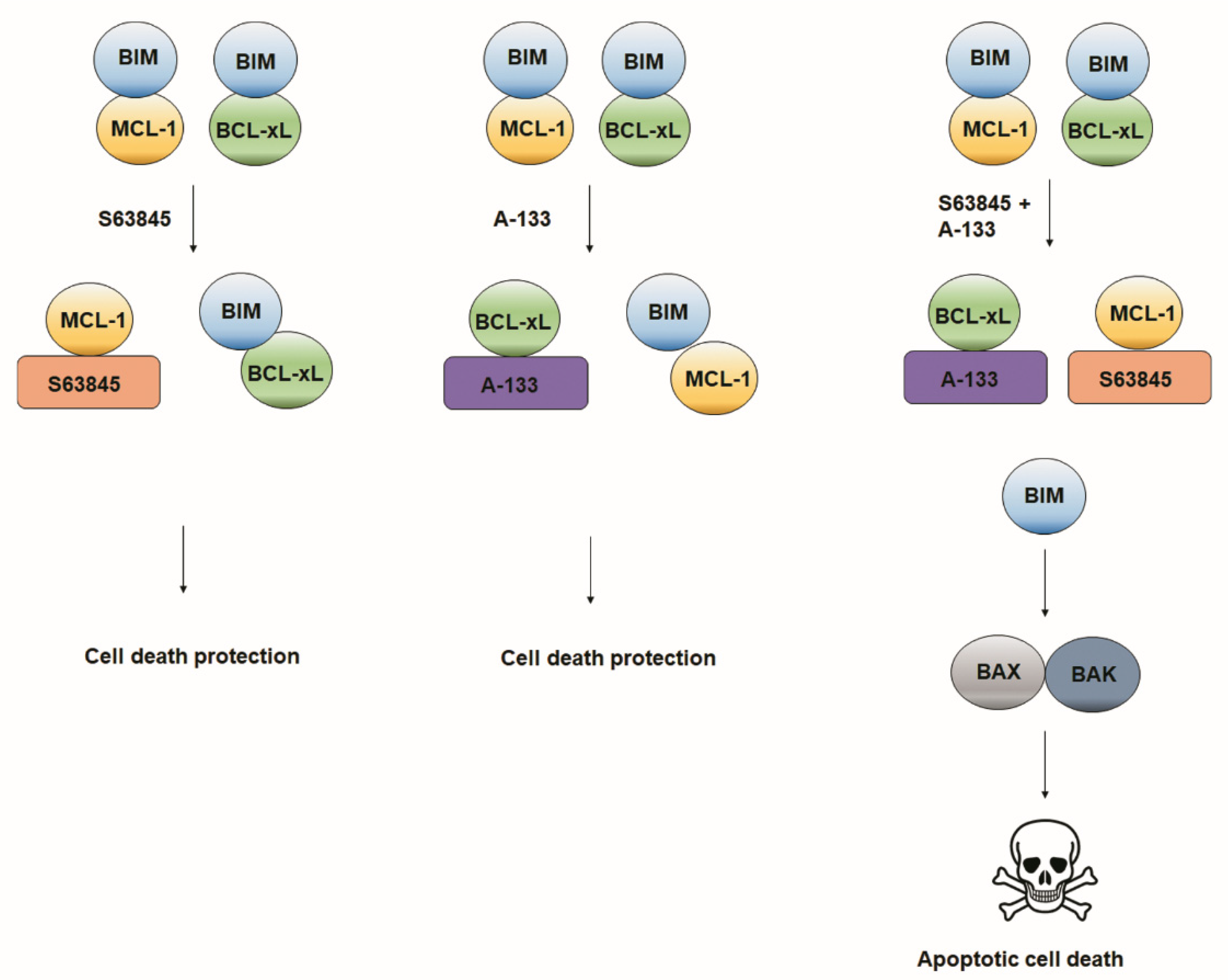
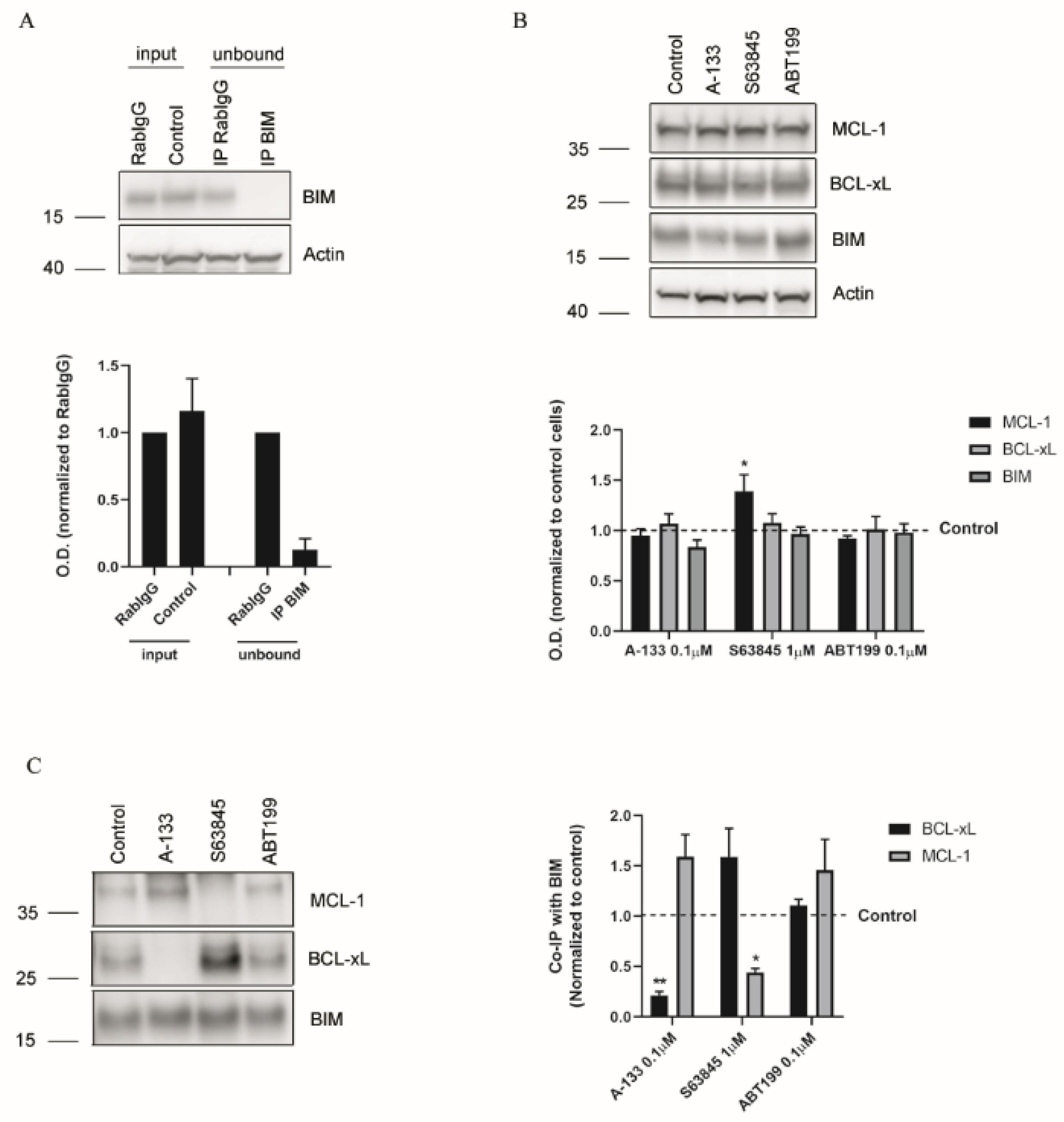
3.3. Resistance to Treatments Relies on BCL-xL and MCL-1 Binding to BIM
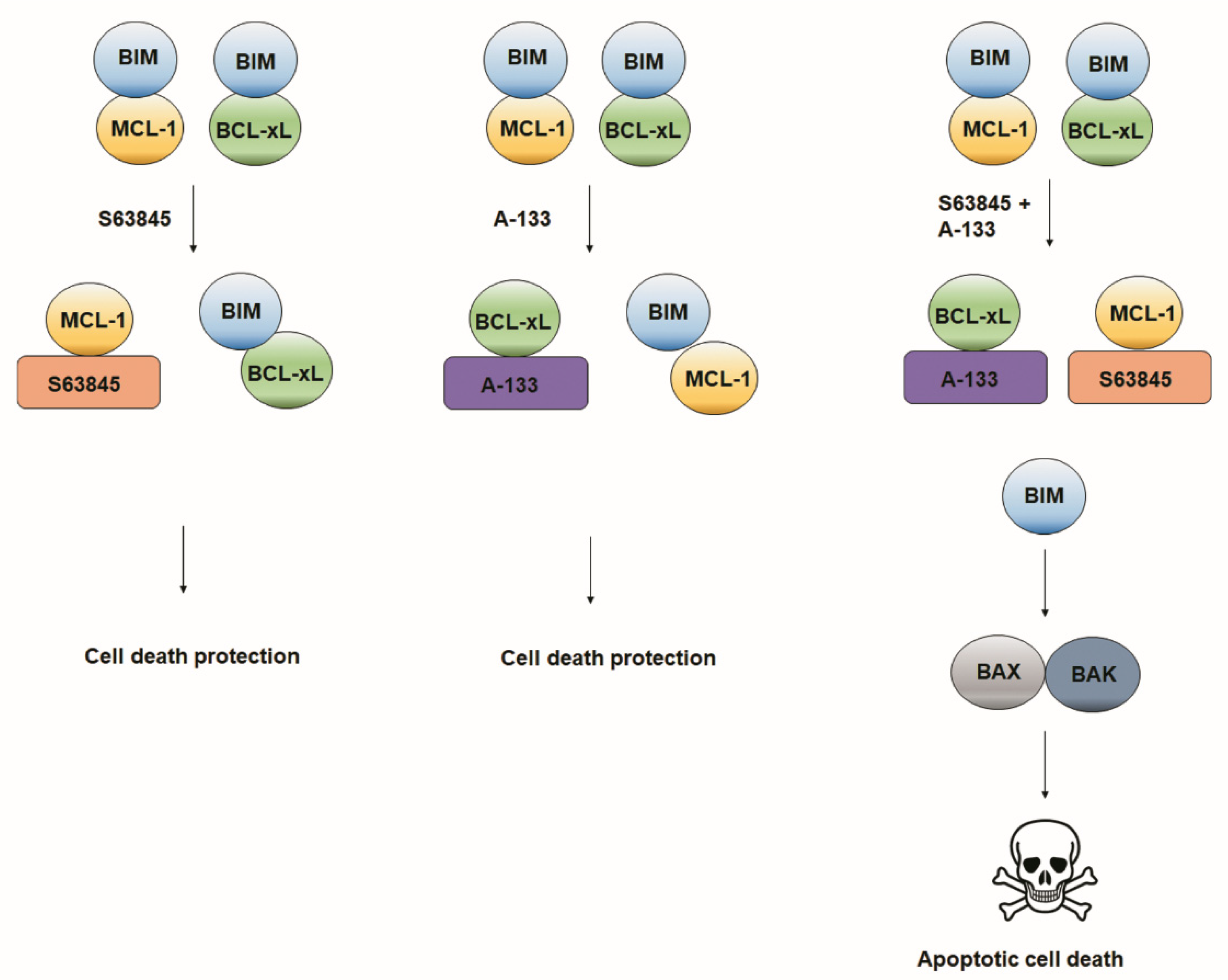
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Murawa, P.; Murawa, D.; Adamczyk, B.; Połom, K. Breast cancer: Actual methods of treatment and future trends. Rep. Pract. Oncol. Radiother. 2014, 19, 165–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, X.; Li, T.; Bai, Z.; Yang, Y.; Liu, X.; Zhan, J.; Shi, B. Breast cancer intrinsic subtype classification, clinical use and future trends. Am. J. Cancer Res. 2015, 5, 2929–2943. [Google Scholar]
- Higgins, M.J.; Baselga, J. Targeted therapies for breast cancer. J. Clin. Investig. 2011, 121, 3797–3803. [Google Scholar] [CrossRef]
- Faber, A.C.; Corcoran, R.B.; Ebi, H.; Sequist, L.V.; Waltman, B.A.; Chung, E.; Incio, J.; Digumarthy, S.R.; Pollack, S.F.; Song, Y.; et al. BIM Expression in Treatment-Naïve Cancers Predicts Responsiveness to Kinase Inhibitors. Cancer Discov. 2011, 1, 352–365. [Google Scholar] [CrossRef] [Green Version]
- Sequist, L.V.; Martins, R.G.; Spigel, D.; Grunberg, S.M.; Spira, A.; Janne, P.A.; Joshi, V.A.; McCollum, D.; Evans, T.L.; Muzikansky, A.; et al. First-line gefitinib in patients with advanced non-small-cell lung cancer harboring somatic EGFR mutations. J. Clin. Oncol. 2008, 26, 2442–2449. [Google Scholar] [CrossRef]
- Brachmann, S.M.; Hofmann, I.; Schnell, C.; Fritsch, C.; Wee, S.; Lane, H.; Wang, S.; Garcia-Echeverria, C.; Maira, S.-M. Specific apoptosis induction by the dual PI3K/mTor inhibitor NVP-BEZ235 in HER2 amplified and PIK3CA mutant breast cancer cells. Proc. Natl. Acad. Sci. USA 2009, 106, 22299–22304. [Google Scholar] [CrossRef] [Green Version]
- Zardavas, D.; Baselga, J.; Piccart-Gebhart, M. Emerging targeted agents in metastatic breast cancer. Nat. Rev. Clin. Oncol. 2013, 10, 191–210. [Google Scholar] [CrossRef]
- Burstein, H.J.; Keshaviah, A.; Baron, A.D.; Hart, R.D.; Lambert-Falls, R.; Marcom, P.K.; Gelman, R.; Winer, E.P. Trastuzumab plus vinorelbine or taxane chemotherapy for HER2-overexpressing metastatic breast cancer: The trastuzumab and vinorelbine or taxane study. Cancer 2007, 110, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Faber, A.C.; Li, D.; Song, Y.; Liang, M.-C.; Yeap, B.Y.; Bronson, R.T.; Lifshits, E.; Chen, Z.; Maira, S.-M.; García-Echeverría, C.; et al. Differential induction of apoptosis in HER2 and EGFR addicted cancers following PI3K inhibition. Proc. Natl. Acad. Sci. USA 2009, 106, 19503–19508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- E Konecny, G.; Venkatesan, N.; Yang, G.; Dering, J.; Ginther, C.; Finn, R.; Rahmeh, M.; Fejzo, M.S.; Toft, D.; Jiang, S.-W.; et al. Activity of lapatinib a novel HER2 and EGFR dual kinase inhibitor in human endometrial cancer cells. Br. J. Cancer 2008, 98, 1076–1084. [Google Scholar] [CrossRef] [Green Version]
- Baselga, J.; Swain, S.M. Novel anticancer targets: Revisiting ERBB2 and discovering ERBB3. Nat. Rev. Cancer 2009, 9, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of Chemotherapy plus a Monoclonal Antibody against HER2 for Metastatic Breast Cancer That Overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef]
- Robson, M.; Im, S.-A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef]
- Carroll, J. EJE PRIZE 2016: Mechanisms of oestrogen receptor (ER) gene regulation in breast cancer. Eur. J. Endocrinol. 2016, 175, R41–R49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, W.; Chang, J.; Fu, P. Endocrine therapy resistance in breast cancer: Current status, possible mechanisms and overcoming strategies. Future Med. Chem. 2015, 7, 1511–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, S. A historic and scientific review of breast cancer: The next global healthcare challenge. Int. J. Gynecol. Obstet. 2015, 131, S36–S39. [Google Scholar] [CrossRef] [Green Version]
- Jordan, V.C. Tamoxifen: A most unlikely pioneering medicine. Nat. Rev. Drug Discov. 2003, 2, 205–213. [Google Scholar] [CrossRef]
- Villanueva, M.T. Targeted therapies: Priming apoptosis. Nat. Rev. Clin. Oncol. 2013, 10, 67. [Google Scholar] [CrossRef]
- Chang, F.; Lee, J.T.; Navolanic, P.M.; Steelman, L.S.; Shelton, J.G.; Blalock, W.L.; Franklin, R.A.; McCubrey, J. Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: A target for cancer chemotherapy. Leukemia 2003, 17, 590–603. [Google Scholar] [CrossRef] [Green Version]
- Cragg, M.S.; Kuroda, J.; Puthalakath, H.; Huang, D.C.; Strasser, A. Gefitinib-Induced Killing of NSCLC Cell Lines Expressing Mutant EGFR Requires BIM and Can Be Enhanced by BH3 Mimetics. PLoS Med. 2007, 4, e316. [Google Scholar] [CrossRef] [Green Version]
- Jacquel, A.; Herrant, M.; Legros, L.; Belhacene, N.; Luciano, F.; Pages, G.; Hofman, P.; Auberger, P. Imatinib induces mitochondria-dependent apoptosis of the Bcr-Abl-positive K562 cell line and its differentiation toward the erythroid lineage 1. FASEB J. 2003, 17, 2160–2162. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Konopleva, M.; Burks, J.K.; Dywer, K.C.; Schober, W.D.; Yang, J.-Y.; McQueen, T.J.; Hung, M.-C.; Andreeff, M. Blockade of Mitogen-Activated Protein Kinase/Extracellular Signal-Regulated Kinase Kinase and Murine Double Minute Synergistically Induces Apoptosis in Acute Myeloid Leukemia via BH3-Only Proteins Puma and Bim. Cancer Res. 2010, 70, 2424–2434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunelle, J.K.; Letai, A. Control of mitochondrial apoptosis by the Bcl-2 family. J. Cell Sci. 2009, 122, 437–441. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.; Shimamura, T.; Perera, S.; Carlson, N.E.; Cai, D.; Shapiro, G.I.; Wong, K.K.; Letai, A. Proapoptotic BH3-Only BCL-2 Family Protein BIM Connects Death Signaling from Epidermal Growth Factor Receptor Inhibition to the Mitochondrion. Cancer Res. 2007, 67, 11867–11875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Certo, M.; Moore, V.D.G.; Nishino, M.; Wei, G.; Korsmeyer, S.; Armstrong, S.A.; Letai, A. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell 2006, 9, 351–365. [Google Scholar] [CrossRef] [Green Version]
- Montero, J.; Sarosiek, K.; DeAngelo, J.; Maertens, O.; Ryan, J.; Ercan, D.; Piao, H.; Horowitz, N.S.; Berkowitz, R.S.; Matulonis, U.; et al. Drug-Induced Death Signaling Strategy Rapidly Predicts Cancer Response to Chemotherapy. Cell 2015, 160, 977–989. [Google Scholar] [CrossRef] [Green Version]
- Montero, J.; Stephansky, J.; Cai, T.; Griffin, G.K.; Cabal-Hierro, L.; Togami, K.; Hogdal, L.J.; Galinsky, I.; Morgan, E.A.; Aster, J.C.; et al. Blastic Plasmacytoid Dendritic Cell Neoplasm Is Dependent on BCL2 and Sensitive to Venetoclax. Cancer Discov. 2017, 7, 156–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef]
- Kotschy, A.; Szlavik, Z.; Murray, J.; Davidson, J.; Maragno, A.L.; Le Toumelin-Braizat, G.; Chanrion, M.; Kelly, G.L.; Gong, J.-N.; Moujalled, D.M.; et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 2016, 538, 477–482. [Google Scholar] [CrossRef]
- Leverson, J.D.; Phillips, D.C.; Mitten, M.J.; Boghaert, E.R.; Diaz, D.; Tahir, S.K.; Belmont, L.D.; Nimmer, P.; Xiao, Y.; Ma, X.M.; et al. Exploiting selective BCL-2 family inhibitors to dissect cell survival dependencies and define improved strategies for cancer therapy. Sci. Transl. Med. 2015, 7, 279ra40. [Google Scholar] [CrossRef]
- Montero, J.; Letai, A. Why do BCL-2 inhibitors work and where should we use them in the clinic? Cell Death Differ. 2018, 25, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef]
- Merino, D.; Whittle, J.R.; Vaillant, F.; Serrano, A.; Gong, J.N.; Giner, G.; Maragno, A.L.; Chanrion, M.; Schneider, E.; Pal, B.; et al. Synergistic action of the MCL-1 inhibitor S63845 with current therapies in preclinical models of triple-negative and HER2-amplified breast cancer. Sci. Transl. Med. 2017, 9, eaam7049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, G.R.; Wardell, S.E.; Cakir, M.; Crawford, L.; Leeds, J.C.; Nussbaum, D.P.; Shankar, P.S.; Soderquist, R.S.; Stein, E.M.; Tingley, J.P.; et al. PIK3CA mutations enable targeting of a breast tumor dependency through mTOR-mediated MCL-1 translation. Sci. Transl. Med. 2016, 8, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Vaillant, F.; Merino, D.; Lee, L.; Breslin, K.; Pal, B.; Ritchie, M.; Smyth, G.K.; Christie, M.; Phillipson, L.; Burns, C.J.; et al. Targeting BCL-2 with the BH3 Mimetic ABT-199 in Estrogen Receptor-Positive Breast Cancer. Cancer Cell 2013, 24, 120–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, J.; Montero, J.; Rocco, J.; Letai, A. iBH3: Simple, fixable BH3 profiling to determine apoptotic priming in primary tissue by flow cytometry. Biol. Chem. 2016, 397, 671–678. [Google Scholar] [CrossRef]
- Montero, J.; Gstalder, C.; Kim, D.J.; Sadowicz, D.; Miles, W.; Manos, M.; Cidado, J.R.; Secrist, J.P.; Tron, A.E.; Flaherty, K.; et al. Destabilization of NOXA mRNA as a common resistance mechanism to targeted therapies. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef]
- Foight, G.W.; Ryan, J.; Gullá, S.V.; Letai, A.; Keating, A.E. Designed BH3 Peptides with High Affinity and Specificity for Targeting Mcl-1 in Cells. ACS Chem. Biol. 2014, 9, 1962–1968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzgerald, J.B.; Schoeberl, B.; Nielsen, U.B.; Sorger, P.K. Systems biology and combination therapy in the quest for clinical efficacy. Nat. Chem. Biol. 2006, 2, 458–466. [Google Scholar] [CrossRef]
- Oakes, S.R.; Vaillant, F.; Lim, E.; Lee, L.; Breslin, K.; Feleppa, F.; Deb, S.; Ritchie, M.E.; Takano, E.; Ward, T.; et al. Sensitization of BCL-2-expressing breast tumors to chemotherapy by the BH3 mimetic ABT-737. Proc. Natl. Acad. Sci. USA 2012, 109, 2766–2771. [Google Scholar] [CrossRef] [Green Version]
- Nitulescu, G.M.; Margina, D.; Juzenas, P.; Peng, Q.; Olaru, O.T.; Saloustros, E.; Fenga, C.; Spandidos, D.; Libra, M.; Tsatsakis, A. Akt inhibitors in cancer treatment: The long journey from drug discovery to clinical use (Review). Int. J. Oncol. 2016, 48, 869–885. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.J.; Tan, T.J.Y.; Dent, R.A. Novel therapeutic avenues in triple-negative breast cancer: PI3K/AKT inhibition, androgen receptor blockade, and beyond. Ther. Adv. Med. Oncol. 2019, 11, 1–11. [Google Scholar] [CrossRef]
- Yi, Z.; Ma, F.; Liu, B.; Guan, X.; Li, L.; Li, C.; Qian, H.; Xu, B. Everolimus in hormone receptor-positive metastatic breast cancer: PIK3CA mutation H1047R was a potential efficacy biomarker in a retrospective study. BMC Cancer 2019, 19, 1–9. [Google Scholar] [CrossRef] [PubMed]
- André, F.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor–Positive Advanced Breast Cancer. N. Engl. J. Med. 2019, 380, 1929–1940. [Google Scholar] [CrossRef] [PubMed]
- Serra, F.; Lapidari, P.; Quaquarini, E.; Tagliaferri, B.; Sottotetti, F.; Palumbo, R. Palbociclib in metastatic breast cancer: Current evidence and real-life data. Drugs Context 2019, 8, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Nathan, M.R.; Schmid, P. A Review of Fulvestrant in Breast Cancer. Oncol. Ther. 2017, 5, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Johnston, S.; Cheung, K. Fulvestrant—A Novel Endocrine Therapy for Breast Cancer. Curr. Med. Chem. 2010, 17, 902–914. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Cheng, H.; Bai, Z.; Li, J. Breast Cancer Cell Line Classification and Its Relevance with Breast Tumor Subtyping. J. Cancer 2017, 8, 3131–3141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foucquier, J.; Guedj, M. Analysis of drug combinations: Current methodological landscape. Pharmacol. Res. Perspect. 2015, 3, e00149. [Google Scholar] [CrossRef] [PubMed]
- Early Breast Cancer Trialists’ Collaborative Group. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: Patient-level meta-analysis of randomised trials. Lancet 2011, 378, 771–784. [Google Scholar] [CrossRef] [Green Version]
- Cree, I.A.; Charlton, P. Molecular chess? Hallmarks of anti-cancer drug resistance. BMC Cancer 2017, 17, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musgrove, E.A.; Sutherland, R.L. Biological determinants of endocrine resistance in breast cancer. Nat. Rev. Cancer 2009, 9, 631–643. [Google Scholar] [CrossRef]
- Campbell, K.J.; Dhayade, S.; Ferrari, N.; Sims, A.; Johnson, E.; Mason, S.M.; Dickson, A.; Ryan, K.M.; Kalna, G.; Edwards, J.; et al. MCL-1 is a prognostic indicator and drug target in breast cancer. Cell Death Dis. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Jänicke, R.U. MCF-7 breast carcinoma cells do not express caspase-3. Breast Cancer Res. Treat. 2008, 117, 219–221. [Google Scholar] [CrossRef] [Green Version]
- Campbell, K.J.; Mason, S.M.; Winder, M.L.; Willemsen, R.B.E.; Cloix, C.; Lawson, H.; Rooney, N.; Dhayade, S.; Sims, A.H.; Blyth, K.; et al. Breast cancer dependence on MCL-1 is due to its canonical anti-apoptotic function. Cell Death Differ. 2021, 1–12. [Google Scholar]
- Hird, A.W.; Tron, A.E. Recent advances in the development of Mcl-1 inhibitors for cancer therapy. Pharmacol. Ther. 2019, 198, 59–67. [Google Scholar] [CrossRef]
- Eichhorn, J.M.; Alford, S.E.; Sakurikar, N.; Chambers, T.C. Molecular analysis of functional redundancy among anti-apoptotic Bcl-2 proteins and its role in cancer cell survival. Exp. Cell Res. 2014, 322, 415–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chipuk, J.E.; Moldoveanu, T.; Llambi, F.; Parsons, M.J.; Green, D.R. The BCL-2 Family Reunion. Mol. Cell 2010, 37, 299–310. [Google Scholar] [CrossRef]
- Fritsch, C.; Huang, A.; Chatenay-Rivauday, C.; Schnell, C.; Reddy, A.; Liu, M.; Kauffmann, A.; Guthy, D.; Erdmann, D.; De Pover, A.; et al. Characterization of the novel and specific PI3Ka inhibitor NVP-BYL719 and development of the patient stratification strategy for clinical trials. Mol. Cancer Ther. 2014, 13, 1117–1129. [Google Scholar] [CrossRef] [Green Version]
- Saura, C.; Roda, D.; Roselló, S.; Oliveira, M.; Macarulla, T.; Pérez-Fidalgo, J.A.; Morales-Barrera, R.; Sanchis-García, J.M.; Musib, L.; Budha, N.; et al. A First-in-Human Phase I Study of the ATP-Competitive AKT Inhibitor Ipatasertib Demonstrates Robust and Safe Targeting of AKT in Patients with Solid Tumors. Cancer Discov. 2017, 7, 102–113. [Google Scholar] [CrossRef] [Green Version]
- Weigelt, B.; Warne, P.H.; Downward, J. PIK3CA mutation, but not PTEN loss of function, determines the sensitivity of breast cancer cells to mTOR inhibitory drugs. Oncogene 2011, 30, 3222–3233. [Google Scholar] [CrossRef] [PubMed]
- Zañudo, J.G.; Scaltriti, M.; Albert, R. A network modeling approach to elucidate drug resistance mechanisms and predict combinatorial drug treatments in breast cancer. Cancer Converg. 2017, 1, 1–25. [Google Scholar]
- Wongchenko, M.J.; Kim, S.-B.; Saura, C.; Oliveira, M.; Lipson, D.; Kennedy, M.; Greene, M.; Breese, V.; Mani, A.; Xu, N.; et al. Circulating Tumor DNA and Biomarker Analyses from the LOTUS Randomized Trial of First-Line Ipatasertib and Paclitaxel for Metastatic Triple-Negative Breast Cancer. JCO Precis. Oncol. 2020, 1012–1024. [Google Scholar] [CrossRef]
- Wu, G.; Xing, M.; Mambo, E.; Huang, X.; Liu, J.; Guo, Z.; Chatterjee, A.; Goldenberg, D.; Gollin, S.M.; Sukumar, S.; et al. Somatic mutation and gain of copy number of PIK3CA in human breast cancer. Breast Cancer Res. 2005, 7, R609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.; Ye, A.S.; Gardino, A.K.; Heijink, A.M.; Sorger, P.K.; MacBeath, G.; Yaffe, M.B. Sequential Application of Anticancer Drugs Enhances Cell Death by Rewiring Apoptotic Signaling Networks. Cell 2012, 149, 780–794. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alcon, C.; Gómez Tejeda Zañudo, J.; Albert, R.; Wagle, N.; Scaltriti, M.; Letai, A.; Samitier, J.; Montero, J. ER+ Breast Cancer Strongly Depends on MCL-1 and BCL-xL Anti-Apoptotic Proteins. Cells 2021, 10, 1659. https://doi.org/10.3390/cells10071659
Alcon C, Gómez Tejeda Zañudo J, Albert R, Wagle N, Scaltriti M, Letai A, Samitier J, Montero J. ER+ Breast Cancer Strongly Depends on MCL-1 and BCL-xL Anti-Apoptotic Proteins. Cells. 2021; 10(7):1659. https://doi.org/10.3390/cells10071659
Chicago/Turabian StyleAlcon, Clara, Jorge Gómez Tejeda Zañudo, Reka Albert, Nikhil Wagle, Maurizio Scaltriti, Anthony Letai, Josep Samitier, and Joan Montero. 2021. "ER+ Breast Cancer Strongly Depends on MCL-1 and BCL-xL Anti-Apoptotic Proteins" Cells 10, no. 7: 1659. https://doi.org/10.3390/cells10071659
APA StyleAlcon, C., Gómez Tejeda Zañudo, J., Albert, R., Wagle, N., Scaltriti, M., Letai, A., Samitier, J., & Montero, J. (2021). ER+ Breast Cancer Strongly Depends on MCL-1 and BCL-xL Anti-Apoptotic Proteins. Cells, 10(7), 1659. https://doi.org/10.3390/cells10071659