
The Role of cAMP in Beta Cell Stimulus–Secretion and Intercellular Coupling

 , ,
, ,  and
and {kind=link}
{kind=link}
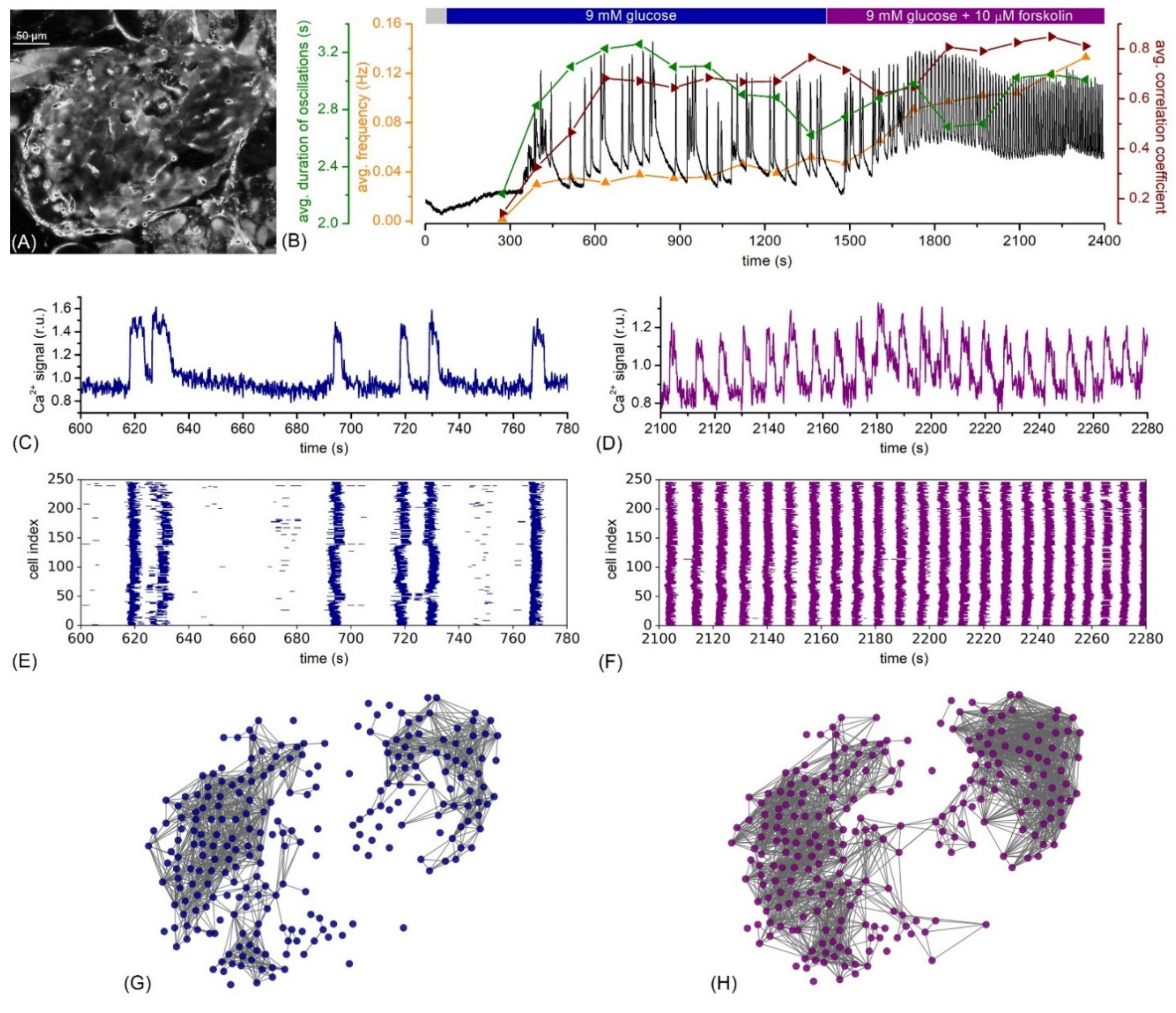{kind=link}
Abstract
1. Introduction
2. The Role of cAMP on Stimulus Secretion Coupling
2.1. The Effect of cAMP on Ion Channels
2.1.1. KATP Channels
2.1.2. Kv Channels
2.1.3. TRP Channels
2.2. The Effect of cAMP on [Ca2+]IC
2.3. The Effect of cAMP on Exocytosis
3. The Role of cAMP in Intercellular Coupling
4. The Role of cAMP during the Development of Insulin Resistance
5. Conclusions
Funding
Conflicts of Interest
References
- IDF Diabetes Atlas, 9th ed.; International Diabetes Federation: Brussels, Belgium, 2019.
- Stratton, I.M.; Adler, A.I.; Neil, H.A.; Matthews, D.R.; Manley, S.E.; Cull, C.A.; Hadden, D.; Turner, R.C.; Holman, R.R. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): Prospective observational study. BMJ 2000, 321, 405–412. [Google Scholar] [CrossRef]
- Weyer, C.; Bogardus, C.; Mott, D.M.; Pratley, R.E. The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. J. Clin. Investig. 1999, 104, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Seino, S.; Takahashi, H.; Takahashi, T.; Shibasaki, T. Treating diabetes today: A matter of selectivity of sulphonylureas. Diabetes Obes. Metab. 2012, 14, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Skelin Klemen, M.; Dolenšek, J.; Slak Rupnik, M.; Stožer, A. The triggering pathway to insulin secretion: Functional similarities and differences between the human and the mouse β cells and their translational relevance. Islets 2017, 9, 109–139. [Google Scholar] [CrossRef]
- Henquin, J.C. Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes 2000, 49, 1751–1760. [Google Scholar] [CrossRef] [PubMed]
- Henquin, J.-C. The dual control of insulin secretion by glucose involves triggering and amplifying pathways in β-cells. Diabetes Res. Clin. Pract. 2011, 93, S27–S31. [Google Scholar] [CrossRef]
- Sturgess, N.C.; Ashford, M.L.; Cook, D.L.; Hales, C.N. The sulphonylurea receptor may be an ATP-sensitive potassium channel. Lancet 1985, 2, 474–475. [Google Scholar] [CrossRef]
- De Wet, H.; Proks, P. Molecular action of sulphonylureas on KATP channels: A real partnership between drugs and nucleotides. Biochem. Soc. Trans. 2015, 43, 901–907. [Google Scholar] [CrossRef]
- Proks, P.; de Wet, H.; Ashcroft, F.M. Molecular mechanism of sulphonylurea block of K(ATP) channels carrying mutations that impair ATP inhibition and cause neonatal diabetes. Diabetes 2013, 62, 3909–3919. [Google Scholar] [CrossRef]
- Pipatpolkai, T.; Usher, S.; Stansfeld, P.J.; Ashcroft, F.M. New insights into KATP channel gene mutations and neonatal diabetes mellitus. Nat. Rev. Endocrinol. 2020, 16, 378–393. [Google Scholar] [CrossRef]
- Efanova, I.B.; Zaitsev, S.V.; Zhivotovsky, B.; Kohler, M.; Efendic, S.; Orrenius, S.; Berggren, P.O. Glucose and tolbutamide induce apoptosis in pancreatic beta-cells. A process dependent on intracellular Ca2+ concentration. J. Biol. Chem. 1998, 273, 33501–33507. [Google Scholar] [CrossRef]
- Maedler, K.; Carr, R.D.; Bosco, D.; Zuellig, R.A.; Berney, T.; Donath, M.Y. Sulfonylurea induced beta-cell apoptosis in cultured human islets. J. Clin. Endocrinol. Metab. 2005, 90, 501–506. [Google Scholar] [CrossRef]
- Elrick, H.; Stimmler, L.; Hlad, C.J., Jr.; Arai, Y. Plasma Insulin Response to Oral and Intravenous Glucose Administration. J. Clin. Endocrinol. Metab. 1964, 24, 1076–1082. [Google Scholar] [CrossRef]
- McIntyre, N.; Holdsworth, C.D.; Turner, D.S. New Interpretation of Oral Glucose Tolerance. Lancet 1964, 2, 20–21. [Google Scholar] [CrossRef]
- Jones, B.; Bloom, S.R.; Buenaventura, T.; Tomas, A.; Rutter, G.A. Control of insulin secretion by GLP-1. Peptides 2018, 100, 75–84. [Google Scholar] [CrossRef]
- Nauck, M.A.; Meier, J.J. Incretin hormones: Their role in health and disease. Diabetes Obes. Metab. 2018, 20, 5–21. [Google Scholar] [CrossRef]
- Nauck, M.A.; Meier, J.J. The incretin effect in healthy individuals and those with type 2 diabetes: Physiology, pathophysiology, and response to therapeutic interventions. Lancet Diabetes Endocrinol. 2016, 4, 525–536. [Google Scholar] [CrossRef]
- Gasbjerg, L.S.; Bergmann, N.C.; Stensen, S.; Christensen, M.B.; Rosenkilde, M.M.; Holst, J.J.; Nauck, M.; Knop, F.K. Evaluation of the incretin effect in humans using GIP and GLP-1 receptor antagonists. Peptides 2020, 125, 170183. [Google Scholar] [CrossRef]
- Holst, J.J. The incretin system in healthy humans: The role of GIP and GLP-1. Metabolism 2019, 96, 46–55. [Google Scholar] [CrossRef]
- Dupre, J.; Ross, S.A.; Watson, D.; Brown, J.C. Stimulation of insulin secretion by gastric inhibitory polypeptide in man. J. Clin. Endocrinol. Metab. 1973, 37, 826–828. [Google Scholar] [CrossRef]
- Herrmann, C.; Goke, R.; Richter, G.; Fehmann, H.C.; Arnold, R.; Goke, B. Glucagon-like peptide-1 and glucose-dependent insulin-releasing polypeptide plasma levels in response to nutrients. Digestion 1995, 56, 117–126. [Google Scholar] [CrossRef]
- Vollmer, K.; Holst, J.J.; Baller, B.; Ellrichmann, M.; Nauck, M.A.; Schmidt, W.E.; Meier, J.J. Predictors of incretin concentrations in subjects with normal, impaired, and diabetic glucose tolerance. Diabetes 2008, 57, 678–687. [Google Scholar] [CrossRef]
- Takeda, J.; Seino, Y.; Tanaka, K.; Fukumoto, H.; Kayano, T.; Takahashi, H.; Mitani, T.; Kurono, M.; Suzuki, T.; Tobe, T.; et al. Sequence of an intestinal cDNA encoding human gastric inhibitory polypeptide precursor. Proc. Nat. Acad. Sci. USA 1987, 84, 7005–7008. [Google Scholar] [CrossRef]
- Mayo, K.E.; Miller, L.J.; Bataille, D.; Dalle, S.; Goke, B.; Thorens, B.; Drucker, D.J. International Union of Pharmacology. XXXV. The glucagon receptor family. Pharmacol. Rev. 2003, 55, 167–194. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, K.; Christensen, L.L.; Holst, J.J.; Orskov, C. GLP-1 and GIP are colocalized in a subset of endocrine cells in the small intestine. Regul. Pept. 2003, 114, 189–196. [Google Scholar] [CrossRef]
- Nauck, M.A.; Heimesaat, M.M.; Orskov, C.; Holst, J.J.; Ebert, R.; Creutzfeldt, W. Preserved incretin activity of glucagon-like peptide 1 [7-36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J. Clin. Investig. 1993, 91, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Toft-Nielsen, M.B.; Damholt, M.B.; Madsbad, S.; Hilsted, L.M.; Hughes, T.E.; Michelsen, B.K.; Holst, J.J. Determinants of the impaired secretion of glucagon-like peptide-1 in type 2 diabetic patients. J. Clin. Endocrinol. Metab. 2001, 86, 3717–3723. [Google Scholar] [CrossRef] [PubMed]
- Toft-Nielsen, M.B.; Madsbad, S.; Holst, J.J. Determinants of the effectiveness of glucagon-like peptide-1 in type 2 diabetes. J. Clin. Endocrinol. Metab. 2001, 86, 3853–3860. [Google Scholar] [CrossRef] [PubMed]
- Gutniak, M.; Orskov, C.; Holst, J.J.; Ahren, B.; Efendic, S. Antidiabetogenic effect of glucagon-like peptide-1 (7-36)amide in normal subjects and patients with diabetes mellitus. N. Engl. J. Med. 1992, 326, 1316–1322. [Google Scholar] [CrossRef]
- Holz, G.G.; Chepurny, O.G. Glucagon-like peptide-1 synthetic analogs: New therapeutic agents for use in the treatment of diabetes mellitus. Curr. Med. Chem. 2003, 10, 2471–2483. [Google Scholar] [CrossRef]
- Mietlicki-Baase, E.G.; Liberini, C.G.; Workinger, J.L.; Bonaccorso, R.L.; Borner, T.; Reiner, D.J.; Koch-Laskowski, K.; McGrath, L.E.; Lhamo, R.; Stein, L.M.; et al. A vitamin B12 conjugate of exendin-4 improves glucose tolerance without associated nausea or hypophagia in rodents. Diabetes Obes. Metab. 2018, 20, 1223–1234. [Google Scholar] [CrossRef]
- Seino, S.; Shibasaki, T.; Minami, K. Dynamics of insulin secretion and the clinical implications for obesity and diabetes. J. Clin. Investig. 2011, 121, 2118–2125. [Google Scholar] [CrossRef]
- Scrocchi, L.A.; Brown, T.J.; MaClusky, N.; Brubaker, P.L.; Auerbach, A.B.; Joyner, A.L.; Drucker, D.J. Glucose intolerance but normal satiety in mice with a null mutation in the glucagon-like peptide 1 receptor gene. Nat. Med. 1996, 2, 1254–1258. [Google Scholar] [CrossRef]
- Salehi, M.; Vahl, T.P.; D’Alessio, D.A. Regulation of islet hormone release and gastric emptying by endogenous glucagon-like peptide 1 after glucose ingestion. J. Clin. Endocrinol. Metab. 2008, 93, 4909–4916. [Google Scholar] [CrossRef]
- Salehi, M.; Aulinger, B.; D’Alessio, D.A. Effect of glycemia on plasma incretins and the incretin effect during oral glucose tolerance test. Diabetes 2012, 61, 2728–2733. [Google Scholar] [CrossRef]
- Aulinger, B.A.; Vahl, T.P.; Wilson-Perez, H.E.; Prigeon, R.L.; D’Alessio, D.A. beta-Cell Sensitivity to GLP-1 in Healthy Humans Is Variable and Proportional to Insulin Sensitivity. J. Clin. Endocrinol. Metab. 2015, 100, 2489–2496. [Google Scholar] [CrossRef]
- Kjems, L.L.; Holst, J.J.; Volund, A.; Madsbad, S. The influence of GLP-1 on glucose-stimulated insulin secretion: Effects on beta-cell sensitivity in type 2 and nondiabetic subjects. Diabetes 2003, 52, 380–386. [Google Scholar] [CrossRef]
- Muscelli, E.; Mari, A.; Casolaro, A.; Camastra, S.; Seghieri, G.; Gastaldelli, A.; Holst, J.J.; Ferrannini, E. Separate impact of obesity and glucose tolerance on the incretin effect in normal subjects and type 2 diabetic patients. Diabetes 2008, 57, 1340–1348. [Google Scholar] [CrossRef]
- Charles, M.A.; Fanska, R.; Schmid, F.G.; Forsham, P.H.; Grodsky, G.M. Adenosine 3′,5′-monophosphate in pancreatic islets: Glucose-induced insulin release. Science 1973, 179, 569–571. [Google Scholar] [CrossRef]
- Charles, M.A.; Lawecki, J.; Pictet, R.; Grodsky, G.M. Insulin secretion. Interrelationships of glucose, cyclic adenosine 3:5-monophosphate, and calcium. J. Biol. Chem. 1975, 250, 6134–6140. [Google Scholar] [CrossRef]
- Saikia, M.; Holter, M.M.; Donahue, L.R.; Lee, I.S.; Zheng, Q.C.; Wise, J.L.; Todero, J.E.; Phuong, D.J.; Garibay, D.; Coch, R.; et al. GLP-1 receptor signaling increases PCSK1 and β cell features in human α cells. JCI Insight 2021, 6. [Google Scholar] [CrossRef] [PubMed]
- Tengholm, A.; Gylfe, E. cAMP signalling in insulin and glucagon secretion. Diabetes Obes. Metab. 2017, 19, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Adame-García, S.R.; Cervantes-Villagrana, R.D.; Orduña-Castillo, L.B.; Del Rio, J.C.; Gutkind, J.S.; Reyes-Cruz, G.; Taylor, S.S.; Vázquez-Prado, J. cAMP-dependent activation of the Rac guanine exchange factor P-REX1 by type I protein kinase A (PKA) regulatory subunits. J. Biol. Chem. 2019, 294, 2232–2246. [Google Scholar] [CrossRef] [PubMed]
- Shibasaki, T.; Takahashi, H.; Miki, T.; Sunaga, Y.; Matsumura, K.; Yamanaka, M.; Zhang, C.; Tamamoto, A.; Satoh, T.; Miyazaki, J.-i.; et al. Essential role of Epac2/Rap1 signaling in regulation of insulin granule dynamics by cAMP. Proc. Natl. Acad. Sci. USA 2007, 104, 19333–19338. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.-L.; Katoh, M.; Shibasaki, T.; Minami, K.; Sunaga, Y.; Takahashi, H.; Yokoi, N.; Iwasaki, M.; Miki, T.; Seino, S. The cAMP Sensor Epac2 Is a Direct Target of Antidiabetic Sulfonylurea Drugs. Science 2009, 325, 607–610. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Shibasaki, T.; Takahashi, H.; Sugawara, K.; Ono, A.; Inoue, N.; Furuya, T.; Seino, S. Antidiabetic sulfonylureas and cAMP cooperatively activate Epac2A. Sci. Signal 2013, 6, ra94. [Google Scholar] [CrossRef]
- Seino, S.; Sugawara, K.; Yokoi, N.; Takahashi, H. beta-Cell signalling and insulin secretagogues: A path for improved diabetes therapy. Diabetes Obes. Metab. 2017, 19, 22–29. [Google Scholar] [CrossRef]
- Song, W.-J.; Seshadri, M.; Ashraf, U.; Mdluli, T.; Mondal, P.; Keil, M.; Azevedo, M.; Kirschner, L.S.; Stratakis, C.A.; Hussain, M.A. Snapin mediates incretin action and augments glucose-dependent insulin secretion. Cell Metab. 2011, 13, 308–319. [Google Scholar] [CrossRef]
- Song, W.J.; Mondal, P.; Li, Y.; Lee, S.E.; Hussain, M.A. Pancreatic beta-cell response to increased metabolic demand and to pharmacologic secretagogues requires EPAC2A. Diabetes 2013, 62, 2796–2807. [Google Scholar] [CrossRef]
- Hwang, M.; Go, Y.; Park, J.H.; Shin, S.K.; Song, S.E.; Oh, B.C.; Im, S.S.; Hwang, I.; Jeon, Y.H.; Lee, I.K.; et al. Epac2a-null mice exhibit obesity-prone nature more susceptible to leptin resistance. Int. J. Obes. 2017, 41, 279–288. [Google Scholar] [CrossRef]
- Jain, R.; Lammert, E. Cell–cell interactions in the endocrine pancreas. Diabetes Obes. Metab. 2009, 11, 159–167. [Google Scholar] [CrossRef]
- Meda, P. The in vivo beta-to-beta-cell chat room: Connexin connections matter. Diabetes 2012, 61, 1656–1658. [Google Scholar] [CrossRef]
- Weitz, J.; Menegaz, D.; Caicedo, A. Deciphering the Complex Communication Networks That Orchestrate Pancreatic Islet Function. Diabetes 2021, 70, 17–26. [Google Scholar] [CrossRef]
- Aslanidi, O.V.; Mornev, O.A.; Skyggebjerg, O.; Arkhammar, P.; Thastrup, O.; Sørensen, M.P.; Christiansen, P.L.; Conradsen, K.; Scott, A.C. Excitation Wave Propagation as a Possible Mechanism for Signal Transmission in Pancreatic Islets of Langerhans. Biophys. J. 2001, 80, 1195–1209. [Google Scholar] [CrossRef]
- Benninger, R.K.; Zhang, M.; Head, W.S.; Satin, L.S.; Piston, D.W. Gap junction coupling and calcium waves in the pancreatic islet. Biophys. J. 2008, 95, 5048–5061. [Google Scholar] [CrossRef]
- Bosco, D.; Haefliger, J.-A.; Meda, P. Connexins: Key Mediators of Endocrine Function. Physiol. Rev. 2011, 91, 1393–1445. [Google Scholar] [CrossRef]
- Stožer, A.; Dolenšek, J.; Rupnik, M.S. Glucose-Stimulated Calcium Dynamics in Islets of Langerhans in Acute Mouse Pancreas Tissue Slices. PLoS ONE 2013, 8, e54638. [Google Scholar] [CrossRef]
- Ravier, M.A.; Güldenagel, M.; Charollais, A.; Gjinovci, A.; Caille, D.; Söhl, G.; Wollheim, C.B.; Willecke, K.; Henquin, J.-C.; Meda, P. Loss of Connexin36 Channels Alters β-Cell Coupling, Islet Synchronization of Glucose-Induced Ca2+ and Insulin Oscillations, and Basal Insulin Release. Diabetes 2005, 54, 1798–1807. [Google Scholar] [CrossRef]
- Hamelin, R.; Allagnat, F.; Haefliger, J.A.; Meda, P. Connexins, diabetes and the metabolic syndrome. Curr. Protein Pept. Sci. 2009, 10, 18–29. [Google Scholar] [CrossRef]
- Head, W.S.; Orseth, M.L.; Nunemaker, C.S.; Satin, L.S.; Piston, D.W.; Benninger, R.K. Connexin-36 gap junctions regulate in vivo first- and second-phase insulin secretion dynamics and glucose tolerance in the conscious mouse. Diabetes 2012, 61, 1700–1707. [Google Scholar] [CrossRef]
- Benninger, R.K.; Piston, D.W. Cellular communication and heterogeneity in pancreatic islet insulin secretion dynamics. Trends Endocrinol. Metab. TEM 2014. [Google Scholar] [CrossRef]
- Satin, L.S.; Butler, P.C.; Ha, J.; Sherman, A.S. Pulsatile insulin secretion, impaired glucose tolerance and type 2 diabetes. Mol. Asp. Med. 2015, 42, 61–77. [Google Scholar] [CrossRef]
- Farnsworth, N.L.; Hemmati, A.; Pozzoli, M.; Benninger, R.K. Fluorescence recovery after photobleaching reveals regulation and distribution of connexin36 gap junction coupling within mouse islets of Langerhans. J. Physiol. 2014, 592, 4431–4446. [Google Scholar] [CrossRef]
- Rutter, G.A.; Hodson, D.J. Beta cell connectivity in pancreatic islets: A type 2 diabetes target? Cell. Mol. Life Sci. CMLS 2015, 72, 453–467. [Google Scholar] [CrossRef]
- Mears, D.; Sheppard, N.F.; Atwater, I.; Rojas, E. Magnitude and modulation of pancreatic β-cell gap junction electrical conductance in situ. J. Membr. Biol. 1995, 146, 163–176. [Google Scholar] [CrossRef]
- Somekawa, S.; Fukuhara, S.; Nakaoka, Y.; Fujita, H.; Saito, Y.; Mochizuki, N. Enhanced functional gap junction neoformation by protein kinase A-dependent and Epac-dependent signals downstream of cAMP in cardiac myocytes. Circ. Res. 2005, 97, 655–662. [Google Scholar] [CrossRef]
- Urschel, S.; Hoher, T.; Schubert, T.; Alev, C.; Sohl, G.; Worsdorfer, P.; Asahara, T.; Dermietzel, R.; Weiler, R.; Willecke, K. Protein kinase A-mediated phosphorylation of connexin36 in mouse retina results in decreased gap junctional communication between AII amacrine cells. J. Biol. Chem. 2006, 281, 33163–33171. [Google Scholar] [CrossRef]
- Benninger, R.K.P.; Head, W.S.; Zhang, M.; Satin, L.S.; Piston, D.W. Gap junctions and other mechanisms of cell–cell communication regulate basal insulin secretion in the pancreatic islet. J. Physiol. 2011, 589, 5453–5466. [Google Scholar] [CrossRef] [PubMed]
- Hodson, D.J.; Mitchell, R.K.; Bellomo, E.A.; Sun, G.; Vinet, L.; Meda, P.; Li, D.; Li, W.H.; Bugliani, M.; Marchetti, P.; et al. Lipotoxicity disrupts incretin-regulated human beta cell connectivity. J. Clin. Investig. 2013, 123, 4182–4194. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.-M.; Lin, S.-Z.; Chang, N.-C. Both PKA and Epac pathways mediate N-acetylcysteine-induced Connexin43 preservation in rats with myocardial infarction. PLoS ONE 2013, 8, e71878. [Google Scholar] [CrossRef] [PubMed]
- Farnsworth, N.L.; Benninger, R.K. New insights into the role of connexins in pancreatic islet function and diabetes. FEBS Lett. 2014, 588, 1278–1287. [Google Scholar] [CrossRef]
- Farnsworth, N.L.; Walter, R.; Piscopio, R.A.; Schleicher, W.E.; Benninger, R.K.P. Exendin-4 overcomes cytokine-induced decreases in gap junction coupling via protein kinase A and Epac2 in mouse and human islets. J. Physiol. 2019, 597, 431–447. [Google Scholar] [CrossRef]
- Bazzigaluppi, P.; Isenia, S.C.; Haasdijk, E.D.; Elgersma, Y.; De Zeeuw, C.I.; van der Giessen, R.S.; de Jeu, M.T.G. Modulation of Murine Olivary Connexin 36 Gap Junctions by PKA and CaMKII. Front. Cell Neurosci. 2017, 11, 397. [Google Scholar] [CrossRef]
- Ni, Q.; Ganesan, A.; Aye-Han, N.N.; Gao, X.; Allen, M.D.; Levchenko, A.; Zhang, J. Signaling diversity of PKA achieved via a Ca2+-cAMP-PKA oscillatory circuit. Nat. Chem. Biol. 2011, 7, 34–40. [Google Scholar] [CrossRef]
- Drucker, D.J.; Nauck, M.A. The incretin system: Glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet 2006, 368, 1696–1705. [Google Scholar] [CrossRef]
- Dessauer, C.W. Adenylyl cyclase--A-kinase anchoring protein complexes: The next dimension in cAMP signaling. Mol. Pharm. 2009, 76, 935–941. [Google Scholar] [CrossRef]
- Tenner, B.; Getz, M.; Ross, B.; Ohadi, D.; Bohrer, C.H.; Greenwald, E.; Mehta, S.; Xiao, J.; Rangamani, P.; Zhang, J. Spatially compartmentalized phase regulation of a Ca(2+)-cAMP-PKA oscillatory circuit. eLife 2020, 9, e55013. [Google Scholar] [CrossRef]
- Inagaki, N.; Gonoi, T.; Clement, J.P.t.; Namba, N.; Inazawa, J.; Gonzalez, G.; Aguilar-Bryan, L.; Seino, S.; Bryan, J. Reconstitution of IKATP: An inward rectifier subunit plus the sulfonylurea receptor. Science 1995, 270, 1166–1170. [Google Scholar] [CrossRef]
- Aguilar-Bryan, L.; Bryan, J.; Nakazaki, M. Of Mice and Men: KATP Channels and Insulin Secretion. Recent. Prog. Horm. Res. 2001, 56, 47–68. [Google Scholar] [CrossRef]
- Speier, S.; Yang, S.B.; Sroka, K.; Rose, T.; Rupnik, M. KATP-channels in beta-cells in tissue slices are directly modulated by millimolar ATP. Mol. Cell. Endocrinol. 2005, 230, 51–58. [Google Scholar] [CrossRef]
- Light, P.E.; Manning Fox, J.E.; Riedel, M.J.; Wheeler, M.B. Glucagon-like peptide-1 inhibits pancreatic ATP-sensitive potassium channels via a protein kinase A- and ADP-dependent mechanism. Mol. Endocrinol. 2002, 16, 2135–2144. [Google Scholar] [CrossRef] [PubMed]
- Beguin, P.; Nagashima, K.; Nishimura, M.; Gonoi, T.; Seino, S. PKA-mediated phosphorylation of the human K(ATP) channel: Separate roles of Kir6.2 and SUR1 subunit phosphorylation. EMBO J. 1999, 18, 4722–4732. [Google Scholar] [CrossRef] [PubMed]
- Kang, G.; Chepurny, O.G.; Malester, B.; Rindler, M.J.; Rehmann, H.; Bos, J.L.; Schwede, F.; Coetzee, W.A.; Holz, G.G. cAMP sensor Epac as a determinant of ATP-sensitive potassium channel activity in human pancreatic beta cells and rat INS-1 cells. J. Physiol. 2006, 573, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, N.; Shibasaki, T.; Kashima, Y.; Miki, T.; Takahashi, K.; Ueno, H.; Sunaga, Y.; Yano, H.; Matsuura, Y.; Iwanaga, T.; et al. cAMP-GEFII is a direct target of cAMP in regulated exocytosis. Nat. Cell Biol. 2000, 2, 805–811. [Google Scholar] [CrossRef]
- Shibasaki, T.; Sunaga, Y.; Fujimoto, K.; Kashima, Y.; Seino, S. Interaction of ATP Sensor, cAMP Sensor, Ca2+ Sensor, and Voltage-dependent Ca2+ Channel in Insulin Granule Exocytosis. J. Biol. Chem. 2004, 279, 7956–7961. [Google Scholar] [CrossRef]
- Shibasaki, T.; Sunaga, Y.; Seino, S. Integration of ATP, cAMP, and Ca2+ signals in insulin granule exocytosis. Diabetes 2004, 53, S59–S62. [Google Scholar] [CrossRef]
- Braun, M.; Ramracheya, R.; Bengtsson, M.; Zhang, Q.; Karanauskaite, J.; Partridge, C.; Johnson, P.R.; Rorsman, P. Voltage-gated ion channels in human pancreatic beta-cells: Electrophysiological characterization and role in insulin secretion. Diabetes 2008, 57, 1618–1628. [Google Scholar] [CrossRef]
- Jacobson, D.A.; Shyng, S.-L. Ion Channels of the Islets in Type 2 Diabetes. J. Mol. Biol. 2020, 432, 1326–1346. [Google Scholar] [CrossRef]
- MacDonald, P.; Wheeler, M. Voltage-dependent K+ channels in pancreatic beta cells: Role, regulation and potential as therapeutic targets. Diabetologia 2003, 46, 1046–1062. [Google Scholar] [CrossRef]
- Yan, L.; Figueroa, D.J.; Austin, C.P.; Liu, Y.; Bugianesi, R.M.; Slaughter, R.S.; Kaczorowski, G.J.; Kohler, M.G. Expression of voltage-gated potassium channels in human and rhesus pancreatic islets. Diabetes 2004, 53, 597–607. [Google Scholar] [CrossRef]
- Segerstolpe, A.; Palasantza, A.; Eliasson, P.; Andersson, E.M.; Andreasson, A.C.; Sun, X.; Picelli, S.; Sabirsh, A.; Clausen, M.; Bjursell, M.K.; et al. Single-Cell Transcriptome Profiling of Human Pancreatic Islets in Health and Type 2 Diabetes. Cell Metab. 2016, 24, 593–607. [Google Scholar] [CrossRef]
- Li, X.N.; Herrington, J.; Petrov, A.; Ge, L.; Eiermann, G.; Xiong, Y.; Jensen, M.V.; Hohmeier, H.E.; Newgard, C.B.; Garcia, M.L.; et al. The role of voltage-gated potassium channels Kv2.1 and Kv2.2 in the regulation of insulin and somatostatin release from pancreatic islets. J. Pharmacol. Exp. Ther. 2013, 344, 407–416. [Google Scholar] [CrossRef]
- Liu, Y.; Zhong, X.; Ding, Y.; Ren, L.; Bai, T.; Liu, M.; Liu, Z.; Guo, Y.; Guo, Q.; Zhang, Y.; et al. Inhibition of voltage-dependent potassium channels mediates cAMP-potentiated insulin secretion in rat pancreatic beta cells. Islets 2017, 9, 11–18. [Google Scholar] [CrossRef]
- Yoshida, M.; Dezaki, K.; Yamato, S.; Aoki, A.; Sugawara, H.; Toyoshima, H.; Ishikawa, S.E.; Kawakami, M.; Nakata, M.; Yada, T.; et al. Regulation of voltage-gated K+ channels by glucose metabolism in pancreatic beta-cells. FEBS Lett. 2009, 583, 2225–2230. [Google Scholar] [CrossRef]
- Lee, C.H.; Chu, C.S.; Tsai, H.J.; Ke, L.Y.; Lee, H.C.; Yeh, J.L.; Chen, C.H.; Wu, B.N. Xanthine-derived KMUP-1 reverses glucotoxicity-activated Kv channels through the cAMP/PKA signaling pathway in rat pancreatic beta cells. Chem. Biol. Interact. 2018, 279, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Choi, W.S.; Han, J.S.; Warnock, G.; Fedida, D.; McIntosh, C.H. A novel mechanism for the suppression of a voltage-gated potassium channel by glucose-dependent insulinotropic polypeptide: Protein kinase A-dependent endocytosis. J. Biol. Chem. 2005, 280, 28692–28700. [Google Scholar] [CrossRef]
- Dai, X.Q.; Manning Fox, J.E.; Chikvashvili, D.; Casimir, M.; Plummer, G.; Hajmrle, C.; Spigelman, A.F.; Kin, T.; Singer-Lahat, D.; Kang, Y.; et al. The voltage-dependent potassium channel subunit Kv2.1 regulates insulin secretion from rodent and human islets independently of its electrical function. Diabetologia 2012, 55, 1709–1720. [Google Scholar] [CrossRef]
- MacDonald, P.E.; Wang, G.; Tsuk, S.; Dodo, C.; Kang, Y.; Tang, L.; Wheeler, M.B.; Cattral, M.S.; Lakey, J.R.; Salapatek, A.M.; et al. Synaptosome-associated protein of 25 kilodaltons modulates Kv2.1 voltage-dependent K(+) channels in neuroendocrine islet beta-cells through an interaction with the channel N terminus. Mol. Endocrinol. 2002, 16, 2452–2461. [Google Scholar] [CrossRef]
- Michaelevski, I.; Chikvashvili, D.; Tsuk, S.; Singer-Lahat, D.; Kang, Y.; Linial, M.; Gaisano, H.Y.; Fili, O.; Lotan, I. Direct interaction of target SNAREs with the Kv2.1 channel. Modal regulation of channel activation and inactivation gating. J. Biol. Chem. 2003, 278, 34320–34330. [Google Scholar] [CrossRef]
- Islam, M.S. Molecular Regulations and Functions of the Transient Receptor Potential Channels of the Islets of Langerhans and Insulinoma Cells. Cells 2020, 9, 685. [Google Scholar] [CrossRef]
- Lange, I.; Yamamoto, S.; Partida-Sanchez, S.; Mori, Y.; Fleig, A.; Penner, R. TRPM2 functions as a lysosomal Ca2+-release channel in beta cells. Sci. Signal 2009, 2, ra23. [Google Scholar] [CrossRef] [PubMed]
- Uchida, K.; Dezaki, K.; Damdindorj, B.; Inada, H.; Shiuchi, T.; Mori, Y.; Yada, T.; Minokoshi, Y.; Tominaga, M. Lack of TRPM2 impaired insulin secretion and glucose metabolisms in mice. Diabetes 2011, 60, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Togashi, K.; Hara, Y.; Tominaga, T.; Higashi, T.; Konishi, Y.; Mori, Y.; Tominaga, M. TRPM2 activation by cyclic ADP-ribose at body temperature is involved in insulin secretion. EMBO J. 2006, 25, 1804–1815. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Roth, B.; Lu, W.; Du, J. Ligand recognition and gating mechanism through three ligand-binding sites of human TRPM2 channel. Elife 2019, 8. [Google Scholar] [CrossRef]
- Du, J.; Xie, J.; Yue, L. Intracellular calcium activates TRPM2 and its alternative spliced isoforms. Proc. Natl. Acad. Sci. USA 2009, 106, 7239–7244. [Google Scholar] [CrossRef]
- Pang, B.; Kim, S.; Li, D.; Ma, Z.; Sun, B.; Zhang, X.; Wu, Z.; Chen, L. Glucagon-like peptide-1 potentiates glucose-stimulated insulin secretion via the transient receptor potential melastatin 2 channel. Exp. Med. 2017, 14, 5219–5227. [Google Scholar] [CrossRef]
- Yosida, M.; Dezaki, K.; Uchida, K.; Kodera, S.; Lam, N.V.; Ito, K.; Rita, R.S.; Yamada, H.; Shimomura, K.; Ishikawa, S.E.; et al. Involvement of cAMP/EPAC/TRPM2 activation in glucose- and incretin-induced insulin secretion. Diabetes 2014, 63, 3394–3403. [Google Scholar] [CrossRef]
- Kim, B.J.; Park, K.H.; Yim, C.Y.; Takasawa, S.; Okamoto, H.; Im, M.J.; Kim, U.H. Generation of nicotinic acid adenine dinucleotide phosphate and cyclic ADP-ribose by glucagon-like peptide-1 evokes Ca2+ signal that is essential for insulin secretion in mouse pancreatic islets. Diabetes 2008, 57, 868–878. [Google Scholar] [CrossRef]
- Ito, K.; Dezaki, K.; Yoshida, M.; Yamada, H.; Miura, R.; Rita, R.S.; Ookawara, S.; Tabei, K.; Kawakami, M.; Hara, K.; et al. Endogenous α2A-Adrenoceptor–Operated Sympathoadrenergic Tones Attenuate Insulin Secretion via cAMP/TRPM2 Signaling. Diabetes 2017, 66, 699. [Google Scholar] [CrossRef]
- Kurashina, T.; Dezaki, K.; Yoshida, M.; Sukma Rita, R.; Ito, K.; Taguchi, M.; Miura, R.; Tominaga, M.; Ishibashi, S.; Kakei, M.; et al. The beta-cell GHSR and downstream cAMP/TRPM2 signaling account for insulinostatic and glycemic effects of ghrelin. Sci. Rep. 2015, 5, 14041. [Google Scholar] [CrossRef]
- Du, J.; Xie, J.; Yue, L. Modulation of TRPM2 by acidic pH and the underlying mechanisms for pH sensitivity. J. Gen. Physiol. 2009, 134, 471–488. [Google Scholar] [CrossRef]
- Holz, G.G.; Leech, C.A.; Habener, J.F. Activation of a cAMP-regulated Ca-Signaling Pathway in Pancreatic -Cells by the Insulinotropic Hormone Glucagon-like Peptide-1. J. Biol. Chem. 1995, 270, 17749–17757. [Google Scholar] [CrossRef]
- Yada, T.; Itoh, K.; Nakata, M. Glucagon-like peptide-1-(7-36)amide and a rise in cyclic adenosine 3′,5′-monophosphate increase cytosolic free Ca2+ in rat pancreatic beta-cells by enhancing Ca2+ channel activity. Endocrinology 1993, 133, 1685–1692. [Google Scholar] [CrossRef]
- Mourad, N.I.; Nenquin, M.; Henquin, J.C. cAMP-mediated and metabolic amplification of insulin secretion are distinct pathways sharing independence of beta-cell microfilaments. Endocrinology 2012, 153, 4644–4654. [Google Scholar] [CrossRef]
- Kang, G.; Joseph, J.W.; Chepurny, O.G.; Monaco, M.; Wheeler, M.B.; Bos, J.L.; Schwede, F.; Genieser, H.-G.; Holz, G.G. Epac-selective Analog 8-pCPT-2′-O-Me-cAMP as a Stimulus for Ca2+-induced Ca2+ Release and Exocytosis in Pancreatic β-Cells. J. Biol. Chem. 2003, 278, 8279–8285. [Google Scholar] [CrossRef]
- Yada, T.; Hamakawa, N.; Yaekura, K. Two distinct modes of Ca2+ signalling by ACh in rat pancreatic beta-cells: Concentration, glucose dependence and Ca2+ origin. J. Physiol. 1995, 488, 13–24. [Google Scholar] [CrossRef]
- Weir, G.C.; Mojsov, S.; Hendrick, G.K.; Habener, J.F. Glucagonlike peptide I (7-37) actions on endocrine pancreas. Diabetes 1989, 38, 338–342. [Google Scholar] [CrossRef]
- Lu, M.; Wheeler, M.B.; Leng, X.H.; Boyd, A.E., III. The role of the free cytosolic calcium level in beta-cell signal transduction by gastric inhibitory polypeptide and glucagon-like peptide I(7-37). Endocrinology 1993, 132, 94–100. [Google Scholar] [CrossRef]
- Gromada, J.; Bokvist, K.; Ding, W.G.; Holst, J.J.; Nielsen, J.H.; Rorsman, P. Glucagon-like peptide 1 (7-36) amide stimulates exocytosis in human pancreatic beta-cells by both proximal and distal regulatory steps in stimulus-secretion coupling. Diabetes 1998, 47, 57–65. [Google Scholar] [CrossRef]
- Holz, G.G.; Leech, C.A.; Heller, R.S.; Castonguay, M.; Habener, J.F. cAMP-dependent mobilization of intracellular Ca2+ stores by activation of ryanodine receptors in pancreatic beta-cells: A Ca2+ signaling system stimulated by the insulinotropic hormone glucagon-like peptide-1-(7-37). J. Biol. Chem. 1999, 274, 14147–14156. [Google Scholar] [CrossRef]
- Chepurny, O.G.; Kelley, G.G.; Dzhura, I.; Leech, C.A.; Roe, M.W.; Dzhura, E.; Li, X.; Schwede, F.; Genieser, H.G.; Holz, G.G. PKA-dependent potentiation of glucose-stimulated insulin secretion by Epac activator 8-pCPT-2′-O-Me-cAMP-AM in human islets of Langerhans. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E622–E633. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, P.E.; El-Kholy, W.; Riedel, M.J.; Salapatek, A.M.; Light, P.E.; Wheeler, M.B. The multiple actions of GLP-1 on the process of glucose-stimulated insulin secretion. Diabetes 2002, 51, S434–S442. [Google Scholar] [CrossRef] [PubMed]
- Salapatek, A.M.; MacDonald, P.E.; Gaisano, H.Y.; Wheeler, M.B. Mutations to the third cytoplasmic domain of the glucagon-like peptide 1 (GLP-1) receptor can functionally uncouple GLP-1-stimulated insulin secretion in HIT-T15 cells. Mol. Endocrinol. 1999, 13, 1305–1317. [Google Scholar] [CrossRef] [PubMed]
- Jacobo, S.M.; Guerra, M.L.; Hockerman, G.H. Cav1.2 and Cav1.3 are differentially coupled to glucagon-like peptide-1 potentiation of glucose-stimulated insulin secretion in the pancreatic beta-cell line INS-1. J. Pharmacol. Exp. Ther. 2009, 331, 724–732. [Google Scholar] [CrossRef]
- Liu, G.; Jacobo, S.M.; Hilliard, N.; Hockerman, G.H. Differential modulation of Cav1.2 and Cav1.3-mediated glucose-stimulated insulin secretion by cAMP in INS-1 cells: Distinct roles for exchange protein directly activated by cAMP 2 (Epac2) and protein kinase A. J. Pharmacol. Exp. Ther. 2006, 318, 152–160. [Google Scholar] [CrossRef]
- Ammala, C.; Ashcroft, F.M.; Rorsman, P. Calcium-independent potentiation of insulin release by cyclic AMP in single [beta]-cells. Nature 1993, 363, 356–358. [Google Scholar] [CrossRef]
- Kanno, T.; Suga, S.; Wu, J.; Kimura, M.; Wakui, M. Intracellular cAMP potentiates voltage-dependent activation of L-type Ca2+ channels in rat islet beta-cells. Pflug. Arch. Eur. J. Physiol. 1998, 435, 578–580. [Google Scholar] [CrossRef]
- Kang, G.; Chepurny, O.G.; Rindler, M.J.; Collis, L.; Chepurny, Z.; Li, W.-H.; Harbeck, M.; Roe, M.W.; Holz, G.G. A cAMP and Ca2+ coincidence detector in support of Ca2+-induced Ca2+ release in mouse pancreatic β cells. J. Physiol. 2005, 566, 173–188. [Google Scholar] [CrossRef]
- Islam, M.S. Stimulus-Secretion Coupling in Beta-Cells: From Basic to Bedside. In Calcium Signaling; Islam, M.S., Ed.; Springer International Publishing: Cham, Switzerland, 2020; pp. 943–963. [Google Scholar]
- Zhang, Q.; Bengtsson, M.; Partridge, C.; Salehi, A.; Braun, M.; Cox, R.; Eliasson, L.; Johnson, P.R.; Renstrom, E.; Schneider, T.; et al. R-type Ca(2+)-channel-evoked CICR regulates glucose-induced somatostatin secretion. Nat. Cell Biol. 2007, 9, 453–460. [Google Scholar] [CrossRef]
- Postić, S.; Sarikas, S.; Pfabe, J.; Pohorec, V.; Bombek, L.K.; Sluga, N.; Klemen, M.S.; Dolenšek, J.; Korošak, D.; Stožer, A.; et al. Intracellular Ca2+ channels initiate physiological glucose signaling in beta cells examined in situ. bioRxiv 2021. [Google Scholar] [CrossRef]
- Dzhura, I.; Chepurny, O.G.; Kelley, G.G.; Leech, C.A.; Roe, M.W.; Dzhura, E.; Afshari, P.; Malik, S.; Rindler, M.J.; Xu, X.; et al. Epac2-dependent mobilization of intracellular Ca2+ by glucagon-like peptide-1 receptor agonist exendin-4 is disrupted in β-cells of phospholipase C-ε knockout mice. J. Physiol. 2010, 588, 4871–4889. [Google Scholar] [CrossRef]
- Islam, M.S. CICR takes centre stage in {beta}-cells: A cute cascade connects cAMP to CICR. J. Physiol. 2010, 588, 4853. [Google Scholar] [CrossRef]
- Leech, C.A.; Chepurny, O.G.; Holz, G.G. Epac2-dependent rap1 activation and the control of islet insulin secretion by glucagon-like peptide-1. Vitam. Horm. 2010, 84, 279–302. [Google Scholar] [CrossRef]
- Bode, H.-P.; Moormann, B.; Dabew, R.; Göke, B. Glucagon-Like Peptide 1 Elevates Cytosolic Calcium in Pancreaticβ -Cells Independently of Protein Kinase A. Endocrinology 1999, 140, 3919–3927. [Google Scholar] [CrossRef]
- Sasaki, S.; Nakagaki, I.; Kondo, H.; Hori, S. Involvement of the ryanodine-sensitive Ca2+ store in GLP-1-induced Ca2+ oscillations in insulin-secreting HIT cells. Pflug. Arch. Eur. J. Physiol. 2002, 445, 342–351. [Google Scholar] [CrossRef]
- Dyachok, O.; Gylfe, E. Ca2+-induced Ca2+ Release via Inositol 1,4,5-trisphosphate Receptors Is Amplified by Protein Kinase A and Triggers Exocytosis in Pancreatic β-Cells. J. Biol. Chem. 2004, 279, 45455–45461. [Google Scholar] [CrossRef]
- Leech, C.A.; Dzhura, I.; Chepurny, O.G.; Kang, G.; Schwede, F.; Genieser, H.G.; Holz, G.G. Molecular physiology of glucagon-like peptide-1 insulin secretagogue action in pancreatic beta cells. Prog. Biophys. Mol. Biol. 2011, 107, 236–247. [Google Scholar] [CrossRef]
- Nordenskjöld, F.; Andersson, B.; Islam, M.S. Expression of the Inositol 1,4,5-Trisphosphate Receptor and the Ryanodine Receptor Ca2+-Release Channels in the Beta-Cells and Alpha-Cells of the Human Islets of Langerhans. In Calcium Signaling; Islam, M.S., Ed.; Springer International Publishing: Cham, Switzerland, 2020; pp. 271–279. [Google Scholar]
- Dzhura, I.; Chepurny, O.G.; Leech, C.A.; Roe, M.W.; Dzhura, E.; Xu, X.; Lu, Y.; Schwede, F.; Genieser, H.-G.; Smrcka, A.V.; et al. Phospholipase C-ε links Epac2 activation to the potentiation of glucose-stimulated insulin secretion from mouse islets of Langerhans. Islets 2011, 3, 121–128. [Google Scholar] [CrossRef]
- Kang, G.; Chepurny, O.G.; Holz, G.G. cAMP-regulated guanine nucleotide exchange factor II (Epac2) mediates Ca2+-induced Ca2+ release in INS-1 pancreatic beta-cells. J. Physiol. 2001, 536, 375–385. [Google Scholar] [CrossRef]
- Gromada, J.; Anker, C.; Bokvist, K.; Knudsen, L.B.; Wahl, P. Glucagon-like peptide-1 receptor expression in Xenopus oocytes stimulates inositol trisphosphate-dependent intracellular Ca2+ mobilization. FEBS Lett. 1998, 425, 277–280. [Google Scholar] [CrossRef]
- Liu, Y.J.; Tengholm, A.; Grapengiesser, E.; Hellman, B.; Gylfe, E. Origin of slow and fast oscillations of Ca2+ in mouse pancreatic islets. J. Physiol. 1998, 508, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Baltrusch, S.; Lenzen, S. Regulation of [Ca2+]i oscillations in mouse pancreatic islets by adrenergic agonists. Biochem Biophys Res Commun 2007, 363, 1038–1043. [Google Scholar] [CrossRef] [PubMed]
- Cane, M.C.; Parrington, J.; Rorsman, P.; Galione, A.; Rutter, G.A. The two pore channel TPC2 is dispensable in pancreatic beta-cells for normal Ca(2)(+) dynamics and insulin secretion. Cell Calcium 2016, 59, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Šterk, M.; Križančić Bombek, L.; Skelin Klemen, M.; Slak Rupnik, M.; Marhl, M.; Stožer, A.; Gosak, M. NMDA receptor inhibition increases, synchronizes, and stabilizes the collective pancreatic beta cell activity: Insights through multilayer network analysis. PLoS Comput. Biol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Tengholm, A. Cyclic AMP dynamics in the pancreatic beta-cell. Ups. J. Med. Sci. 2012, 117, 355–369. [Google Scholar] [CrossRef]
- Tian, G.; Sandler, S.; Gylfe, E.; Tengholm, A. Glucose- and hormone-induced cAMP oscillations in α- and β-cells within intact pancreatic islets. Diabetes 2011, 60, 1535–1543. [Google Scholar] [CrossRef]
- Tengholm, A.; Gylfe, E. Oscillatory control of insulin secretion. Mol. Cell. Endocrinol. 2009, 297, 58–72. [Google Scholar] [CrossRef]
- Flamez, D.; Gilon, P.; Moens, K.; Van Breusegem, A.; Delmeire, D.; Scrocchi, L.A.; Henquin, J.C.; Drucker, D.J.; Schuit, F. Altered cAMP and Ca2+ signaling in mouse pancreatic islets with glucagon-like peptide-1 receptor null phenotype. Diabetes 1999, 48, 1979–1986. [Google Scholar] [CrossRef]
- Bruton, J.; Cheng, A.J.; Westerblad, H. Measuring Ca2+ in Living Cells. In Calcium Signaling; Islam, M.S., Ed.; Springer International Publishing: Cham, Switzerland, 2020; pp. 7–26. [Google Scholar]
- Gillis, K.D.; Misler, S. Enhancers of cytosolic cAMP augment depolarization-induced exocytosis from pancreatic B-cells: Evidence for effects distal to Ca2+ entry. Pflug. Arch. Eur. J. Physiol. 1993, 424, 195–197. [Google Scholar] [CrossRef]
- Trogden, K.P.; Zhu, X.; Lee, J.S.; Wright, C.V.E.; Gu, G.; Kaverina, I. Regulation of Glucose-Dependent Golgi-Derived Microtubules by cAMP/EPAC2 Promotes Secretory Vesicle Biogenesis in Pancreatic beta Cells. Curr. Biol. CB 2019, 29, 2339–2350.e2335. [Google Scholar] [CrossRef]
- Ying, Y.; Li, L.; Cao, W.; Yan, D.; Zeng, Q.; Kong, X.; Lu, L.; Yan, M.; Xu, X.; Qu, J.; et al. The microtubule associated protein syntabulin is required for glucose-stimulated and cAMP-potentiated insulin secretion. FEBS Lett. 2012, 586, 3674–3680. [Google Scholar] [CrossRef]
- Henquin, J.C.; Nenquin, M. Activators of PKA and Epac distinctly influence insulin secretion and cytosolic Ca2+ in female mouse islets stimulated by glucose and tolbutamide. Endocrinology 2014, 155, 3274–3287. [Google Scholar] [CrossRef][Green Version]
- Alenkvist, I.; Gandasi, N.R.; Barg, S.; Tengholm, A. Recruitment of Epac2A to Insulin Granule Docking Sites Regulates Priming for Exocytosis. Diabetes 2017, 66, 2610–2622. [Google Scholar] [CrossRef]
- Yasuda, T.; Shibasaki, T.; Minami, K.; Takahashi, H.; Mizoguchi, A.; Uriu, Y.; Numata, T.; Mori, Y.; Miyazaki, J.-i.; Miki, T.; et al. Rim2α Determines Docking and Priming States in Insulin Granule Exocytosis. Cell Metab. 2010, 12, 117–129. [Google Scholar] [CrossRef]
- Eliasson, L.; Ma, X.; Renstrom, E.; Barg, S.; Berggren, P.-O.; Galvanovskis, J.; Gromada, J.; Jing, X.; Lundquist, I.; Salehi, A.; et al. SUR1 Regulates PKA-independent cAMP-induced Granule Priming in Mouse Pancreatic B-cells. J. Gen. Physiol. 2003, 121, 181–197. [Google Scholar] [CrossRef]
- Renström, E.; Eliasson, L.; Rorsman, P. Protein kinase A-dependent and -independent stimulation of exocytosis by cAMP in mouse pancreatic B-cells. J. Physiol. 1997, 502, 105–118. [Google Scholar] [CrossRef]
- Vikman, J.; Svensson, H.; Huang, Y.C.; Kang, Y.; Andersson, S.A.; Gaisano, H.Y.; Eliasson, L. Truncation of SNAP-25 reduces the stimulatory action of cAMP on rapid exocytosis in insulin-secreting cells. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E452–E461. [Google Scholar] [CrossRef]
- Leung, Y.M.; Kwan, E.P.; Ng, B.; Kang, Y.; Gaisano, H.Y. SNAREing voltage-gated K+ and ATP-sensitive K+ channels: Tuning beta-cell excitability with syntaxin-1A and other exocytotic proteins. Endocr. Rev. 2007, 28, 653–663. [Google Scholar] [CrossRef]
- Skelin, M.; Rupnik, M. cAMP increases the sensitivity of exocytosis to Ca2+ primarily through protein kinase A in mouse pancreatic beta cells. Cell Calcium 2011, 49, 89–99. [Google Scholar] [CrossRef]
- Wan, Q.F.; Dong, Y.; Yang, H.; Lou, X.; Ding, J.; Xu, T. Protein kinase activation increases insulin secretion by sensitizing the secretory machinery to Ca2+. J. Gen. Physiol. 2004, 124, 653–662. [Google Scholar] [CrossRef]
- Dolensek, J.; Skelin, M.; Rupnik, M.S. Calcium Dependencies of Regulated Exocytosis in Different Endocrine Cells. Physiol. Res. 2011, 60, S29–S38. [Google Scholar] [CrossRef] [PubMed]
- Dolensek, J.; Rupnik, M.S.; Stozer, A. Structural similarities and differences between the human and the mouse pancreas. Islets 2015, 7, e1024405. [Google Scholar] [CrossRef] [PubMed]
- Briant, L.J.B.; Reinbothe, T.M.; Spiliotis, I.; Miranda, C.; Rodriguez, B.; Rorsman, P. delta-cells and beta-cells are electrically coupled and regulate alpha-cell activity via somatostatin. J. Physiol. 2018, 596, 197–215. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Yang, T.; Zhang, Q. delta-Cells: The Neighborhood Watch in the Islet Community. Biology 2021, 10, 74. [Google Scholar] [CrossRef]
- Benninger, R.K.; Hutchens, T.; Head, W.S.; McCaughey, M.J.; Zhang, M.; Le Marchand, S.J.; Satin, L.S.; Piston, D.W. Intrinsic islet heterogeneity and gap junction coupling determine spatiotemporal Ca(2)(+) wave dynamics. Biophys. J. 2014, 107, 2723–2733. [Google Scholar] [CrossRef]
- Benninger, R.K.P.; Hodson, D.J. New Understanding of beta-Cell Heterogeneity and In Situ Islet Function. Diabetes 2018, 67, 537–547. [Google Scholar] [CrossRef]
- Gosak, M.; Stozer, A.; Markovic, R.; Dolensek, J.; Marhl, M.; Rupnik, M.S.; Perc, M. The relationship between node degree and dissipation rate in networks of diffusively coupled oscillators and its significance for pancreatic beta cells. Chaos 2015, 25, 07311. [Google Scholar] [CrossRef]
- Zmazek, J.; Klemen, M.S.; Markovič, R.; Dolenšek, J.; Marhl, M.; Stožer, A.; Gosak, M. Assessing Different Temporal Scales of Calcium Dynamics in Networks of Beta Cell Populations. Front. Physiol. 2021, 12, 612233. [Google Scholar] [CrossRef]
- Šterk, M.; Dolenšek, J.; Bombek, L.K.; Markovič, R.; Zakelšek, D.; Perc, M.; Pohorec, V.; Stožer, A.; Gosak, M. Assessing the origin and velocity of Ca2+ waves in three-dimensional tissue: Insights from a mathematical model and confocal imaging in mouse pancreas tissue slices. Commun. Nonlinear Sci. Numer. Simul. 2021, 93, 105495. [Google Scholar] [CrossRef]
- Stožer, A.; Gosak, M.; Dolenšek, J.; Perc, M.; Marhl, M.; Rupnik, M.S.; Korošak, D. Functional Connectivity in Islets of Langerhans from Mouse Pancreas Tissue Slices. PLoS Comput. Biol. 2013, 9, e1002923. [Google Scholar] [CrossRef]
- Stožer, A.; Gosak, M.; Korošak, D.; Yakubo, K.; Dolenšek, J.; Marhl, M.; Rupnik, M.S. Correlations between beta-cells’ calcium dynamics reveal differences in functional connectivity patterns in islets of Langerhans from pancreas tissue slices under low and high levels of glucose. AIP Conf. Proc. 2012, 1468, 332–339. [Google Scholar]
- Cappon, G.; Pedersen, M.G. Heterogeneity and nearest-neighbor coupling can explain small-worldness and wave properties in pancreatic islets. Chaos 2016, 26, 053103. [Google Scholar] [CrossRef]
- Johnston, N.R.; Mitchell, R.K.; Haythorne, E.; Pessoa, M.P.; Semplici, F.; Ferrer, J.; Piemonti, L.; Marchetti, P.; Bugliani, M.; Bosco, D.; et al. Beta Cell Hubs Dictate Pancreatic Islet Responses to Glucose. Cell Metab. 2016, 24, 389–401. [Google Scholar] [CrossRef]
- Gosak, M.; Markovic, R.; Dolensek, J.; Slak Rupnik, M.; Marhl, M.; Stozer, A.; Perc, M. Network science of biological systems at different scales: A review. Phys Life Rev. 2018, 24, 118–135. [Google Scholar] [CrossRef]
- Charpantier, E.; Cancela, J.; Meda, P. Beta cells preferentially exchange cationic molecules via connexin 36 gap junction channels. Diabetologia 2007, 50, 2332–2341. [Google Scholar] [CrossRef]
- Bukauskas, F.F. Neurons and beta-cells of the pancreas express connexin36, forming gap junction channels that exhibit strong cationic selectivity. J. Membr. Biol. 2012, 245, 243–253. [Google Scholar] [CrossRef]
- Meda, P.; Amherdt, M.; Perrelet, A.; Orci, L. Metabolic coupling between cultured pancreatic b-cells. Exp. Cell Res. 1981, 133, 421–430. [Google Scholar] [CrossRef]
- Meda, P.; Kohen, E.; Kohen, C.; Orci, L. Heterocellular coupling in cultures of endocrine pancreatic cells. C. R. Seances Acad. Sci. III 1981, 293, 607–610. [Google Scholar]
- Pedersen, M.G.; Bertram, R.; Sherman, A. Intra- and inter-islet synchronization of metabolically driven insulin secretion. Biophys. J. 2005, 89, 107–119. [Google Scholar] [CrossRef]
- Cigliola, V.; Chellakudam, V.; Arabieter, W.; Meda, P. Connexins and β-cell functions. Diabetes Res. Clin. Pract. 2013, 99, 250–259. [Google Scholar] [CrossRef]
- Rao, V.P.; Rizzo, M.A. Diffusion of metabolites across gap junctions mediates metabolic coordination of β-islet cells. bioRxiv 2020. [Google Scholar] [CrossRef]
- Carvalho, C.P.F.; Oliveira, R.B.; Britan, A.; Santos-Silva, J.C.; Boschero, A.C.; Meda, P.; Collares-Buzato, C.B. Impaired β-cell-β-cell coupling mediated by Cx36 gap junctions in prediabetic mice. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E144–E151. [Google Scholar] [CrossRef]
- Allagnat, F.; Martin, D.; Condorelli, D.F.; Waeber, G.; Haefliger, J.-A. Glucose represses connexin36 in insulin-secreting cells. J. Cell Sci. 2005, 118, 5335–5344. [Google Scholar] [CrossRef]
- Irles, E.; Neco, P.; Lluesma, M.; Villar-Pazos, S.; Santos-Silva, J.C.; Vettorazzi, J.F.; Alonso-Magdalena, P.; Carneiro, E.M.; Boschero, A.C.; Nadal, A.; et al. Enhanced glucose-induced intracellular signaling promotes insulin hypersecretion: Pancreatic beta-cell functional adaptations in a model of genetic obesity and prediabetes. Mol. Cell Endocrinol. 2015, 404, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Farnsworth, N.L.; Walter, R.L.; Hemmati, A.; Westacott, M.J.; Benninger, R.K.P. Low Level Pro-inflammatory Cytokines Decrease Connexin36 Gap Junction Coupling in Mouse and Human Islets through Nitric Oxide-mediated Protein Kinase Cδ. J. Biol. Chem. 2016, 291, 3184–3196. [Google Scholar] [CrossRef]
- Perez-Armendariz, E.M. Connexin 36, a key element in pancreatic beta cell function. Neuropharmacology 2013, 75, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Rutter, G.A.; Georgiadou, E.; Martinez-Sanchez, A.; Pullen, T.J. Metabolic and functional specialisations of the pancreatic beta cell: Gene disallowance, mitochondrial metabolism and intercellular connectivity. Diabetologia 2020, 63, 1990–1998. [Google Scholar] [CrossRef] [PubMed]
- Corezola do Amaral, M.E.; Kravets, V.; Dwulet, J.M.; Farnsworth, N.L.; Piscopio, R.; Schleicher, W.E.; Miranda, J.G.; Benninger, R.K.P. Caloric restriction recovers impaired β-cell-β-cell gap junction coupling, calcium oscillation coordination, and insulin secretion in prediabetic mice. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E709–E720. [Google Scholar] [CrossRef]
- Pérez-Armendariz, M.; Roy, C.; Spray, D.C.; Bennett, M.V. Biophysical properties of gap junctions between freshly dispersed pairs of mouse pancreatic beta cells. Biophys. J. 1991, 59, 76–92. [Google Scholar] [CrossRef]
- Allagnat, F.; Alonso, F.; Martin, D.; Abderrahmani, A.; Waeber, G.; Haefliger, J.A. ICER-1gamma overexpression drives palmitate-mediated connexin36 down-regulation in insulin-secreting cells. J. Biol. Chem. 2008, 283, 5226–5234. [Google Scholar] [CrossRef]
- Haefliger, J.-A.; Martin, D.; Favre, D.; Petremand, Y.; Mazzolai, L.; Abderrahmani, A.; Meda, P.; Waeber, G.; Allagnat, F. Reduction of Connexin36 Content by ICER-1 Contributes to Insulin-Secreting Cells Apoptosis Induced by Oxidized LDL Particles. PLoS ONE 2013, 8, e55198. [Google Scholar] [CrossRef]
- Kothmann, W.W.; Massey, S.C.; O’Brien, J. Dopamine-stimulated dephosphorylation of connexin 36 mediates AII amacrine cell uncoupling. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 14903–14911. [Google Scholar] [CrossRef]
- Calabrese, A.; Zhang, M.; Serre-Beinier, V.; Caton, D.; Mas, C.; Satin, L.S.; Meda, P. Connexin 36 Controls Synchronization of Ca2+ Oscillations and Insulin Secretion in MIN6 Cells. Diabetes 2003, 52, 417–424. [Google Scholar] [CrossRef]
- Moreno, A.P.; Lau, A.F. Gap junction channel gating modulated through protein phosphorylation. Prog. Biophys. Mol. Biol. 2007, 94, 107–119. [Google Scholar] [CrossRef][Green Version]
- Skelin Klemen, M.; Dolenšek, J.; Križančić Bombek, L.; Pohorec, V.; Gosak, M.; Slak Rupnik, M.; Stožer, A. The Effect of cAMP and the Role of Epac2A During Activation, Activity, and Deactivation of Beta Cell Networks. Preprints 2021. [Google Scholar] [CrossRef]
- Stožer, A.; Dolenšek, J.; Križančić Bombek, L.; Pohorec, V.; Slak Rupnik, M.; Klemen, M.S. Confocal Laser Scanning Microscopy of Calcium Dynamics in Acute Mouse Pancreatic Tissue Slices. JoVE 2021, e62293. [Google Scholar] [CrossRef]
- Stozer, A.; Hojs, R.; Dolensek, J. Beta Cell Functional Adaptation and Dysfunction in Insulin Resistance and the Role of Chronic Kidney Disease. Nephron 2019, 143, 33–37. [Google Scholar] [CrossRef]
- Kahn, S.E.; Cooper, M.E.; Del Prato, S. Pathophysiology and treatment of type 2 diabetes: Perspectives on the past, present, and future. Lancet 2014, 383, 1068–1083. [Google Scholar] [CrossRef]
- Kargar, C.; Ktorza, A. Anatomical versus functional beta-cell mass in experimental diabetes. Diabetes Obes. Metab. 2008, 10, 43–53. [Google Scholar] [CrossRef]
- Saisho, Y.; Butler, A.E.; Manesso, E.; Elashoff, D.; Rizza, R.A.; Butler, P.C. beta-cell mass and turnover in humans: Effects of obesity and aging. Diabetes Care 2013, 36, 111–117. [Google Scholar] [CrossRef]
- Rahier, J.; Guiot, Y.; Goebbels, R.M.; Sempoux, C.; Henquin, J.C. Pancreatic β-cell mass in European subjects with type 2 diabetes. Diabetes Obes. Metab. 2008, 10, 32–42. [Google Scholar] [CrossRef]
- Kou, K.; Saisho, Y.; Satoh, S.; Yamada, T.; Itoh, H. Change in beta-cell mass in Japanese nondiabetic obese individuals. J. Clin. Endocrinol. Metab. 2013, 98, 3724–3730. [Google Scholar] [CrossRef]
- Karaca, M.; Magnan, C.; Kargar, C. Functional pancreatic beta-cell mass: Involvement in type 2 diabetes and therapeutic intervention. Diabetes Amp. Metab. 2009, 35, 77–84. [Google Scholar] [CrossRef]
- Shen, C.-a.; Fagan, S.; Fischman, A.J.; Carter, E.E.; Chai, J.-K.; Lu, X.-M.; Yu, Y.-M.; Tompkins, R.G. Effects of glucagon-like peptide 1 on glycemia control and its metabolic consequence after severe thermal injury-studies in an animal model. Surgery 2011, 149, 635–644. [Google Scholar] [CrossRef]
- Sah, S.P.; Singh, B.; Choudhary, S.; Kumar, A. Animal models of insulin resistance: A review. Pharmacol. Rep. 2016, 68, 1165–1177. [Google Scholar] [CrossRef]
- Butler, A.E.; Janson, J.; Bonner-Weir, S.; Ritzel, R.; Rizza, R.A.; Butler, P.C. beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 2003, 52, 102–110. [Google Scholar] [CrossRef]
- Wang, Q.; Zhao, C.; Jin, L.; Zhang, H.; Miao, Q.; Liu, H.; Zhang, D. AWRK6, a Novel GLP-1 Receptor Agonist, Attenuates Diabetes by Stimulating Insulin Secretion. Int. J. Mol. Sci. 2018, 19, 3053. [Google Scholar] [CrossRef] [PubMed]
- Hull, R.L.; Kodama, K.; Utzschneider, K.M.; Carr, D.B.; Prigeon, R.L.; Kahn, S.E. Dietary-fat-induced obesity in mice results in beta cell hyperplasia but not increased insulin release: Evidence for specificity of impaired beta cell adaptation. Diabetologia 2005, 48, 1350–1358. [Google Scholar] [CrossRef] [PubMed]
- Grapengiesser, E.; Gylfe, E.; Hellman, B. Glucose-induced oscillations of cytoplasmic Ca2+ in the pancreatic β-cell. Biochem. Biophys. Res. Commun. 1988, 151, 1299–1304. [Google Scholar] [CrossRef]
- Gylfe, E.; Grapengiesser, E.; Hellman, B. Propagation of cytoplasmic Ca2+ oscillations in clusters of pancreatic β-cells exposed to glucose. Cell Calcium 1991, 12, 229–240. [Google Scholar] [CrossRef]
- Santos, R.M.; Rosario, L.M.; Nadal, A.; Garcia-Sancho, J.; Soria, B.; Valdeolmillos, M. Widespread synchronous Ca oscillations due to bursting electrical activity in single pancreatic islets. Pflüg. Arch. Eur. J. Physiol. 1991, 418, 417–422. [Google Scholar] [CrossRef]
- Gopel, S.; Zhang, Q.; Eliasson, L.; Ma, X.S.; Galvanovskis, J.; Kanno, T.; Salehi, A.; Rorsman, P. Capacitance measurements of exocytosis in mouse pancreatic alpha-, beta- and delta-cells within intact islets of Langerhans. J. Physiol. 2004, 556, 711–726. [Google Scholar] [CrossRef]
- Gonzalez, A.; Merino, B.; Marroqui, L.; Neco, P.; Alonso-Magdalena, P.; Caballero-Garrido, E.; Vieira, E.; Soriano, S.; Gomis, R.; Nadal, A.; et al. Insulin hypersecretion in islets from diet-induced hyperinsulinemic obese female mice is associated with several functional adaptations in individual beta-cells. Endocrinology 2013, 154, 3515–3524. [Google Scholar] [CrossRef]
- Westacott, M.J.; Farnsworth, N.L.; St Clair, J.R.; Poffenberger, G.; Heintz, A.; Ludin, N.W.; Hart, N.J.; Powers, A.C.; Benninger, R.K.P. Age-Dependent Decline in the Coordinated [Ca(2+)] and Insulin Secretory Dynamics in Human Pancreatic Islets. Diabetes 2017, 66, 2436–2445. [Google Scholar] [CrossRef]
- Holz, G.G.; Chepurny, O.G.; Leech, C.A. Epac2A makes a new impact in beta-cell biology. Diabetes 2013, 62, 2665–2666. [Google Scholar] [CrossRef]
- Idevall-Hagren, O.; Jakobsson, I.; Xu, Y.; Tengholm, A. Spatial control of Epac2 activity by cAMP and Ca2+-mediated activation of Ras in pancreatic beta cells. Sci. Signal 2013, 6, ra29. [Google Scholar] [CrossRef]
- Park, J.H.; Shim, H.M.; Na, A.Y.; Bae, J.H.; Im, S.S.; Song, D.K. Orexin A regulates plasma insulin and leptin levels in a time-dependent manner following a glucose load in mice. Diabetologia 2015, 58, 1542–1550. [Google Scholar] [CrossRef][Green Version]
- Shen, Y.; Zhao, Y.; Zheng, D.; Chang, X.; Ju, S.; Guo, L. Effects of orexin A on GLUT4 expression and lipid content via MAPK signaling in 3T3-L1 adipocytes. J. Steroid Biochem. Mol. Biol. 2013, 138, 376–383. [Google Scholar] [CrossRef]
- Arafat, A.M.; Kaczmarek, P.; Skrzypski, M.; Pruszynska-Oszmalek, E.; Kolodziejski, P.; Adamidou, A.; Ruhla, S.; Szczepankiewicz, D.; Sassek, M.; Billert, M.; et al. Glucagon regulates orexin A secretion in humans and rodents. Diabetologia 2014, 57, 2108–2116. [Google Scholar] [CrossRef]
- Baranowska, B.; Wolinska-Witort, E.; Martynska, L.; Chmielowska, M.; Baranowska-Bik, A. Plasma orexin A, orexin B, leptin, neuropeptide Y (NPY) and insulin in obese women. Neuro Endocrinol. Lett. 2005, 26, 293–296. [Google Scholar]
- Blanchet, E.; Van de Velde, S.; Matsumura, S.; Hao, E.; LeLay, J.; Kaestner, K.; Montminy, M. Feedback inhibition of CREB signaling promotes beta cell dysfunction in insulin resistance. Cell Rep. 2015, 10, 1149–1157. [Google Scholar] [CrossRef]
- Takahashi, H.; Shibasaki, T.; Park, J.H.; Hidaka, S.; Takahashi, T.; Ono, A.; Song, D.K.; Seino, S. Role of Epac2A/Rap1 signaling in interplay between incretin and sulfonylurea in insulin secretion. Diabetes 2015, 64, 1262–1272. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stožer, A.; Paradiž Leitgeb, E.; Pohorec, V.; Dolenšek, J.; Križančić Bombek, L.; Gosak, M.; Skelin Klemen, M. The Role of cAMP in Beta Cell Stimulus–Secretion and Intercellular Coupling. Cells 2021, 10, 1658. https://doi.org/10.3390/cells10071658
Stožer A, Paradiž Leitgeb E, Pohorec V, Dolenšek J, Križančić Bombek L, Gosak M, Skelin Klemen M. The Role of cAMP in Beta Cell Stimulus–Secretion and Intercellular Coupling. Cells. 2021; 10(7):1658. https://doi.org/10.3390/cells10071658
Chicago/Turabian StyleStožer, Andraž, Eva Paradiž Leitgeb, Viljem Pohorec, Jurij Dolenšek, Lidija Križančić Bombek, Marko Gosak, and Maša Skelin Klemen. 2021. "The Role of cAMP in Beta Cell Stimulus–Secretion and Intercellular Coupling" Cells 10, no. 7: 1658. https://doi.org/10.3390/cells10071658
APA StyleStožer, A., Paradiž Leitgeb, E., Pohorec, V., Dolenšek, J., Križančić Bombek, L., Gosak, M., & Skelin Klemen, M. (2021). The Role of cAMP in Beta Cell Stimulus–Secretion and Intercellular Coupling. Cells, 10(7), 1658. https://doi.org/10.3390/cells10071658