
Targeting Canonical and Non-Canonical STAT Signaling Pathways in Renal Diseases


Abstract
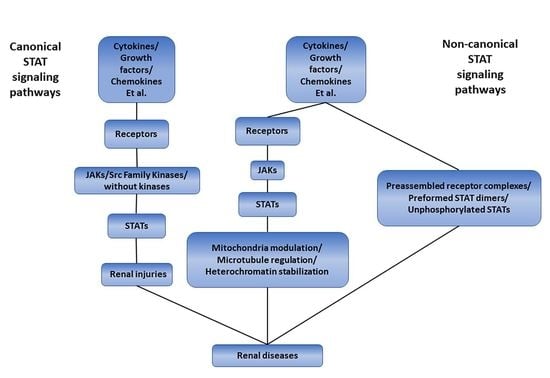
1. Introduction
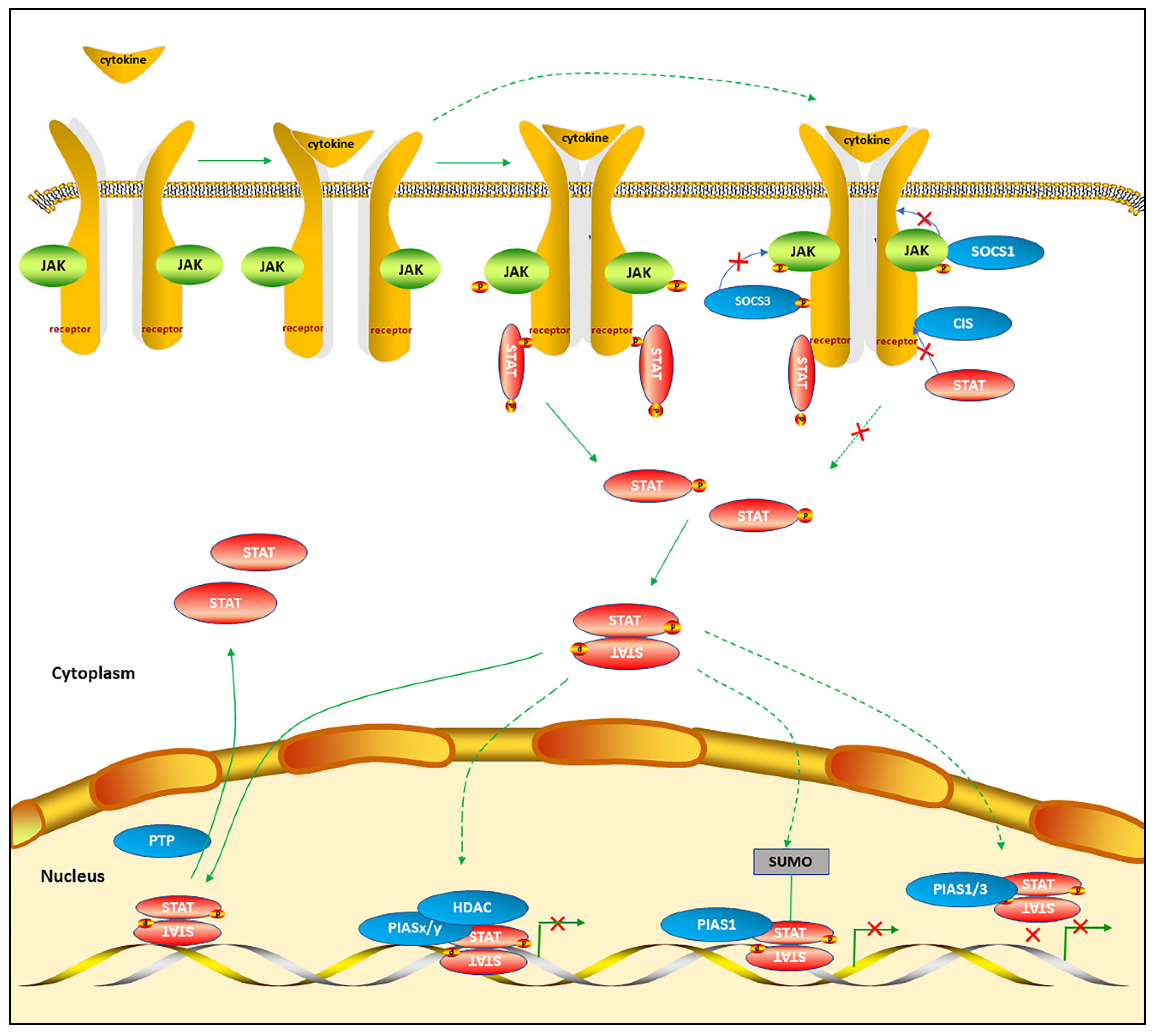
2. The Canonical STAT Signaling Pathways
2.1. The Mechanisms of Canonical STAT Signaling Pathways
2.2. The Alternative STAT Post-Translational Modifications
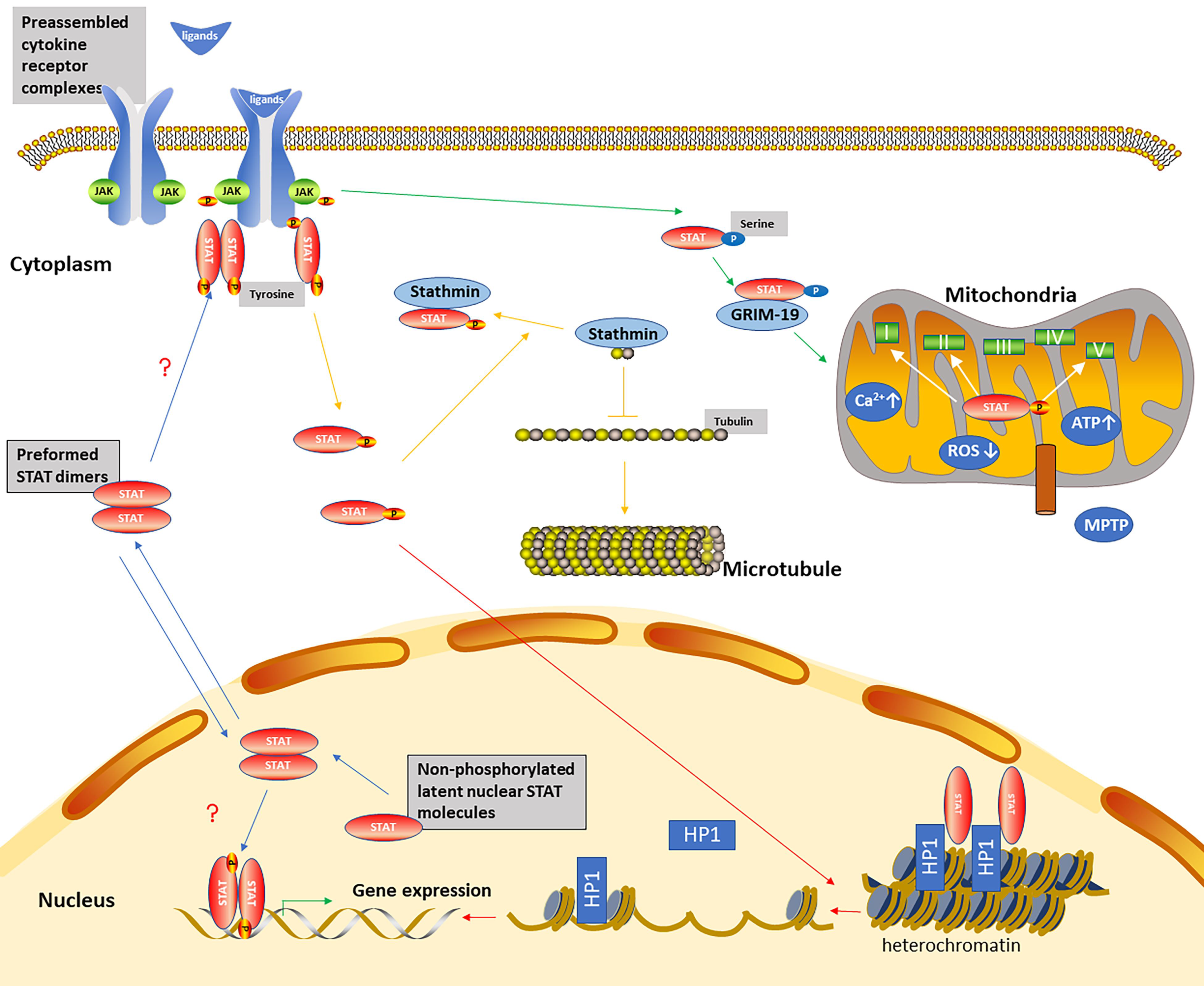
3. The Non-Canonical STAT Signaling Pathways
The Mechanisms of Non-Canonical STAT Signaling Pathways
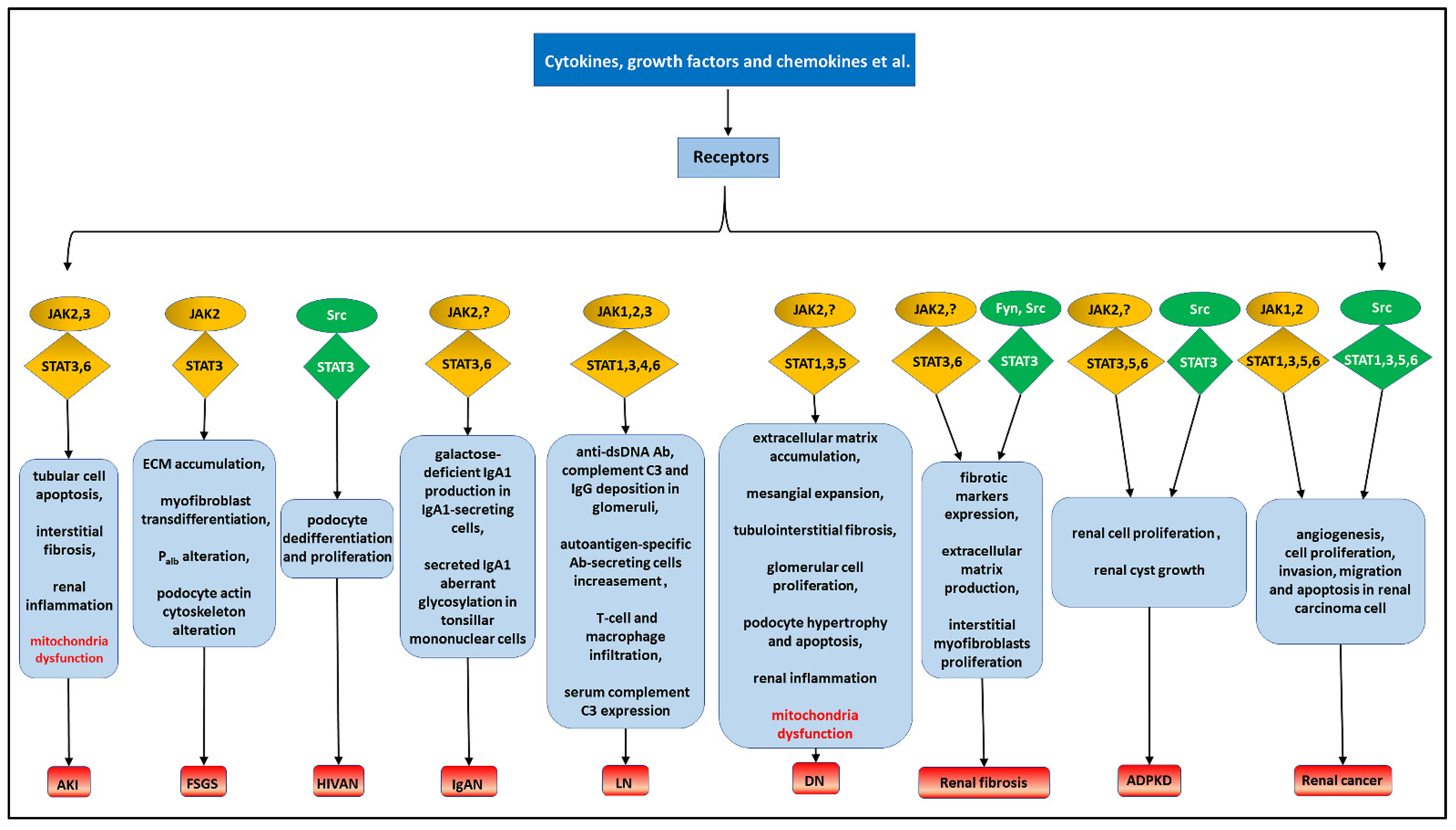
4. The STAT Signaling Pathways and Renal Diseases
4.1. Acute Kidney Injury
4.2. Focal Segmental Glomerulosclerosis
4.3. IgA Nephropathy
4.4. Lupus Nephritis
4.5. Diabetic Nephropathy
4.6. Renal Fibrosis
4.7. Autosomal Dominant Polycystic Kidney Disease
4.8. Renal Cancers
4.9. Other Renal Diseases
5. Endogenous Inhibitors of STAT Signaling Pathways
6. Pharmacological Inhibition of STAT Signaling Pathways
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lim, C.P.; Cao, X. Structure, function, and regulation of STAT proteins. Mol. Biosyst. 2006, 2, 536–550. [Google Scholar] [CrossRef]
- Mudter, J.; Weigmann, B.; Bartsch, B.; Kiesslich, R.; Strand, D.; Galle, P.R.; Lehr, H.A.; Schmidt, J.; Neurath, M.F. Activation pattern of signal transducers and activators of transcription (STAT) factors in inflammatory bowel diseases. Am. J. Gastroenterol. 2005, 100, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Goropevsek, A.; Holcar, M.; Avcin, T. The Role of STAT Signaling Pathways in the Pathogenesis of Systemic Lupus Erythematosus. Clin. Rev. Allergy Immunol. 2017, 52, 164–181. [Google Scholar] [CrossRef] [PubMed]
- Kitcharoensakkul, M.; Cooper, M.A. Rheumatologic and autoimmune manifestations in primary immune deficiency. Curr Opin Allergy Clin. Immunol. 2019, 19, 545–552. [Google Scholar] [CrossRef]
- Verhoeven, Y.; Tilborghs, S.; Jacobs, J.; De Waele, J.; Quatannens, D.; Deben, C.; Prenen, H.; Pauwels, P.; Trinh, X.B.; Wouters, A.; et al. The potential and controversy of targeting STAT family members in cancer. Semin. Cancer Biol. 2020, 60, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Platanitis, E.; Decker, T. Regulatory Networks Involving STATs, IRFs, and NFkappaB in Inflammation. Front. Immunol. 2018, 9, 2542. [Google Scholar] [CrossRef]
- Bromberg, J.F. Activation of STAT proteins and growth control. Bioessays 2001, 23, 161–169. [Google Scholar] [CrossRef]
- Leonard, W.J. Role of Jak kinases and STATs in cytokine signal transduction. Int. J. Hematol. 2001, 73, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.J.; An, H.J.; Cho, E.S.; Kang, H.C.; Lee, J.Y.; Lee, H.S.; Cho, Y.Y. Stat2 stability regulation: An intersection between immunity and carcinogenesis. Exp. Mol. Med. 2020, 52, 1526–1536. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.M.; Ouyang, W.; Szabo, S.J.; Jacobson, N.G.; Guler, M.L.; Gorham, J.D.; Gubler, U.; Murphy, T.L. T helper differentiation proceeds through Stat1-dependent, Stat4-dependent and Stat4-independent phases. Curr. Top. Microbiol. Immunol. 1999, 238, 13–26. [Google Scholar] [CrossRef]
- Martens, N.; Uzan, G.; Wery, M.; Hooghe, R.; Hooghe-Peters, E.L.; Gertler, A. Suppressor of cytokine signaling 7 inhibits prolactin, growth hormone, and leptin signaling by interacting with STAT5 or STAT3 and attenuating their nuclear translocation. J. Biol. Chem. 2005, 280, 13817–13823. [Google Scholar] [CrossRef]
- Baik, M.; Yu, J.H.; Hennighausen, L. Growth hormone-STAT5 regulation of growth, hepatocellular carcinoma, and liver metabolism. Ann. N. Y. Acad. Sci. 2011, 1229, 29–37. [Google Scholar] [CrossRef]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer. 2009, 9, 798–809. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Holland, S.M.; Staudt, L.M. JAKs and STATs in immunity, immunodeficiency, and cancer. N. Engl. J. Med. 2013, 368, 161–170. [Google Scholar] [CrossRef]
- Garcia, R.; Bowman, T.L.; Niu, G.; Yu, H.; Minton, S.; Muro-Cacho, C.A.; Cox, C.E.; Falcone, R.; Fairclough, R.; Parsons, S.; et al. Constitutive activation of Stat3 by the Src and JAK tyrosine kinases participates in growth regulation of human breast carcinoma cells. Oncogene 2001, 20, 2499–2513. [Google Scholar] [CrossRef]
- Amiri, F.; Shaw, S.; Wang, X.; Tang, J.; Waller, J.L.; Eaton, D.C.; Marrero, M.B. Angiotensin II activation of the JAK/STAT pathway in mesangial cells is altered by high glucose. Kidney Int. 2002, 61, 1605–1616. [Google Scholar] [CrossRef]
- Patel, A.; Sabbineni, H.; Clarke, A.; Somanath, P.R. Novel roles of Src in cancer cell epithelial-to-mesenchymal transition, vascular permeability, microinvasion and metastasis. Life Sci. 2016, 157, 52–61. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Src protein-tyrosine kinase structure, mechanism, and small molecule inhibitors. Pharmacol. Res. 2015, 94, 9–25. [Google Scholar] [CrossRef]
- Decker, T.; Kovarik, P. Serine phosphorylation of STATs. Oncogene 2000, 19, 2628–2637. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, S. Regulation of STAT signaling by acetylation. Cell Signal 2013, 25, 1924–1931. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Huang, J.; Dasgupta, M.; Sears, N.; Miyagi, M.; Wang, B.; Chance, M.R.; Chen, X.; Du, Y.; Wang, Y.; et al. Reversible methylation of promoter-bound STAT3 by histone-modifying enzymes. Proc. Natl. Acad. Sci. USA 2010, 107, 21499–21504. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Feng, Q.; Jin, L.; Huang, F.; Miao, Y.; Liu, J.; Xu, Y.; Chen, X.; Zhang, H.; Guo, T.; et al. Regulation of the linear ubiquitination of STAT1 controls antiviral interferon signaling. Nat. Commun. 2020, 11, 1146. [Google Scholar] [CrossRef]
- Jakobs, A.; Koehnke, J.; Himstedt, F.; Funk, M.; Korn, B.; Gaestel, M.; Niedenthal, R. Ubc9 fusion–directed SUMOylation (UFDS): A method to analyze function of protein SUMOylation. Nat. Methods 2007, 4, 245–250. [Google Scholar] [CrossRef]
- Zhou, Z.; Wang, M.; Li, J.; Xiao, M.; Chin, Y.E.; Cheng, J.; Yeh, E.T.; Yang, J.; Yi, J. SUMOylation and SENP3 regulate STAT3 activation in head and neck cancer. Oncogene 2016, 35, 5826–5838. [Google Scholar] [CrossRef]
- Rauth, M.; Freund, P.; Orlova, A.; Grunert, S.; Tasic, N.; Han, X.; Ruan, H.B.; Neubauer, H.A.; Moriggl, R. Cell Metabolism Control Through O-GlcNAcylation of STAT5: A Full or Empty Fuel Tank Makes a Big Difference for Cancer Cell Growth and Survival. Int. J. Mol. Sci. 2019, 20, 1028. [Google Scholar] [CrossRef]
- Mohr, A.; Chatain, N.; Domoszlai, T.; Rinis, N.; Sommerauer, M.; Vogt, M.; Muller-Newen, G. Dynamics and non-canonical aspects of JAK/STAT signalling. Eur. J. Cell Biol. 2012, 91, 524–532. [Google Scholar] [CrossRef]
- Livnah, O.; Stura, E.A.; Middleton, S.A.; Johnson, D.L.; Jolliffe, L.K.; Wilson, I.A. Crystallographic evidence for preformed dimers of erythropoietin receptor before ligand activation. Science 1999, 283, 987–990. [Google Scholar] [CrossRef]
- Braunstein, J.; Brutsaert, S.; Olson, R.; Schindler, C. STATs dimerize in the absence of phosphorylation. J. Biol. Chem. 2003, 278, 34133–34140. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Stark, G.R. Roles of unphosphorylated STATs in signaling. Cell Res. 2008, 18, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Li, W.X. Canonical and non-canonical JAK-STAT signaling. Trends Cell Biol. 2008, 18, 545–551. [Google Scholar] [CrossRef]
- Gough, D.J.; Corlett, A.; Schlessinger, K.; Wegrzyn, J.; Larner, A.C.; Levy, D.E. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science 2009, 324, 1713–1716. [Google Scholar] [CrossRef]
- Tammineni, P.; Anugula, C.; Mohammed, F.; Anjaneyulu, M.; Larner, A.C.; Sepuri, N.B. The import of the transcription factor STAT3 into mitochondria depends on GRIM-19, a component of the electron transport chain. J. Biol. Chem. 2013, 288, 4723–4732. [Google Scholar] [CrossRef]
- Boengler, K.; Hilfiker-Kleiner, D.; Heusch, G.; Schulz, R. Inhibition of permeability transition pore opening by mitochondrial STAT3 and its role in myocardial ischemia/reperfusion. Basic Res. Cardiol. 2010, 105, 771–785. [Google Scholar] [CrossRef]
- Xu, Y.S.; Liang, J.J.; Wang, Y.; Zhao, X.J.; Xu, L.; Xu, Y.Y.; Zou, Q.C.; Zhang, J.M.; Tu, C.E.; Cui, Y.G.; et al. STAT3 Undergoes Acetylation-dependent Mitochondrial Translocation to Regulate Pyruvate Metabolism. Sci. Rep. 2016, 6, 39517. [Google Scholar] [CrossRef]
- Yang, R.; Lirussi, D.; Thornton, T.M.; Jelley-Gibbs, D.M.; Diehl, S.A.; Case, L.K.; Madesh, M.; Taatjes, D.J.; Teuscher, C.; Haynes, L.; et al. Mitochondrial Ca(2)(+) and membrane potential, an alternative pathway for Interleukin 6 to regulate CD4 cell effector function. Elife 2015, 4. [Google Scholar] [CrossRef]
- Meier, J.A.; Hyun, M.; Cantwell, M.; Raza, A.; Mertens, C.; Raje, V.; Sisler, J.; Tracy, E.; Torres-Odio, S.; Gispert, S.; et al. Stress-induced dynamic regulation of mitochondrial STAT3 and its association with cyclophilin D reduce mitochondrial ROS production. Sci. Signal 2017, 10, 472. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Too, H.P. Mitochondrial localized STAT3 is involved in NGF induced neurite outgrowth. PLoS ONE 2011, 6, e21680. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Ribeiro, M.; Bray, E.R.; Lee, D.H.; Yungher, B.J.; Mehta, S.T.; Thakor, K.A.; Diaz, F.; Lee, J.K.; Moraes, C.T.; et al. Enhanced Transcriptional Activity and Mitochondrial Localization of STAT3 Co-induce Axon Regrowth in the Adult Central Nervous System. Cell Rep. 2016, 15, 398–410. [Google Scholar] [CrossRef]
- Park, K.W.; Lin, C.Y.; Benveniste, E.N.; Lee, Y.S. Mitochondrial STAT3 is negatively regulated by SOCS3 and upregulated after spinal cord injury. Exp. Neurol. 2016, 284, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Rincon, M.; Pereira, F.V. A New Perspective: Mitochondrial Stat3 as a Regulator for Lymphocyte Function. Int. J. Mol. Sci. 2018, 19, 1656. [Google Scholar] [CrossRef]
- Ng, D.C.; Lin, B.H.; Lim, C.P.; Huang, G.; Zhang, T.; Poli, V.; Cao, X. Stat3 regulates microtubules by antagonizing the depolymerization activity of stathmin. J. Cell Biol. 2006, 172, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Fani, F.; Regolisti, G.; Delsante, M.; Cantaluppi, V.; Castellano, G.; Gesualdo, L.; Villa, G.; Fiaccadori, E. Recent advances in the pathogenetic mechanisms of sepsis-associated acute kidney injury. J. Nephrol. 2018, 31, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Dube, S.; Matam, T.; Yen, J.; Mang, H.E.; Dagher, P.C.; Hato, T.; Sutton, T.A. Endothelial STAT3 Modulates Protective Mechanisms in a Mouse Ischemia-Reperfusion Model of Acute Kidney Injury. J. Immunol. Res. 2017, 2017, 4609502. [Google Scholar] [CrossRef]
- Zhang, L.; Lu, P.; Guo, X.; Liu, T.; Luo, X.; Zhu, Y.T. Inhibition of JAK2/STAT3 signaling pathway protects mice from the DDP-induced acute kidney injury in lung cancer. Inflamm. Res. 2019, 68, 751–760. [Google Scholar] [CrossRef]
- Chen, J.; Shetty, S.; Zhang, P.; Gao, R.; Hu, Y.; Wang, S.; Li, Z.; Fu, J. Aspirin-triggered resolvin D1 down-regulates inflammatory responses and protects against endotoxin-induced acute kidney injury. Toxicol. Appl. Pharmacol. 2014, 277, 118–123. [Google Scholar] [CrossRef]
- Xiao, Y.; Yang, N.; Zhang, Q.; Wang, Y.; Yang, S.; Liu, Z. Pentraxin 3 inhibits acute renal injury-induced interstitial fibrosis through suppression of IL-6/Stat3 pathway. Inflammation 2014, 37, 1895–1901. [Google Scholar] [CrossRef]
- Hsu, Y.H.; Li, H.H.; Sung, J.M.; Chen, W.T.; Hou, Y.C.; Chang, M.S. Interleukin-19 mediates tissue damage in murine ischemic acute kidney injury. PLoS ONE 2013, 8, e56028. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.Z.; Wang, X.; Wang, Y.; Niu, A.; Wang, S.; Zou, C.; Harris, R.C. IL-4/IL-13-mediated polarization of renal macrophages/dendritic cells to an M2a phenotype is essential for recovery from acute kidney injury. Kidney Int. 2017, 91, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.J.; Feng, D.; Wang, H.; Guan, Y.; Yan, X.; Gao, B. IL-22 ameliorates renal ischemia-reperfusion injury by targeting proximal tubule epithelium. J. Am. Soc. Nephrol. 2014, 25, 967–977. [Google Scholar] [CrossRef]
- Zhou, Y.; Xu, W.; Zhu, H. CXCL8(3-72) K11R/G31P protects against sepsis-induced acute kidney injury via NF-kappaB and JAK2/STAT3 pathway. Biol. Res. 2019, 52, 29. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Li, X.; Zhang, F.; Wu, L.; Dong, Z.; Zhang, D. EGFR drives the progression of AKI to CKD through HIPK2 overexpression. Theranostics 2019, 9, 2712–2726. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zhang, E.; Ren, X.; Bai, X.; Wang, D.; Bai, L.; Luo, D.; Guo, Z.; Wang, Q.; Yang, J. Edaravone alleviates cell apoptosis and mitochondrial injury in ischemia-reperfusion-induced kidney injury via the JAK/STAT pathway. Biol. Res. 2020, 53, 28. [Google Scholar] [CrossRef]
- Fogo, A.B. Causes and pathogenesis of focal segmental glomerulosclerosis. Nat. Rev. Nephrol. 2015, 11, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Mariani, L.; Eddy, S.; Maecker, H.; Kambham, N.; Mehta, K.; Hartman, J.; Wang, W.; Kretzler, M.; Lafayette, R.A. JAK-STAT signaling is activated in the kidney and peripheral blood cells of patients with focal segmental glomerulosclerosis. Kidney Int. 2018, 94, 795–808. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Jin, Y.; Li, Y. Expression of JAKs/STATs pathway molecules in rat model of rapid focal segmental glomerulosclerosis. Pediatr. Nephrol. 2009, 24, 1661–1671. [Google Scholar] [CrossRef]
- Savin, V.J.; Sharma, M.; Zhou, J.; Genochi, D.; Sharma, R.; Srivastava, T.; Ilahe, A.; Budhiraja, P.; Gupta, A.; McCarthy, E.T. Multiple Targets for Novel Therapy of FSGS Associated with Circulating Permeability Factor. BioMed Res. Int. 2017, 2017, 6232616. [Google Scholar] [CrossRef]
- Sharma, M.; Zhou, J.; Gauchat, J.F.; Sharma, R.; McCarthy, E.T.; Srivastava, T.; Savin, V.J. Janus kinase 2/signal transducer and activator of transcription 3 inhibitors attenuate the effect of cardiotrophin-like cytokine factor 1 and human focal segmental glomerulosclerosis serum on glomerular filtration barrier. Transl. Res. 2015, 166, 384–398. [Google Scholar] [CrossRef]
- He, J.C.; Husain, M.; Sunamoto, M.; D’Agati, V.D.; Klotman, M.E.; Iyengar, R.; Klotman, P.E. Nef stimulates proliferation of glomerular podocytes through activation of Src-dependent Stat3 and MAPK1,2 pathways. J. Clin. Investig. 2004, 114, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Dai, Y.; Xu, J.; Mallipattu, S.; Kaufman, L.; Klotman, P.E.; He, J.C.; Chuang, P.Y. Deletion of podocyte STAT3 mitigates the entire spectrum of HIV-1-associated nephropathy. Aids 2013, 27, 1091–1098. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Lu, T.C.; Chuang, P.Y.; Fang, W.; Ratnam, K.; Xiong, H.; Ouyang, X.; Shen, Y.; Levy, D.E.; Hyink, D.; et al. Reduction of Stat3 activity attenuates HIV-induced kidney injury. J. Am. Soc. Nephrol. 2009, 20, 2138–2146. [Google Scholar] [CrossRef]
- Wang, X.; Liu, R.; Zhang, W.; Hyink, D.P.; Das, G.C.; Das, B.; Li, Z.; Wang, A.; Yuan, W.; Klotman, P.E.; et al. Role of SIRT1 in HIV-associated kidney disease. Am. J. Physiol. Renal Physiol. 2020, 319, F335–F344. [Google Scholar] [CrossRef] [PubMed]
- Roberts, I.S. Pathology of IgA nephropathy. Nat. Rev. Nephrol. 2014, 10, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, T.; Masaki, T.; Hirai, T.; Doi, S.; Kuratsune, M.; Arihiro, K.; Kohno, N.; Yorioka, N. Activation of signal transducer and activator of transcription 3 correlates with cell proliferation and renal injury in human glomerulonephritis. Nephrol. Dial. Transplant. 2008, 23, 3418–3426. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Mariani, L.; Eddy, S.; Maecker, H.; Kambham, N.; Mehta, K.; Hartman, J.; Wang, W.; Kretzler, M.; Lafayette, R.A. JAK-STAT Activity in Peripheral Blood Cells and Kidney Tissue in IgA Nephropathy. Clin. J. Am. Soc. Nephrol. 2020, 15, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Huang, Z.Q.; Raska, M.; Reily, C.; Anderson, J.C.; Suzuki, H.; Ueda, H.; Moldoveanu, Z.; Kiryluk, K.; Suzuki, Y.; et al. Inhibition of STAT3 Signaling Reduces IgA1 Autoantigen Production in IgA Nephropathy. Kidney Int. Rep. 2017, 2, 1194–1207. [Google Scholar] [CrossRef]
- He, L.; Peng, Y.; Liu, H.; Yin, W.; Chen, X.; Peng, X.; Shao, J.; Liu, Y.; Liu, F. Activation of the interleukin-4/signal transducer and activator of transcription 6 signaling pathway and homeodomain-interacting protein kinase 2 production by tonsillar mononuclear cells in IgA nephropathy. Am. J. Nephrol. 2013, 38, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Almaani, S.; Meara, A.; Rovin, B.H. Update on Lupus Nephritis. Clin. J. Am. Soc. Nephrol. 2017, 12, 825–835. [Google Scholar] [CrossRef]
- Aljaberi, N.; Bennett, M.; Brunner, H.I.; Devarajan, P. Proteomic profiling of urine: Implications for lupus nephritis. Expert Rev. Proteom. 2019, 16, 303–313. [Google Scholar] [CrossRef]
- Lu, L.D.; Stump, K.L.; Wallace, N.H.; Dobrzanski, P.; Serdikoff, C.; Gingrich, D.E.; Dugan, B.J.; Angeles, T.S.; Albom, M.S.; Mason, J.L.; et al. Depletion of autoreactive plasma cells and treatment of lupus nephritis in mice using CEP-33779, a novel, orally active, selective inhibitor of JAK2. J. Immunol. 2011, 187, 3840–3853. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.D.; Liang, D.; Wu, X.N.; Li, Y.; Niu, J.W.; Zhou, C.; Wang, L.; Chen, H.; Zheng, W.J.; Fei, Y.Y.; et al. Contribution and underlying mechanisms of CXCR4 overexpression in patients with systemic lupus erythematosus. Cell Mol. Immunol. 2017, 14, 842–849. [Google Scholar] [CrossRef]
- Ripoll, E.; de Ramon, L.; Draibe Bordignon, J.; Merino, A.; Bolanos, N.; Goma, M.; Cruzado, J.M.; Grinyo, J.M.; Torras, J. JAK3-STAT pathway blocking benefits in experimental lupus nephritis. Arthritis Res. Ther. 2016, 18, 134. [Google Scholar] [CrossRef]
- Du, Y.; Zhang, W.; Liu, S.; Feng, X.; Gao, F.; Liu, Q. S3I-201 ameliorates tubulointerstitial lesion of the kidneys in MRL/lpr mice. Biochem. Biophys. Res. Commun. 2018, 503, 177–180. [Google Scholar] [CrossRef]
- Edwards, L.J.; Mizui, M.; Kyttaris, V. Signal transducer and activator of transcription (STAT) 3 inhibition delays the onset of lupus nephritis in MRL/lpr mice. Clin. Immunol. 2015, 158, 221–230. [Google Scholar] [CrossRef]
- Yoshida, N.; He, F.; Kyttaris, V.C.J.L. T cell–specific STAT3 deficiency abrogates lupus nephritis. Lupus 2019, 28, 1468–1472. [Google Scholar] [CrossRef]
- Singh, R.R.; Saxena, V.; Zang, S.; Li, L.; Finkelman, F.D.; Witte, D.P.; Jacob, C.O. Differential contribution of IL-4 and STAT6 vs STAT4 to the development of lupus nephritis. J. Immunol. 2003, 170, 4818–4825. [Google Scholar] [CrossRef]
- Dang, W.Z.; Li, H.; Jiang, B.; Nandakumar, K.S.; Liu, K.F.; Liu, L.X.; Yu, X.C.; Tan, H.J.; Zhou, C. Therapeutic effects of artesunate on lupus-prone MRL/lpr mice are dependent on T follicular helper cell differentiation and activation of JAK2-STAT3 signaling pathway. Phytomedicine 2019, 62, 152965. [Google Scholar] [CrossRef]
- Ahmad, R.; Raina, D.; Meyer, C.; Kufe, D. Triterpenoid CDDO-methyl ester inhibits the Janus-activated kinase-1 (JAK1)-->signal transducer and activator of transcription-3 (STAT3) pathway by direct inhibition of JAK1 and STAT3. Cancer Res. 2008, 68, 2920–2926. [Google Scholar] [CrossRef]
- Alicic, R.Z.; Rooney, M.T.; Tuttle, K.R. Diabetic Kidney Disease: Challenges, Progress, and Possibilities. Clin. J. Am. Soc. Nephrol. 2017, 12, 2032–2045. [Google Scholar] [CrossRef]
- Berthier, C.C.; Zhang, H.; Schin, M.; Henger, A.; Nelson, R.G.; Yee, B.; Boucherot, A.; Neusser, M.A.; Cohen, C.D.; Carter-Su, C.; et al. Enhanced expression of Janus kinase-signal transducer and activator of transcription pathway members in human diabetic nephropathy. Diabetes 2009, 58, 469–477. [Google Scholar] [CrossRef]
- Cooker, L.A.; Peterson, D.; Rambow, J.; Riser, M.L.; Riser, R.E.; Najmabadi, F.; Brigstock, D.; Riser, B.L. TNF-alpha, but not IFN-gamma, regulates CCN2 (CTGF), collagen type I, and proliferation in mesangial cells: Possible roles in the progression of renal fibrosis. Am. J. Physiol. Renal Physiol. 2007, 293, F157–F165. [Google Scholar] [CrossRef]
- Du, J.; Wang, L.; Liu, L.; Fan, Q.; Yao, L.; Cui, Y.; Kang, P.; Zhao, H.; Feng, X.; Gao, H. IFN-gamma suppresses the high glucose-induced increase in TGF-beta1 and CTGF synthesis in mesangial cells. Pharmacol. Rep. 2011, 63, 1137–1144. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Dong, W.; Li, H.; Li, B.; Liu, X.; Kong, Q.; Sun, W.; Sun, T.; Ma, P.; Cui, Y.; et al. Protective effects of IFN-gamma on the kidney of type- 2 diabetic KKAy mice. Pharmacol. Rep. 2018, 70, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, R.; Jayakumar, C.; Chen, F.; Fulton, D.; Stepp, D.; Gansevoort, R.T.; Ramesh, G. Low-Dose IL-17 Therapy Prevents and Reverses Diabetic Nephropathy, Metabolic Syndrome, and Associated Organ Fibrosis. J. Am. Soc. Nephrol. 2016, 27, 745–765. [Google Scholar] [CrossRef] [PubMed]
- Feigerlova, E.; Battaglia-Hsu, S.F. IL-6 signaling in diabetic nephropathy: From pathophysiology to therapeutic perspectives. Cytokine Growth Factor Rev. 2017, 37, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Xing, L.; Wang, L.; Yao, F.; Liu, S.; Hao, J.; Liu, W.; Duan, H. Therapeutic effects of suppressors of cytokine signaling in diabetic nephropathy. J. Histochem. Cytochem. 2014, 62, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Liu, S.; Shi, Y.; Li, H.; Hao, J.; Xing, L.; Cao, Y.; Duan, H. Suppressors of cytokine signaling inhibit tubular epithelial cell-myofibroblast transdifferentiation. Am. J. Nephrol. 2011, 34, 142–151. [Google Scholar] [CrossRef]
- Yang, M.; Shen, Z.; Chen, D.; Gan, H.; Shen, Q.; Yang, B.; Du, X. Effects of 1,25-(OH)(2)D (3) on the expressions of vitamin D receptor, STAT5 and cytoskeletal rearrangement in human monocytes incubated with sera from type 2 diabetes patients and diabetic nephropathy patients with uremia. Inflamm. Res. 2012, 61, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Sanz, L.; Bernal, S.; Recio, C.; Lazaro, I.; Oguiza, A.; Melgar, A.; Jimenez-Castilla, L.; Egido, J.; Gomez-Guerrero, C. SOCS1-targeted therapy ameliorates renal and vascular oxidative stress in diabetes via STAT1 and PI3K inhibition. Lab. Investig. 2018, 98, 1276–1290. [Google Scholar] [CrossRef]
- Shaw, S.; Wang, X.; Redd, H.; Alexander, G.D.; Isales, C.M.; Marrero, M.B. High glucose augments the angiotensin II-induced activation of JAK2 in vascular smooth muscle cells via the polyol pathway. J. Biol. Chem. 2003, 278, 30634–30641. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Wu, H.; Liu, Z.Y.; Zhu, Q.; Shan, C.; Zhang, K.Q. Advanced glycation end products induce the apoptosis of and inflammation in mouse podocytes through CXCL9-mediated JAK2/STAT3 pathway activation. Int. J. Mol. Med. 2017, 40, 1185–1193. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Zhong, Y.; Li, X.; Chen, H.; Jim, B.; Zhou, M.M.; Chuang, P.Y.; He, J.C. Role of transcription factor acetylation in diabetic kidney disease. Diabetes 2014, 63, 2440–2453. [Google Scholar] [CrossRef] [PubMed]
- Banes-Berceli, A.K.; Shaw, S.; Ma, G.; Brands, M.; Eaton, D.C.; Stern, D.M.; Fulton, D.; Caldwell, R.W.; Marrero, M.B. Effect of simvastatin on high glucose- and angiotensin II-induced activation of the JAK/STAT pathway in mesangial cells. Am. J. Physiol. Renal Physiol. 2006, 291, F116–F121. [Google Scholar] [CrossRef][Green Version]
- Huang, J.-S.; Guh, J.-Y.; Hung, W.-C.; Yang, M.-L.; Lai, Y.-H.; Chen, H.-C.; Chuang, L.-Y. Role of the Janus kinase (JAK)/signal transducters and activators of transcription (STAT) cascade in advanced glycation end-product-induced cellular mitogenesis in NRK-49F cells. Biochem. J. 1999, 342, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.C.; Wang, Z.H.; Feng, X.; Chuang, P.Y.; Fang, W.; Shen, Y.; Levy, D.E.; Xiong, H.; Chen, N.; He, J.C. Knockdown of Stat3 activity in vivo prevents diabetic glomerulopathy. Kidney Int. 2009, 76, 63–71. [Google Scholar] [CrossRef]
- Zhang, Q.; He, L.; Dong, Y.; Fei, Y.; Wen, J.; Li, X.; Guan, J.; Liu, F.; Zhou, T.; Li, Z.; et al. Sitagliptin ameliorates renal tubular injury in diabetic kidney disease via STAT3-dependent mitochondrial homeostasis through SDF-1alpha/CXCR4 pathway. FASEB J. 2020, 34, 7500–7519. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Klimova, E.; Aparicio-Trejo, O.E.; Tapia, E.; Pedraza-Chaverri, J. Unilateral Ureteral Obstruction as a Model to Investigate Fibrosis-Attenuating Treatments. Biomolecules 2019, 9, 141. [Google Scholar] [CrossRef] [PubMed]
- Kuratsune, M.; Masaki, T.; Hirai, T.; Kiribayashi, K.; Yokoyama, Y.; Arakawa, T.; Yorioka, N.; Kohno, N.J.N. Signal transducer and activator of transcription 3 involvement in the development of renal interstitial fibrosis after unilateral ureteral obstruction. Nephrology 2007, 12, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Pang, M.; Ma, L.; Gong, R.; Tolbert, E.; Mao, H.; Ponnusamy, M.; Chin, Y.E.; Yan, H.; Dworkin, L.D.; Zhuang, S. A novel STAT3 inhibitor, S3I-201, attenuates renal interstitial fibroblast activation and interstitial fibrosis in obstructive nephropathy. Kidney Int. 2010, 78, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.-Y.; Jeon, J.-H.; Jung, Y.-A.; Jung, G.-S.; Lee, E.J.; Choi, Y.-K.; Park, K.-G.; Choe, M.S.; Jang, B.K.; Kim, M.-K.; et al. Fyn deficiency attenuates renal fibrosis by inhibition of phospho-STAT3. Kidney Int. 2016, 90, 1285–1297. [Google Scholar] [CrossRef] [PubMed]
- Hassan, N.M.E.; Shehatou, G.S.G.; Kenawy, H.I.; Said, E. Dasatinib mitigates renal fibrosis in a rat model of UUO via inhibition of Src/STAT-3/NF-kappaB signaling. Environ. Toxicol. Pharmacol. 2021, 84, 103625. [Google Scholar] [CrossRef]
- Hassan, N.M.E.; Said, E.; Shehatou, G.S.G. Nifuroxazide suppresses UUO-induced renal fibrosis in rats via inhibiting STAT-3/NF-kappaB signaling, oxidative stress and inflammation. Life Sci. 2021, 272, 119241. [Google Scholar] [CrossRef] [PubMed]
- Makitani, K.; Ogo, N.; Asai, A.J.P.R. STX---0119, a novel STAT3 dimerization inhibitor, prevents fibrotic gene expression in a mouse model of kidney fibrosis by regulating Cxcr4 and Ccr1 expression. Physiol. Rep. 2020, 8, e14627. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Shen, Y.; Wang, Z.; Shao, D.C.; Liu, J.; Fu, L.J.; Kong, Y.L.; Zhou, L.; Xue, H.; Huang, Y.; et al. Inhibition of STAT3 acetylation is associated with angiotesin renal fibrosis in the obstructed kidney. Acta Pharmacol. Sin. 2014, 35, 1045–1054. [Google Scholar] [CrossRef]
- Koike, K.; Ueda, S.; Yamagishi, S.-i.; Yasukawa, H.; Kaida, Y.; Yokoro, M.; Fukami, K.; Yoshimura, A.; Okuda, S.J.C.I. Protective role of JAK/STAT signaling against renal fibrosis in mice with unilateral ureteral obstruction. Clin. Immunol. 2014, 150, 78–87. [Google Scholar] [CrossRef]
- Yukawa, K.; Kishino, M.; Goda, M.; Liang, X.M.; Kimura, A.; Tanaka, T.; Bai, T.; Owada-Makabe, K.; Tsubota, Y.; Ueyama, T.; et al. STAT6 deficiency inhibits tubulointerstitial fibrosis in obstructive nephropathy. Int. J. Mol. Med. 2005, 15, 225–230. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhu, X.; Wang, X.; Peng, Y.; Du, J.; Yin, H.; Yang, H.; Ni, X.; Zhang, W. H2S alleviates renal injury and fibrosis in response to unilateral ureteral obstruction by regulating macrophage infiltration via inhibition of NLRP3 signaling. Exp. Cell Res. 2020, 387. [Google Scholar] [CrossRef]
- Cornec-Le Gall, E.; Alam, A.; Perrone, R.D. Autosomal dominant polycystic kidney disease. Lancet 2019, 393, 919–935. [Google Scholar] [CrossRef]
- Weimbs, T.; Olsan, E.E.; Talbot, J.J. Regulation of STATs by polycystin-1 and their role in polycystic kidney disease. Jakstat 2013, 2, e23650. [Google Scholar] [CrossRef]
- Talbot, J.J.; Song, X.; Wang, X.; Rinschen, M.M.; Doerr, N.; LaRiviere, W.B.; Schermer, B.; Pei, Y.P.; Torres, V.E.; Weimbs, T. The cleaved cytoplasmic tail of polycystin-1 regulates Src-dependent STAT3 activation. J. Am. Soc. Nephrol. 2014, 25, 1737–1748. [Google Scholar] [CrossRef]
- Takakura, A.; Nelson, E.A.; Haque, N.; Humphreys, B.D.; Zandi-Nejad, K.; Frank, D.A.; Zhou, J. Pyrimethamine inhibits adult polycystic kidney disease by modulating STAT signaling pathways. Hum. Mol. Genet. 2011, 20, 4143–4154. [Google Scholar] [CrossRef]
- Li, L.X.; Fan, L.X.; Zhou, J.X.; Grantham, J.J.; Calvet, J.P.; Sage, J.; Li, X. Lysine methyltransferase SMYD2 promotes cyst growth in autosomal dominant polycystic kidney disease. J. Clin. Investig. 2017, 127, 2751–2764. [Google Scholar] [CrossRef]
- Fragiadaki, M.; Lannoy, M.; Themanns, M.; Maurer, B.; Leonhard, W.N.; Peters, D.J.; Moriggl, R.; Ong, A.C. STAT5 drives abnormal proliferation in autosomal dominant polycystic kidney disease. Kidney Int. 2017, 91, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Buettner, R.; Mora, L.B.; Jove, R. Activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention. Clin. Cancer Res. Clin. Cancer Res. 2002, 8, 945–954. [Google Scholar] [PubMed]
- Kim, C.; Baek, S.H.; Um, J.Y.; Shim, B.S.; Ahn, K.S. Resveratrol attenuates constitutive STAT3 and STAT5 activation through induction of PTPepsilon and SHP-2 tyrosine phosphatases and potentiates sorafenib-induced apoptosis in renal cell carcinoma. BMC Nephrol. 2016, 17, 19. [Google Scholar] [CrossRef]
- Pak, S.; Kim, W.; Kim, Y.; Song, C.; Ahn, H. Dihydrotestosterone promotes kidney cancer cell proliferation by activating the STAT5 pathway via androgen and glucocorticoid receptors. J. Cancer Res. Clin. Oncol. 2019, 145, 2293–2301. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, M.K.; Rajendran, P.; Li, F.; Kim, C.; Sikka, S.; Siveen, K.S.; Kumar, A.P.; Ahn, K.S.; Sethi, G. Abrogation of STAT3 signaling cascade by zerumbone inhibits proliferation and induces apoptosis in renal cell carcinoma xenograft mouse model. Mol. Carcinog. 2015, 54, 971–985. [Google Scholar] [CrossRef]
- Aoki, Y.; Feldman, G.M.; Tosato, G. Inhibition of STAT3 signaling induces apoptosis and decreases survivin expression in primary effusion lymphoma. Blood 2003, 101, 1535–1542. [Google Scholar] [CrossRef]
- de Vivar Chevez, A.R.; Finke, J.; Bukowski, R. The role of inflammation in kidney cancer. Adv. Exp. Med. Biol. 2014, 816, 197–234. [Google Scholar] [CrossRef]
- Oguro, T.; Ishibashi, K.; Sugino, T.; Hashimoto, K.; Tomita, S.; Takahashi, N.; Yanagida, T.; Haga, N.; Aikawa, K.; Suzutani, T.; et al. Humanised antihuman IL-6R antibody with interferon inhibits renal cell carcinoma cell growth in vitro and in vivo through suppressed SOCS3 expression. Eur. J. Cancer 2013, 49, 1715–1724. [Google Scholar] [CrossRef]
- Zhu, J.; Zhu, D.Q.; Zhang, Y.; Liu, Q.M.; Wang, P.C.; Li, H.Z.; Ma, X.; Zhang, X. MicroRNA-363 inhibits angiogenesis, proliferation, invasion, and migration of renal cell carcinoma via inactivation of the Janus tyrosine kinases 2-signal transducers and activators of transcription 3 axis by suppressing growth hormone receptor gene. J. Cell Physiol. 2019, 234, 2581–2592. [Google Scholar] [CrossRef]
- Lei, J.; Xiao, J.H.; Zhang, S.H.; Liu, Z.Q.; Huang, K.; Luo, Z.P.; Xiao, X.L.; Hong, Z.D. Non-coding RNA 886 promotes renal cell carcinoma growth and metastasis through the Janus kinase 2/signal transducer and activator of transcription 3 signaling pathway. Mol. Med. Rep. 2017, 16, 4273–4278. [Google Scholar] [CrossRef]
- Lue, H.W.; Cole, B.; Rao, S.A.; Podolak, J.; Van Gaest, A.; King, C.; Eide, C.A.; Wilmot, B.; Xue, C.; Spellman, P.T.; et al. Src and STAT3 inhibitors synergize to promote tumor inhibition in renal cell carcinoma. Oncotarget 2015, 6, 44675–44687. [Google Scholar] [CrossRef][Green Version]
- Hui, Z.; Tretiakova, M.; Zhang, Z.; Li, Y.; Wang, X.; Zhu, J.X.; Gao, Y.; Mai, W.; Furge, K.; Qian, C.N.; et al. Radiosensitization by inhibiting STAT1 in renal cell carcinoma. Int. J. Radiat. Oncol. Biol. Phys. 2009, 73, 288–295. [Google Scholar] [CrossRef]
- Pawlus, M.R.; Wang, L.; Hu, C.J. STAT3 and HIF1alpha cooperatively activate HIF1 target genes in MDA-MB-231 and RCC4 cells. Oncogene 2014, 33, 1670–1679. [Google Scholar] [CrossRef]
- Kim, H.D.; Yu, S.J.; Kim, H.S.; Kim, Y.J.; Choe, J.M.; Park, Y.G.; Kim, J.; Sohn, J. Interleukin-4 induces senescence in human renal carcinoma cell lines through STAT6 and p38 MAPK. J. Biol. Chem. 2013, 288, 28743–28754. [Google Scholar] [CrossRef]
- Chen, Y.; Zhu, Y.; Sheng, Y.; Xiao, J.; Xiao, Y.; Cheng, N.; Chai, Y.; Wu, X.; Zhang, S.; Xiang, T. SIRT1 downregulated FGB expression to inhibit RCC tumorigenesis by destabilizing STAT3. Exp. Cell Res. 2019, 382, 111466. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Wu, L.; Chen, X.; Wang, L.; Li, Q.; Zhang, Y.; Wu, J.; Cai, G.; Chen, X. Suppressor of Cytokine Signaling-1/STAT1 Regulates Renal Inflammation in Mesangial Proliferative Glomerulonephritis Models. Front. Immunol. 2018, 9, 1982. [Google Scholar] [CrossRef] [PubMed]
- Kalechman, Y.; Gafter, U.; Weinstein, T.; Chagnac, A.; Freidkin, I.; Tobar, A.; Albeck, M.; Sredni, B. Inhibition of Interleukin-10 by the Immunomodulator AS101 Reduces Mesangial Cell Proliferation in Experimental Mesangioproliferative Glomerulonephritis Association with Dephosphorylation of Stat3. J. Biol. Chem. 2004, 279, 24724–24732. [Google Scholar] [CrossRef]
- Baan, C.C.; Kannegieter, N.M.; Felipe, C.R.; Tedesco Silva, H., Jr. Targeting JAK/STAT Signaling to Prevent Rejection After Kidney Transplantation: A Reappraisal. Transplantation 2016, 100, 1833–1839. [Google Scholar] [CrossRef]
- Li, Y.; Xiong, Y.; Zhang, H.; Li, J.; Wang, D.; Chen, W.; Yuan, X.; Su, Q.; Li, W.; Huang, H.; et al. Ginkgo biloba extract EGb761 attenuates brain death-induced renal injury by inhibiting pro-inflammatory cytokines and the SAPK and JAK-STAT signalings. Sci. Rep. 2017, 7, 45192. [Google Scholar] [CrossRef]
- Zheng, X.; Zhao, Y.; Yang, L. Acute Kidney Injury in COVID-19: The Chinese Experience. Semin Nephrol. 2020, 40, 430–442. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, T.; Kubli, S.P.; Yoshinaga, S.K.; Pfeffer, K.; Mak, T.W. An aberrant STAT pathway is central to COVID-19. Cell Death Differ. 2020, 27, 3209–3225. [Google Scholar] [CrossRef]
- Wiede, F.; Lu, K.H.; Du, X.; Liang, S.; Hochheiser, K.; Dodd, G.T.; Goh, P.K.; Kearney, C.; Meyran, D.; Beavis, P.A.; et al. PTPN2 phosphatase deletion in T cells promotes anti-tumour immunity and CAR T-cell efficacy in solid tumours. EMBO J. 2020, 39, e103637. [Google Scholar] [CrossRef]
- Song, Z.; Wang, M.; Ge, Y.; Chen, X.P.; Xu, Z.; Sun, Y.; Xiong, X.F. Tyrosine phosphatase SHP2 inhibitors in tumor-targeted therapies. Acta Pharm. Sin. B 2021, 11, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhou, H.; Li, Y.; Han, L.; Song, M.; Chen, F.; Shang, G.; Wang, D.; Wang, Z.; Zhang, W.; et al. PTPN2 improved renal injury and fibrosis by suppressing STAT-induced inflammation in early diabetic nephropathy. J. Cell Mol. Med. 2019, 23, 4179–4195. [Google Scholar] [CrossRef] [PubMed]
- Krebs, D.L.; Hilton, D.J. SOCS proteins: Negative regulators of cytokine signaling. Stem Cells 2001, 19, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Susnik, N.; Sorensen-Zender, I.; Rong, S.; von Vietinghoff, S.; Lu, X.; Rubera, I.; Tauc, M.; Falk, C.S.; Alexander, W.S.; Melk, A.; et al. Ablation of proximal tubular suppressor of cytokine signaling 3 enhances tubular cell cycling and modifies macrophage phenotype during acute kidney injury. Kidney Int. 2014, 85, 1357–1368. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Yang, J.; Tong, F.; Duan, Z.; Liu, X.; Xia, L.; Li, K.; Xia, Y. Anti-Double-Stranded DNA IgG Participates in Renal Fibrosis through Suppressing the Suppressor of Cytokine Signaling 1 Signals. Front. Immunol. 2017, 8, 610. [Google Scholar] [CrossRef]
- Recio, C.; Lazaro, I.; Oguiza, A.; Lopez-Sanz, L.; Bernal, S.; Blanco, J.; Egido, J.; Gomez-Guerrero, C. Suppressor of Cytokine Signaling-1 Peptidomimetic Limits Progression of Diabetic Nephropathy. J. Am. Soc. Nephrol. 2017, 28, 575–585. [Google Scholar] [CrossRef]
- Shuai, K. Regulation of cytokine signaling pathways by PIAS proteins. Cell Res. 2006, 16, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Heppler, L.N.; Frank, D.A. Targeting Oncogenic Transcription Factors: Therapeutic Implications of Endogenous STAT Inhibitors. Trends Cancer 2017, 3, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Xu, A.; Lv, J.; Liu, Y. Expression of STAT3 and PIAS3 in renal tissues of MRL/lpr mice. Nan Fang Yi Ke Da Xue Xue Bao 2012, 32, 821–825. [Google Scholar]
- Nafar, M.; Kalantari, S.; Samavat, S.; Omrani, M.D.; Arsang-Jang, S.; Taheri, M.; Ghafouri-Fard, S. Downregulation of Protein Inhibitor of Activated STAT (PIAS) 1 Is Possibly Involved in the Process of Allograft Rejection. Transplant. Proc. 2020, 52, 414–418. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Pal, S.K.; Escudier, B.J.; Atkins, M.B.; Hutson, T.E.; Porta, C.; Verzoni, E.; Needle, M.N.; McDermott, D.F. Tivozanib versus sorafenib in patients with advanced renal cell carcinoma (TIVO-3): A phase 3, multicentre, randomised, controlled, open-label study. Lancet Oncol. 2020, 21, 95–104. [Google Scholar] [CrossRef]
- Song, L.; Morris, M.; Bagui, T.; Lee, F.Y.; Jove, R.; Haura, E.B. Dasatinib (BMS-354825) selectively induces apoptosis in lung cancer cells dependent on epidermal growth factor receptor signaling for survival. Cancer Res. 2006, 66, 5542–5548. [Google Scholar] [CrossRef]
- Wang, J.; Zhuang, S. Src family kinases in chronic kidney disease. Am. J. Physiol. Renal Physiol. 2017, 313, F721–F728. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Dong, Z.; Wang, F.; Peng, H.; Liu, J.Y.; Zhang, J.T. A small molecule compound targeting STAT3 DNA-binding domain inhibits cancer cell proliferation, migration, and invasion. ACS Chem. Biol. 2014, 9, 1188–1196. [Google Scholar] [CrossRef] [PubMed]
- Reilley, M.J.; McCoon, P.; Cook, C.; Lyne, P.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N.; et al. STAT3 antisense oligonucleotide AZD9150 in a subset of patients with heavily pretreated lymphoma: Results of a phase 1b trial. J. Immunother. Cancer 2018, 6, 119. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, G.G.; Huang, L.; Alston, N.; Ouyang, N.; Vrankova, K.; Mattheolabakis, G.; Constantinides, P.P.; Rigas, B. Targeting mitochondrial STAT3 with the novel phospho-valproic acid (MDC-1112) inhibits pancreatic cancer growth in mice. PLoS ONE 2013, 8, e61532. [Google Scholar] [CrossRef]
- Genini, D.; Brambilla, L.; Laurini, E.; Merulla, J.; Civenni, G.; Pandit, S.; D’Antuono, R.; Perez, L.; Levy, D.E.; Pricl, S.; et al. Mitochondrial dysfunction induced by a SH2 domain-targeting STAT3 inhibitor leads to metabolic synthetic lethality in cancer cells. Proc. Natl. Acad. Sci. USA 2017, 114, E4924–E4933. [Google Scholar] [CrossRef] [PubMed]
- Vo, A.A.; Choi, J.; Kim, I.; Louie, S.; Cisneros, K.; Kahwaji, J.; Toyoda, M.; Ge, S.; Haas, M.; Puliyanda, D.; et al. A Phase I/II Trial of the Interleukin-6 Receptor-Specific Humanized Monoclonal (Tocilizumab) + Intravenous Immunoglobulin in Difficult to Desensitize Patients. Transplantation 2015, 99, 2356–2363. [Google Scholar] [CrossRef] [PubMed]
- Vincenti, F.; Tedesco Silva, H.; Busque, S.; O’Connell, P.; Friedewald, J.; Cibrik, D.; Budde, K.; Yoshida, A.; Cohney, S.; Weimar, W.; et al. Randomized phase 2b trial of tofacitinib (CP-690,550) in de novo kidney transplant patients: Efficacy, renal function and safety at 1 year. Am. J. Transplant. 2012, 12, 2446–2456. [Google Scholar] [CrossRef] [PubMed]
- Krishnaswami, S.; Chow, V.; Boy, M.; Wang, C.; Chan, G. Pharmacokinetics of tofacitinib, a janus kinase inhibitor, in patients with impaired renal function and end-stage renal disease. J. Clin. Pharmacol. 2014, 54, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, K.R.; Brosius, F.C., 3rd; Adler, S.G.; Kretzler, M.; Mehta, R.L.; Tumlin, J.A.; Tanaka, Y.; Haneda, M.; Liu, J.; Silk, M.E.; et al. JAK1/JAK2 inhibition by baricitinib in diabetic kidney disease: Results from a Phase 2 randomized controlled clinical trial. Nephrol. Dial. Transplant. 2018, 33, 1950–1959. [Google Scholar] [CrossRef]
- Eisen, T.; Shparyk, Y.; Macleod, N.; Jones, R.; Wallenstein, G.; Temple, G.; Khder, Y.; Dallinger, C.; Studeny, M.; Loembe, A.B.; et al. Effect of small angiokinase inhibitor nintedanib (BIBF 1120) on QT interval in patients with previously untreated, advanced renal cell cancer in an open-label, phase II study. Investig. New Drugs 2013, 31, 1283–1293. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| STATs | Main Ligands | Biological Events |
|---|---|---|
| STAT1 | INFα/β, INFγ. | Anti-virus and anti-bacteria response; Cell growth; Cell apoptosis; Oncogenesis [1,8]. |
| STAT2 | INFα/β. | Anti-virus response; Oncogenesis [1,9]. |
| STAT3 | IL-6 family members: IL-6, IL-11, OSM, LIF, CLCF1, CNTF and erythropoietin, et al. Growth factors: EGF and HGF, et al. | Cell mitogenesis; Cell apoptosis; Oncogenesis; Cell proliferation; Th17 differentiation [1,7]. |
| STAT4 | IL-12. | Th1 development [1,10]. |
| STAT5a | Prolactin, IL-2, GM-CSF, erythropoietin and other hormonelike cytokines. | Prolactin signaling; Treg cells differentiation [1,11]. |
| STAT5b | Growth hormone, IL-2 and other hormonelike cytokines. | Growth hormone signaling [1,12]. |
| STAT6 | IL-4/13. | Th2 development [1]. |
| Targets of STAT Pathways | AKI Exacerbation | AKI Amelioration |
|---|---|---|
| Ligands | IL-6, IL-19, CXCL8 [45,46,47,50] | IL-4/IL-13, IL-22 [48,49] |
| Receptor inhibitors | IL-6 inhibitors, CXCR1/2 antagonist: G31P, EGFR inhibitor: gefitinib [45,46,50,51] | |
| JAK inhibitors | JAK3 inhibitor: AG490 [48] | JAK2 inhibitor: tofacitinib [44] |
| STAT inhibitors | STAT3 inhibitors [43] | STAT3 inhibitors [44] |
| Targeted Sites | Drug in Clinical Trials or Approved by FDA | Related Renal Diseases |
|---|---|---|
| Cytokine and cytokine receptor inhibitors | IL-6R inhibitors: tocilizumab, sarilumab | Tocilizumab in kidney transplantation [151] |
| VEGFR& EGFR inhibitors: Sorafenib | Sorafenib in renal cell carcinoma [144] | |
| Kinase inhibitors | JAK inhibitors: tofacitinib, ruxolitinib, baricitinib, fedratinib, upadacitinib (ClinicalTrials.gov) AZD1480 (NCT00910728) | Tofacitinib in kidney transplantation [152] Tofacitinib in end-stage renal disease undergoing hemodialysis [153] Baricitinib in type 2 diabetes and diabetic nephropathy [154] |
| Src inhibitors: dasatinib [145], Nintedanib [146] | Dasatinib in senescence of Chronic Kidney Disease (NCT02848131) Nintedanib in renal cell carcinoma [155] | |
| STAT inhibitors | Inhibitors targeted SH2 domain: STA-21(NCT01047943), OPB-31121 (NCT01406574), TTI-101(NCT03195699), OPB-51602 (NCT02058017) | |
| Antisense oligonucleotide: danvatirsen [148] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gai, L.; Zhu, Y.; Zhang, C.; Meng, X. Targeting Canonical and Non-Canonical STAT Signaling Pathways in Renal Diseases. Cells 2021, 10, 1610. https://doi.org/10.3390/cells10071610
Gai L, Zhu Y, Zhang C, Meng X. Targeting Canonical and Non-Canonical STAT Signaling Pathways in Renal Diseases. Cells. 2021; 10(7):1610. https://doi.org/10.3390/cells10071610
Chicago/Turabian StyleGai, Lili, Yuting Zhu, Chun Zhang, and Xianfang Meng. 2021. "Targeting Canonical and Non-Canonical STAT Signaling Pathways in Renal Diseases" Cells 10, no. 7: 1610. https://doi.org/10.3390/cells10071610
APA StyleGai, L., Zhu, Y., Zhang, C., & Meng, X. (2021). Targeting Canonical and Non-Canonical STAT Signaling Pathways in Renal Diseases. Cells, 10(7), 1610. https://doi.org/10.3390/cells10071610