
The Heritability of Replication Problems


{kind=link}
Abstract
1. Introduction
2. Progression of Replication Problems into Mitosis
2.1. Stimulation of Mitotic DNA Synthesis in Early Mitosis
2.2. Generation of UFB in Mitosis
3. Consequences of the Inheritability of Under-Replicated DNA in Daughter Cells
3.1. Transmitted DNA Damage Are Protected within 53BP1 NBs in G1 Phase and Resolved in S Phase of Daughter Cells
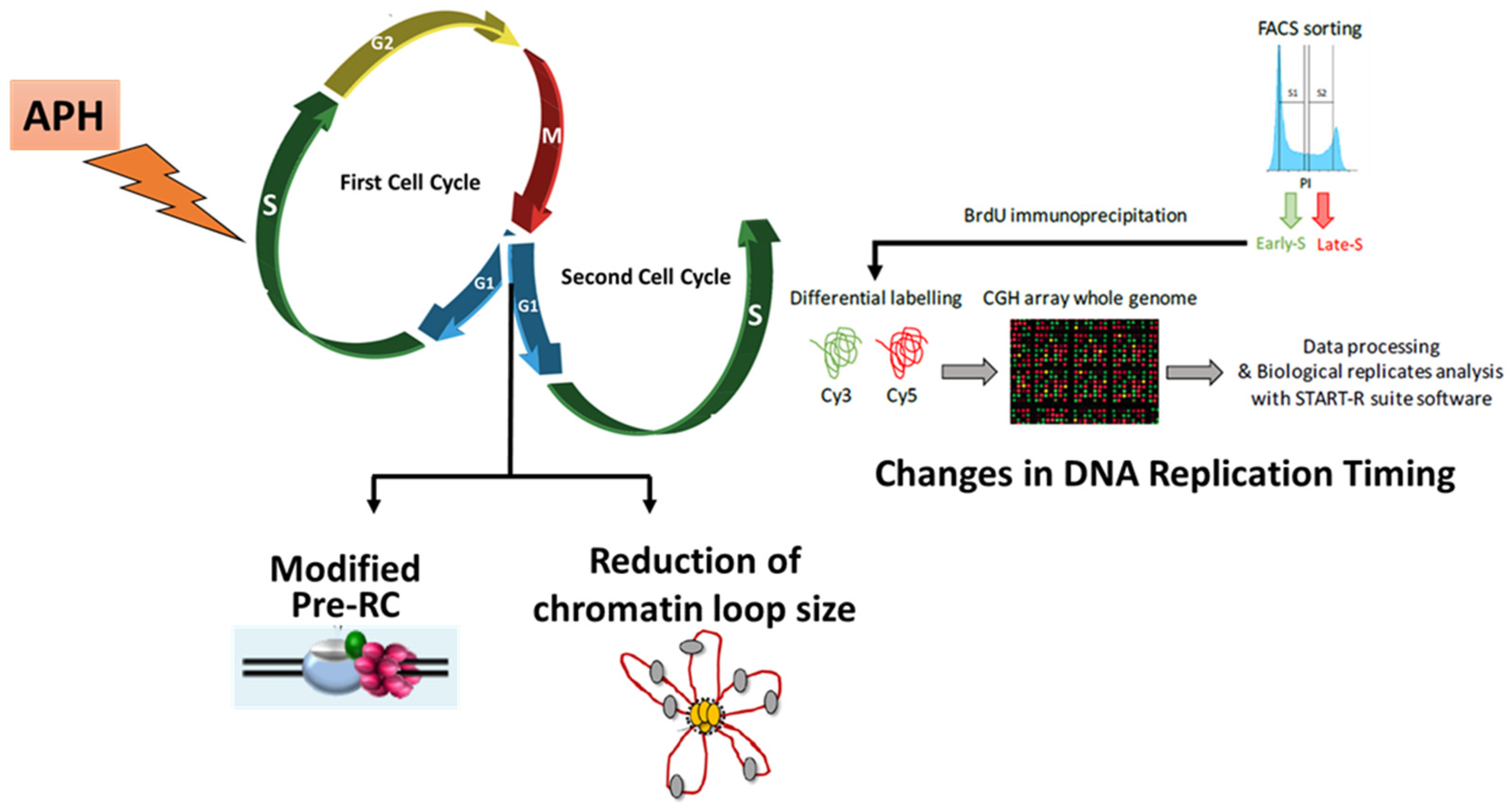
3.2. RS modifies Pre-RC Formation and Spatial Organization of Replication Origins in the Next Generation of Cells
4. RS Modifies the Replication Timing at a Fraction of Chromosomal Domains in Unstressed Daughter Cells
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Yeeles, J.T.P.; Janska, A.; Early, A.; Diffley, J.F.X. How the eukaryotic replisome achieves rapid and efficient DNA replication. Mol. Cell 2017, 65, 105–116. [Google Scholar] [CrossRef]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Courtot, L.; Hoffmann, J.-S.; Bergoglio, V. The protective role of dormant origins in response to replicative stress. Int. J. Mol. Sci. 2018, 19, 3569. [Google Scholar] [CrossRef] [PubMed]
- Bertolin, A.P.; Hoffmann, J.-S.; Gottifredi, V. Under-Replicated DNA: The byproduct of large genomes? Cancers 2020, 12, 2764. [Google Scholar] [CrossRef] [PubMed]
- Boyer, A.-S.; Grgurevic, S.; Cazaux, C.; Hoffmann, J.-S. The human specialized DNA polymerases and Non-B DNA: Vital relationships to preserve genome integrity. J. Mol. Biol. 2013, 425, 4767–4781. [Google Scholar] [CrossRef] [PubMed]
- Hecht, F.; Glover, T.W. Cancer chromosome breakpoints and common fragile sites induced by aphidicolin. Cancer Genet. Cytogenet. 1984, 13, 185–188. [Google Scholar] [CrossRef]
- Gaillard, H.; García-Muse, T.; Aguilera, A. Replication stress and cancer. Nat. Rev. Cancer 2015, 15, 276–289. [Google Scholar] [CrossRef]
- Macheret, M.; Halazonetis, T.D. DNA replication stress as a hallmark of cancer. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 425–448. [Google Scholar] [CrossRef]
- Saldivar, J.C.; Cortez, D.; Cimprich, K.A. The essential kinase ATR: Ensuring faithful duplication of a challenging genome. Nat. Rev. Mol. Cell Biol. 2017, 18, 622–636. [Google Scholar] [CrossRef]
- Franchet, C.; Hoffmann, J.-S. When RAD52 allows mitosis to accept unscheduled DNA synthesis. Cancers 2020, 12, 26. [Google Scholar] [CrossRef] [PubMed]
- Bergoglio, V.; Boyer, A.S.; Walsh, E.; Naim, V.; Legube, G.; Lee, M.Y.; Rey, L.; Rosselli, F.; Cazaux, C.; Eckert, K.A.; et al. DNA synthesis by Pol eta promotes fragile site stability by preventing under-replicated DNA in mitosis. J. Cell Biol. 2013, 201, 395–408. [Google Scholar] [CrossRef]
- Minocherhomji, S.; Ying, S.; Bjerregaard, V.A.; Bursomanno, S.; Aleliunaite, A.; Wu, W.; Mankouri, H.W.; Shen, H.; Liu, Y.; Hickson, I.D. Replication stress activates DNA repair synthesis in mitosis. Nature 2015, 528, 286–290. [Google Scholar] [CrossRef]
- Lai, X.; Broderick, R.; Bergoglio, V.; Zimmer, J.; Badie, S.; Niedzwiedz, W.; Hoffmann, J.S.; Tarsounas, M. MUS81 nuclease activity is essential for replication stress tolerance and chromosome segregation in BRCA2-deficient cells. Nat. Commun. 2017, 8, 15983. [Google Scholar] [CrossRef]
- Garcia-Exposito, L.; Bournique, E.; Bergoglio, V.; Bose, A.; Barroso-Gonzalez, J.; Zhang, S.; Roncaioli, J.L.; Lee, M.; Wallace, C.T.; Watkins, S.C.; et al. Proteomic profiling reveals a specific role for translesion DNA polymerase eta in the alternative lengthening of telomeres. Cell Rep. 2016, 17, 1858–1871. [Google Scholar] [CrossRef]
- Ozer, O.; Bhowmick, R.; Liu, Y.; Hickson, I.D. Human cancer cells utilize mitotic DNA synthesis to resist replication stress at telomeres regardless of their telomere maintenance mechanism. Oncotarget 2018, 9, 15836–15846. [Google Scholar] [CrossRef] [PubMed]
- Sonneville, R.; Bhowmick, R.; Hoffmann, S.; Mailand, N.; Hickson, I.D.; Labib, K. TRAIP drives replisome disassembly and mitotic DNA repair synthesis at sites of incomplete DNA replication. eLife 2019, 8, e48686. [Google Scholar] [CrossRef]
- Chan, K.L.; Hickson, I.D. New insights into the formation and resolution of ultra-fne anaphase bridges. Sem. Cell Dev. Biol. 2011, 22, 906–912. [Google Scholar] [CrossRef]
- Liu, Y.; Nielsen, C.F.; Yao, Q.; Hickson, I.D. The origins and processing of ultra fne anaphase DNA bridges. Curr. Opin. Genet. Dev. 2014, 26, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Mankouri, H.W.; Huttner, D.; Hickson, I.D. How unfinished business from S-phase affects mitosis and beyond. EMBO J. 2013, 32, 2661–2671. [Google Scholar] [CrossRef] [PubMed]
- Lukas, C.; Savic, V.; Bekker-Jensen, S.; Doil, C.; Neumann, B.; Pedersen, R.S.; Grøfte, M.; Chan, K.L.; Hickson, I.D.; Bartek, J.; et al. 53BP1 nuclear bodies form around DNA lesions generated by mitotic. Nat. Cell Biol. 2011, 13, 243–253. [Google Scholar] [CrossRef]
- Harrigan, J.A.; Belotserkovskaya, R.; Coates, J.; Dimitrova, D.S.; Polo, S.E.; Bradshaw, C.R.; Fraser, P.; Jackson, S.P. Replication stress induces 53BP1-containing OPT domains in G1 cells. J. Cell Biol. 2011, 193, 97–108. [Google Scholar] [CrossRef]
- Bothmer, A.; Robbiani, D.F.; Feldhahn, N.; Gazumyan, A.; Nussenzweig, A.; Nussenzweig, M.C. 53BP1 regulates DNA resection and the choice between classical and alternative end joining during class switch recombination. J. Exp. Med. 2010, 207, 855–865. [Google Scholar] [CrossRef]
- Mansilla, S.F.; Bertolin, A.P.; Bergoglio, V.; Pillaire, M.J.; Besteiro, M.A.G.; Luzzani, C.; Miriuka, S.G.; Cazaux, C.; Hoffmann, J.S.; Gottifredi, V. Cyclin Kinase-independent role of p21CDKN1A in the promotion of nascent DNA elongation in unstressed cells. eLife 2016, 5, e18020. [Google Scholar] [CrossRef]
- Spies, J.; Lukas, C.; Somyajit, K.; Rask, M.B.; Lukas, J.; Neelsen, K.J. 53BP1 nuclear bodies enforce replication timing at under-replicated DNA to limit heritable DNA damage. Nat. Cell Biol. 2019, 21, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Mechali, M. Eukaryotic DNA replication origins: Many choices for appropriate answers. Nat. Rev. Mol. Cell Biol. 2010, 11, 728–738. [Google Scholar] [CrossRef] [PubMed]
- Courtot, L.; Bournique, E.; Maric, C.; Guitton-Sert, L.; Madrid-Mencía, M.; Pancaldi, V.; Cadoret, J.-C.; Hoffmann, J.-S.; Bergoglio, V. Low replicative stress triggers cell-type specific inheritable advanced replication timing. Int. J. Mol. Sci. 2021, 22, 4959. [Google Scholar] [CrossRef] [PubMed]
- Guillou, E.; Ibarra, A.; Coulon, V.; Casado-Vela, J.; Rico, D.; Casal, I.; Schwob, E.; Losada, A.; Méndez, J. Cohesin organizes chromatin loops at DNA replication factories. Genes Dev. 2010, 24, 2812–2822. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Vidal, A.; Guitton-Sert, L.; Cadoret, J.C.; Drac, M.; Schwob, E.; Baldacci, J.; Cazaux, C.; Hoffmann, J.S. A role for DNA polymerase theta in the timing of DNA replication. Nat. Commun. 2014, 5, 4285. [Google Scholar] [CrossRef] [PubMed]
- Kermi, C.; Aze, A.; Maiorano, D. Preserving genome integrity during the early embryonic DNA replication cycles. Genes 2019, 10, 398. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoffmann, J.-S. The Heritability of Replication Problems. Cells 2021, 10, 1464. https://doi.org/10.3390/cells10061464
Hoffmann J-S. The Heritability of Replication Problems. Cells. 2021; 10(6):1464. https://doi.org/10.3390/cells10061464
Chicago/Turabian StyleHoffmann, Jean-Sébastien. 2021. "The Heritability of Replication Problems" Cells 10, no. 6: 1464. https://doi.org/10.3390/cells10061464
APA StyleHoffmann, J.-S. (2021). The Heritability of Replication Problems. Cells, 10(6), 1464. https://doi.org/10.3390/cells10061464