
Lung Transplantation, Pulmonary Endothelial Inflammation, and Ex-Situ Lung Perfusion: A Review


Abstract
1. Introduction
2. Hostile Environments, Risk Factors, Clinical Manifestations and Treatment of Primary Graft Dysfunction in Lung Transplantation
2.1. Pretransplant/Donor Risk Factors for PGD
2.2. Procurement and Cold Static Preservation: Gold Standard Preservation Strategies Contribute to Primary Graft Dysfunction
2.3. Posttransplant/Recipient Risk Factors for PGD
2.4. Clinical Manifestation of PGD Post LTx
2.5. Postoperative Care and Current Treatment of Lungs with PGD
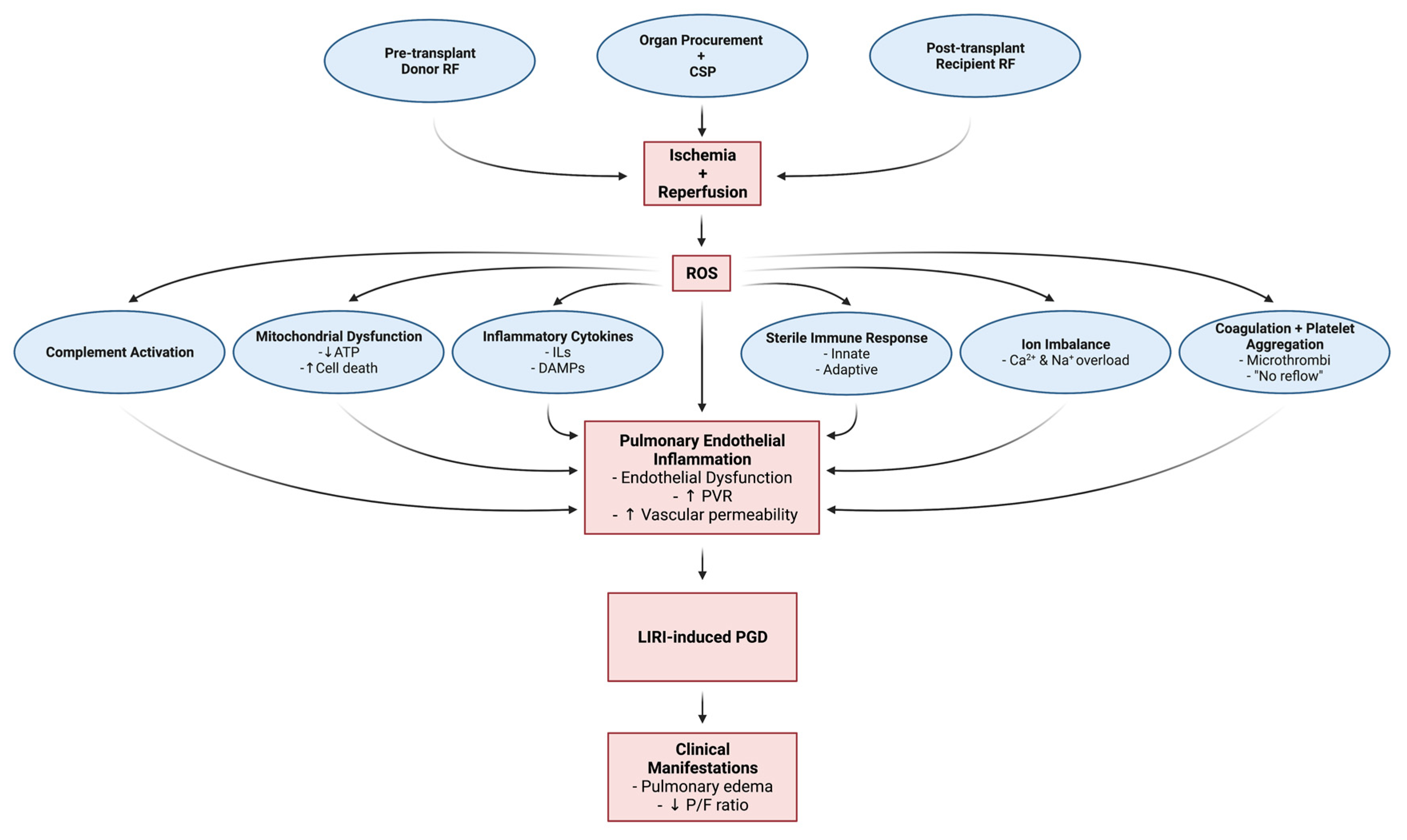
3. Pathophysiology of Ischemic Reperfusion Injury, Pulmonary Endothelial Inflammation, and Primary Graft Dysfunction
3.1. Pathophysiologic and Cellular Mechanisms of LIRI, Pulmonary Inflammation and PGD
3.1.1. Oxidative Stress Response: Reactive Oxygen Species Generation and Downstream Signaling
3.1.2. Inflammation: Cytokines, Chemokines, and Damage-Associated Molecular Patterns (DAMPs)
3.1.3. Sterile Immunologic Injury: Innate and Adaptive Immune Responses
3.1.4. Multifactorial Cellular Mechanisms: Anaerobic Metabolism, Ion Imbalances and Mitochondrial Dysfunction
3.1.5. Complement Activation, Coagulation, and Increased Platelet Aggregation
3.1.6. Endothelial Cell Dysfunction, Pulmonary Vascular Resistance, and Vascular Permeability
3.1.7. Summary
4. Ex-Situ Lung Perfusion for the Prevention and Treatment of PGD
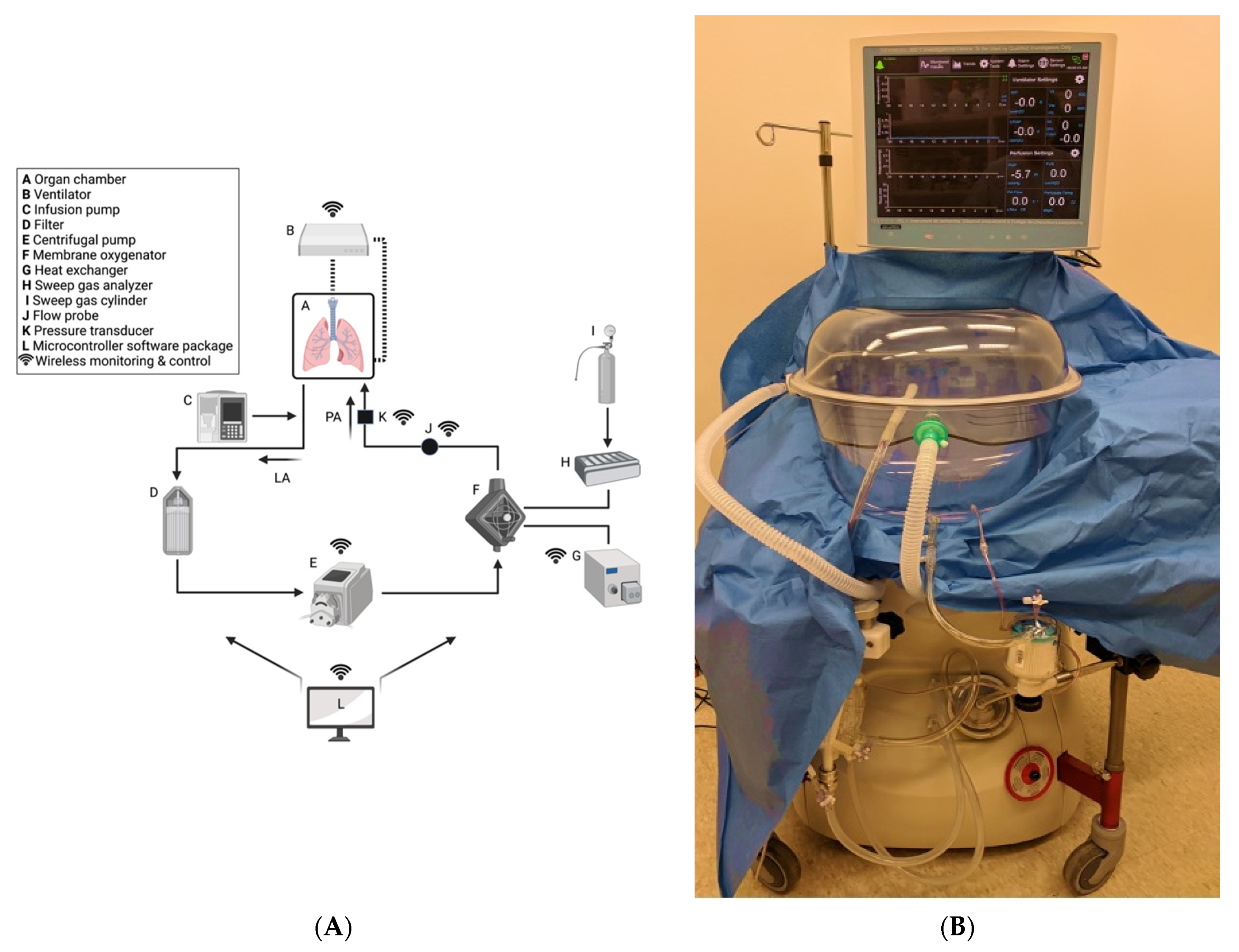
4.1. Overview of ESLP
4.2. ESLP: LIRI and Pulmonary Inflammation
ESLP: Pulmonary and Bronchial Circulation
4.3. Negative Pressure Ventilation (NPV) versus Positive Pressure Ventilation (PPV) ESLP
4.4. NPV-ESLP and Extended Criteria Donors
4.5. ESLP as a Therapeutic Vehicle
4.6. Animal Models of ESLP Therapeutics
4.6.1. Gene Therapy
4.6.2. Mesenchymal Stem Cells
4.6.3. A2AR Agonists
4.6.4. Inhaled Agents
4.7. The Future of ESLP
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cooper, J.; Pearson, F.; Patterson, G.; Todd, T.R.; Ginsberg, R.J.; Goldberg, M.; DeMajo, W.A. Technique of successful lung transplantation in humans. J. Thorac. Cardiovasc. Surg. 1987, 93, 173–181. [Google Scholar] [CrossRef]
- Chambers, D.C.; Yusen, R.D.; Cherikh, W.S.; Goldfarb, S.B.; Kucheryavaya, A.Y.; Khusch, K.; Levvey, B.J.; Lund, L.H.; Meiser, B.; Rossano, J.W.; et al. The Registry of the International Society for Heart and Lung Transplantation: Thirty-fourth Adult Lung and Heart-Lung Transplantation Report-2017; Focus Theme: Allograft ischemic time. J. Heart Lung Transplant. 2017, 36, 1047–1059. [Google Scholar] [CrossRef] [PubMed]
- Israni, A.K.; Zaun, D.; Bolch, C.; Rosendale, J.D.; Schaffhausen, C.; Snyder, J.J.; Kasiske, B.L. OPTN/SRTR 2015 Annual Data Report: Deceased Organ Donation. Am. J. Transplant. 2017, 17, 503–542. [Google Scholar] [CrossRef] [PubMed]
- Snell, G.I.; Paraskeva, M.; Westall, G.P. Donor selection and management. Semin. Respir. Crit. Care Med. 2013, 34, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Orens, J.B.; Boehler, A.; Perrot, M.D.; Estenne, M.; Glanville, A.R.; Keshavjee, S.; Kotloff, R.; Morton, J.; Studer, S.M.; Van Raemdonck, D.; et al. A review of lung transplant donor acceptability criteria. J. Heart Lung Transplant. 2003, 22, 1183–1200. [Google Scholar] [CrossRef]
- De Perrot, M.; Weder, W.; Patterson, G.A.; Keshavjee, S. Strategies to increase limited donor resources. Eur. Respir. J. 2004, 23, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Snell, G.I.; Griffiths, A.; Levvey, B.J.; Oto, T. Availability of lungs for transplantation: Exploring the real potential of the donor pool. J. Heart Lung Transplant. 2008, 27, 662–667. [Google Scholar] [CrossRef]
- Bhorade, S.M.; Vigneswaran, W.; McCabe, M.A.; Garrity, E.R. Liberalization of donor criteria may expand the donor pool without adverse consequence in lung transplantation. J. Heart Lung Transplant. 2000, 19, 1199–1204. [Google Scholar] [CrossRef]
- Botha, P.; Trivedi, D.; Weir, C.J.; Searl, C.P.; Corris, P.A.; Dark, J.H.; Schueler, S.V. Extended donor criteria in lung transplantation: Impact on organ allocation. J. Thorac. Cardiovasc. Surg. 2006, 131, 1154–1160. [Google Scholar] [CrossRef]
- De Perrot, M.; Liu, M.; Waddell, T.K.; Keshavjee, S. Ischemia-reperfusion-induced lung injury. Am. J. Respir. Crit. Care Med. 2003, 167, 490–511. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Cantu, E.; Christie, J.D. Primary graft dysfunction. Semin. Respir. Crit. Care Med. 2013, 34, 305–319. [Google Scholar] [CrossRef] [PubMed]
- Lardinois, D.; Banysch, M.; Korom, S.; Hillinger, S.; Rousson, V.; Boehler, A.; Speich, R.; Weder, W. Extended donor lungs: Eleven years experience in a consecutive series. Eur. J. Cardiothorac. Surg. 2005, 27, 762–767. [Google Scholar] [CrossRef] [PubMed]
- Steen, S.; Sjoberg, T.; Pierre, L.; Liao, Q.; Eriksson, L.; Algotsson, L. Transplantation of lungs from a non-heart-beating donor. Lancet 2001, 357, 825–829. [Google Scholar] [CrossRef]
- Talaie, T.; Dichiacchio, L.; Prasad, N.K.; Pasrija, C.; Julliard, W.; Kaczorowski, D.J.; Zhao, Y.; Lau, C.L. Ischemia-reperfusion injury in the transplanted lung: A literature review. Transplant. Direct 2021, 7, e652. [Google Scholar] [CrossRef] [PubMed]
- De Perrot, M.; Bonser, R.S.; Dark, J.; Kelly, R.F.; McGiffin, D.; Menza, R.; Pajaro, O.; Schueler, S.; Verleden, G.M.; ISHLT Working Group on Primary Lung Graft Dysfunction. Report of the ISHLT Working Group on Primary Lung Graft Dysfunction part III: Donor-related risk factors and markers. J. Heart Lung Transplant. 2005, 24, 1460–1467. [Google Scholar] [CrossRef] [PubMed]
- Barr, M.L.; Kawut, S.M.; Whelan, T.P.; Girgis, R.; Böttcher, H.; Sonett, J.; Vigneswaran, W.; Follette, D.M.; Corris, P.A.; ISHLT Working Group on Primary Lung Graft Dysfunction. Report of the ISHLT Working Group on Primary Lung Graft Dysfunction part IV: Recipient-related risk factors and markers. J. Heart Lung Transplant. 2005, 24, 1468–1482. [Google Scholar] [CrossRef]
- Christie, J.D.; Kotloff, R.M.; Pochettino, A.; Arcasoy, S.M.; Rosengard, B.R.; Landis, J.R.; Kimmel, S.E. Clinical risk factors for primary graft failure following lung transplantation. Chest 2003, 124, 1232–1241. [Google Scholar] [CrossRef] [PubMed]
- Shaver, C.M.; Ware, L.B. Primary graft dysfunction: Pathophysiology to guide new preventive therapies. Expert Rev. Respir. Med. 2017, 11, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Oto, T.; Excell, L.; Griffiths, A.P.; Levvey, B.J.; Snell, G.I. The implications of pulmonary embolism in a multiorgan donor for subsequent pulmonary, renal, and cardiac transplantation. J. Heart Lung Transplant. 2008, 27, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Porteous, M.K.; Lee, J.C. Primary graft dysfunction after lung transplantation. Clin. Chest Med. 2017, 38, 641–654. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Busuttil, R.W.; Kupiec-Weglinski, J.W. Liver ischemia and reperfusion injury: New insights into mechanisms of innate-adaptive immune-mediated tissue inflammation. Am. J. Transplant. 2011, 11, 1563–1569. [Google Scholar] [CrossRef] [PubMed]
- Cinel, I.; Opal, S.M. Molecular biology of inflammation and sepsis: A primer. Crit. Care Med. 2009, 37, 291–304. [Google Scholar] [CrossRef]
- Babiker, M.A.; Obeid, H.A.; Ashong, E.F. Acute reversible pulmonary ischemia. A cause of the acute chest syndrome in sickle cell disease. Clin. Pediatr. 1985, 24, 716–718. [Google Scholar] [CrossRef]
- Templeton, A.W.; Garrotto, L.J. Acquired extracardiac causes of pulmonary ischemia. Dis. Chest 1967, 51, 166–171. [Google Scholar] [CrossRef]
- Apostolakis, E.; Filos, K.S.; Koletsis, E.; Dougenis, D. Lung dys- function following cardiopulmonary bypass. J. Card. Surg. 2010, 25, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Weyker, P.D.; Webb, C.A.; Kiamanesh, D.; Flynn, B.C. Lung ischemia reperfusion injury: A bench- to-bedside review. Semin. Cardiothorac. Vasc. Anesth. 2013, 17, 28–43. [Google Scholar] [CrossRef] [PubMed]
- Petrenko, A.; Carnevale, M.; Somov, A.; Osorio, J.; Rodríguez, J.; Guibert, E.; Fuller, B.; Froghi, F. Organ preservation into the 2020s: The era of dynamic intervention. Transfus. Med. Hemother. 2019, 46, 151–172. [Google Scholar] [CrossRef]
- Courtwright, A.; Cantu, E. Evaluation and management of the potential lung donor. Clin. Chest Med. 2017, 38, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Lederer, D.J.; Kawut, S.M.; Wickersham, N.; Winterbottom, C.; Bhorade, S.; Palmer, S.M.; Lee, J.; Diamond, J.M.; Wille, K.M.; Weinacker, A.; et al. Obesity and primary graft dysfunction after lung transplantation: The Lung Transplant Outcomes Group Obesity Study. Am. J. Respir. Crit. Care Med. 2011, 184, 1055–1061. [Google Scholar] [CrossRef]
- Goodwin, J.; Tinckam, K.; denHollander, N.; Haroon, A.; Keshavjee, S.; Cserti-Gazdewich, C.M. Transfusion-related acute lung injury (TRALI) in graft by blood donor antibodies against host leukocytes. J. Heart Lung Transplant. 2010, 29, 1067–1070. [Google Scholar] [CrossRef] [PubMed]
- Pierre, A.F.; De Campos, K.N.; Liu, M.; Edwards, V.; Cutz, E.; Slutsky, A.S.; Keshavjee, S.H. Rapid reperfusion causes stress failure in ischemic rat lungs. J. Thorac. Cardiovasc. Surg. 1998, 116, 932–942. [Google Scholar] [CrossRef]
- Bhabra, M.S.; Hopkinson, D.N.; Shaw, T.E.; Onwu, N.; Hooper, T.L. Controlled reperfusion protects lung grafts during a transient early increase in permeability. Ann. Thorac. Surg. 1998, 65, 187–192. [Google Scholar] [CrossRef]
- Halldorsson, A.; Kronon, M.; Allen, B.S.; Bolling, K.S.; Wang, T.; Rahman, S.; Feinberg, H. Controlled reperfusion after lung ischemia: Implications for improved function after lung transplantation. J. Thorac. Cardiovasc. Surg. 1998, 115, 415–424. [Google Scholar] [CrossRef][Green Version]
- Dos Santos, C.C.; Slutsky, A.S. Invited review: Mechanisms of ventilator-induced lung injury: A perspective. J. Appl. Physiol. 2000, 89, 1645–1655. [Google Scholar] [CrossRef]
- De Perrot, M.; Imai, Y.; Volgyesi, G.A.; Waddell, T.K.; Liu, M.; Mullen, J.B.; McRae, K.; Zhang, H.; Slutsky, A.S.; Ranieri, V.M.; et al. Effect of ventilator-induced lung injury on the development of reperfusion injury in a rat lung transplant model. J. Thorac. Cardiovasc. Surg. 2002, 124, 1137–1144. [Google Scholar] [CrossRef] [PubMed]
- McRae, K.M. Pulmonary transplantation. Curr. Opin. Anaesthesiol. 2000, 13, 53–59. [Google Scholar] [CrossRef]
- Christie, J.D.; Carby, M.; Bag, R.; Corris, P.; Hertz, M.; Weill, D.; ISHLT Working Group on Primary Lung Graft Dysfunction. Report of the ISHLT Working Group on Primary Lung Graft Dysfunction part II: Definition. A consensus statement of the International Society for Heart and Lung Transplantation. J. Heart Lung Transplant. 2005, 24, 1454–1459. [Google Scholar]
- Beer, A.; Reed, R.M.; Bolukbas, S.; Budev, M.; Chaux, G.; Zamora, M.R.; Snell, G.; Orens, J.B.; Klesney-Tait, J.A.; Schmidt, G.A.; et al. Mechanical ventilation after lung transplantation. An international survey of practices and preferences. Ann. Am. Thorac. Soc. 2014, 11, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Verbeek, G.L.; Myles, P.S. Intraoperative protective ventilation strategies in lung transplantation. Transplant. Rev. 2013, 27, 30–35. [Google Scholar] [CrossRef]
- Goudarzi, B.M.; Bonvino, S. Critical care issues in lung and heart transplantation. Crit. Care Clin. 2003, 19, 209–231. [Google Scholar] [CrossRef]
- Porteous, M.K.; Diamond, J.M.; Christie, J.D. Primary graft dysfunction: Lessons learned about the first 72 h after lung transplantation. Curr. Opin. Organ. Transplant. 2015, 20, 506–514. [Google Scholar] [CrossRef]
- Jin, Z.; Suen, K.C.; Wang, Z.; Ma, D. Review 2: Primary graft dysfunction after lung transplant-pathophysiology, clinical considerations and therapeutic targets. J. Anesth. 2020, 34, 729–740. [Google Scholar] [CrossRef]
- Christie, J.D.; Bellamy, S.; Ware, L.B.; Lederer, D.; Hadjiliadis, D.; Lee, J.; Robinson, N.; Localio, A.R.; Wille, K.; Lama, V.; et al. Construct validity of the definition of primary graft dysfunction after lung transplantation. J. Heart Lung Transplant. 2010, 29, 1231–1239. [Google Scholar] [CrossRef] [PubMed]
- Snell, G.I.; Yusen, R.D.; Weill, D.; Strueber, M.; Garrity, E.; Reed, A.; Pelaez, A.; Whelan, T.P.; Perch, M.; Bag, R.; et al. Report of the ISHLT Working Group on Primary Lung Graft Dysfunction, part I: Definition and grading-A 2016 Consensus Group statement of the International Society for Heart and Lung Transplantation. J. Heart Lung Transplant. 2017, 36, 1097–1103. [Google Scholar] [CrossRef]
- Lucangelo, U.; Del Sorbo, L.; Boffini, M.; Ranieri, V.M. Protective ventilation for lung transplantation. Curr. Opin. Anaesthesiol. 2012, 25, 170–174. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, S.A.; Robinson, N.; Christie, J.D. Primary graft dysfunction. Curr. Opin. Organ. Transplant. 2007, 12, 473–478. [Google Scholar] [CrossRef]
- Shargall, Y.; Guenther, G.; Ahya, V.N.; Ardehali, A.; Singhal, A.; Keshavjee, S.; ISHLT Working Group on Primary Lung Graft Dysfunction. Report of the ISHLT Working Group on Primary Lung Graft Dysfunction part VI: Treatment. J. Heart Lung Transplant. 2005, 24, 1489–1500. [Google Scholar]
- Meyers, B.F.; Sundt, T.M., 3rd; Henry, S.; Trulock, E.P.; Guthrie, T.; Cooper, J.D.; Patterson, G.A. Selective use of extracorporeal membrane oxygenation is warranted after lung transplantation. J. Thorac. Cardiovasc. Surg. 2000, 120, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, M.G.; Walczak, R.; Lin, S.S.; Davis, R.D. Improved survival but marginal allograft function in patients treated with extracorporeal membrane oxygenation after lung transplantation. Ann. Thorac. Surg. 2012, 93, 366–371. [Google Scholar] [CrossRef]
- Kalogeris, T.; Bao, Y.; Korthuis, R.J. Mitochondrial reactive oxygen species: A double edged sword in ischemia/reperfusion vs. preconditioning. Redox Biol. 2014, 2, 702–714. [Google Scholar] [CrossRef]
- Den Hengst, W.A.; Gielis, J.F.; Lin, J.Y.; Van Schil, P.E.; De Windt, L.J.; Moens, A.L. Lung ischemia-reperfusion injury: A molecular and clinical view on a complex pathophysiological process. American Journal of Physiology. Heart Circ. Physiol. 2010, 299, 1283–1299. [Google Scholar] [CrossRef]
- Girn, H.R.; Ahilathirunayagam, S.; Mavor, A.I.; Homer-Vanniasinkam, S. Reperfusion syndrome: Cellular mechanisms of microvascular dysfunction and potential therapeutic strategies. Vasc. Endovasc. Surg. 2007, 41, 277–293. [Google Scholar]
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion—From mechanism to translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef]
- Forgiarini, L.A., Jr.; Grun, G.; Kretzmann, N.A.; de Muñoz, G.A.; de Almeida, A.; Forgiarini, L.F.; Andrade, C.F. When is injury potentially reversible in a lung ischemia-reperfusion model? J. Surg. Res. 2013, 179, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, R.S.; Andrade, C.F. Oxidative stress and lung ischemia-reperfusion injury. Oxid. Med. Cell. Longev. 2015, 2015, 590987. [Google Scholar] [CrossRef] [PubMed]
- Pak, O.; Sydykov, A.; Kosanovic, D.; Schermuly, R.T.; Dietrich, A.; Schröder, K.; Brandes, R.P.; Gudermann, T.; Sommer, N.; Weissmann, N. Lung ischaemia-reperfusion injury: The role of reactive oxygen species. Adv. Exp. Med. Biol. 2017, 967, 195–225. [Google Scholar] [PubMed]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- D’Autréaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Mailloux, R.J. Teaching the fundamentals of electron transfer reactions in mitochondria and the production and detection of reactive oxygen species. Redox Biol. 2015, 4, 381–398. [Google Scholar] [CrossRef]
- Ovechkin, A.V.; Lominadze, D.; Sedoris, K.C.; Robinson, T.W.; Tyagi, S.C.; Roberts, A.M. Lung ischemia-reperfusion injury: Implications of oxidative stress and platelet-arteriolar wall interactions. Arch. Physiol. Biochem. 2007, 113, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, N.F.; Eickelberg, O. Three is better than one: An improved multiple-hit model of primary graft dysfunction. Am. J. Respir. Cell Mol. Biol. 2019, 61, 141–142. [Google Scholar] [CrossRef]
- Weissmann, N.; Sydykov, A.; Kalwa, H.; Storch, U.; Fuchs, B.; Mederos y Schnitzler, M.; Brandes, R.P.; Grimminger, F.; Meissner, M.; Freichel, M. Activation of TRPC6 channels is essential for lung ischaemia-reperfusion induced oedema in mice. Nat. Commun. 2012, 3, 649. [Google Scholar] [CrossRef]
- Schröder, K. NADPH oxidases in redox regulation of cell adhesion and migration. Antioxid. Redox Signal. 2014, 20, 2043–2058. [Google Scholar] [CrossRef] [PubMed]
- Brigham, K.L. Role of free radicals in lung injury. Chest 1986, 89, 859–863. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, T.P.; Rao, N.V.; Hopkins, C.; Pennington, L.; Tolley, E.; Hoidal, J.R. Role of reactive oxygen species in reperfusion injury of the rabbit lung. J. Clin. Investig. 1989, 83, 1326–1335. [Google Scholar] [CrossRef]
- McCord, J.M. Oxygen-derived free radicals in postischemic tissue injury. N. Engl. J. Med. 1985, 312, 159–163. [Google Scholar] [PubMed]
- Egemnazarov, B.; Sydykov, A.; Schermuly, R.T.; Weissmann, N.; Stasch, J.P.; Sarybaev, A.S.; Seeger, W.; Grimminger, F.; Ghofrani, H.A. Novel soluble guanylyl cyclase stimulator BAY 41-2272 attenuates ischemia-reperfusion-induced lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, 462–469. [Google Scholar] [CrossRef]
- Zhu, X.; Zuo, L. Characterization of oxygen radical formation mechanism at early cardiac ischemia. Cell Death Dis. 2013, 4, e787. [Google Scholar] [CrossRef]
- Zhou, T.; Chuang, C.-C.; Zuo, L. Molecular characterization of reactive oxygen species in myocardial ischemia-reperfusion injury. Biomed. Res. Int. 2015, 2015, 864946. [Google Scholar] [CrossRef]
- Terada, L.S.; Piermattei, D.; Shibao, G.N.; McManaman, J.L.; Wright, R.M. Hypoxia regulates xanthine dehydrogenase activity at pre- and posttranslational levels. Arch. Biochem. Biophys. 1997, 348, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef]
- de Perrot, M.; Sekine, Y.; Fischer, S.; Waddell, T.K.; McRae, K.; Liu, M.; Wigle, D.A.; Keshavjee, S. Interleukin-8 release during early reperfusion predicts graft function in human lung transplantation. Am. J. Respir. Crit. Care Med. 2002, 165, 211–215. [Google Scholar] [CrossRef]
- Smail, H.; Baste, J.M.; Gay, A.; Begueret, H.; Noël, R.; Morin, J.P.; Litzler, P.Y. Role of inflammatory cells and adenosine in lung ischemia reoxygenation injury using a model of lung donation after cardiac death. Exp. Lung Res. 2016, 42, 131–141. [Google Scholar] [CrossRef]
- Zhao, Q.; Wu, J.; Hua, Q.; Lin, Z.; Ye, L.; Zhang, W.; Wu, G.; Du, J.; Xia, J.; Chu, M.; et al. Resolvin D1 mitigates energy metabolism disorder after ischemia-reperfusion of the rat lung. J. Transl. Med. 2016, 14, 81. [Google Scholar] [CrossRef]
- Ng, C.S.; Wan, S.; Yim, A.P. Pulmonary ischaemia-reperfusion injury: Role of apoptosis. Eur. Respir. J. 2005, 25, 356–363. [Google Scholar] [CrossRef]
- Almeida, F.M.; Oliveira-Junior, M.C.; Souza, R.A.; Petroni, R.C.; Soto, S.F.; Soriano, F.G.; Carvalho, P.T.; Albertini, R.; Damaceno-Rodrigues, N.R.; Lopes, F.D.; et al. Creatine supplementation attenuates pulmonary and systemic effects of lung ischemia and reperfusion injury. J. Heart Lung Transplant. 2016, 35, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Zanotti, G.; Casiraghi, M.; Abano, J.B.; Tatreau, J.R.; Sevala, M.; Berlin, H.; Smyth, S.; Funkhouser, W.K.; Burridge, K.; Randell, S.H.; et al. Novel critical role of Toll-like receptor 4 in lung ischemia-reperfusion injury and edema. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L52–L63. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Y.; Nuñez, G. Sterile inflammation: Sensing and reacting to damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef]
- Cantu, E.; Lederer, D.J.; Meyer, K.; Milewski, K.; Suzuki, Y.; Shah, R.J.; Diamond, J.M.; Meyer, N.J.; Tobias, J.W.; Baldwin, D.A.; et al. Gene set enrichment analysis identifies key innate immune pathways in primary graft dysfunction after lung transplantation. Am. J. Transplant. 2013, 13, 1898–1904. [Google Scholar] [CrossRef]
- Diamond, J.M.; Wigfield, C.H. Role of innate immunity in primary graft dysfunction after lung transplantation. Curr. Opin. Organ. Transplant. 2013, 18, 518–523. [Google Scholar] [CrossRef]
- Kreisel, D.; Goldstein, D.R. Innate immunity and organ transplantation: Focus on lung transplantation. Transpl. Int. 2013, 26, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Phelan, P.; Merry, H.E.; Hwang, B.; Mulligan, M.S. Differential toll-like receptor activation in lung ischemia reperfusion injury. J. Thorac. Cardiovasc. Surg. 2015, 149, 1653–1661. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ochando, J.; Ordikhani, F.; Boros, P.; Jordan, S. The innate immune response to allotransplants: Mechanisms and therapeutic potentials. Cell. Mol. Immunol. 2019, 16, 350–356. [Google Scholar] [CrossRef]
- Laubach, V.E.; Sharma, A.K. Mechanisms of lung ischemia-reperfusion injury. Curr. Opin. Organ. Transplant. 2016, 21, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.K.; Fernandez, L.G.; Awad, A.S.; Kron, I.L.; Laubach, V.E. Proinflammatory response of alveolar epithelial cells is enhanced by alveolar macrophage-produced TNF-alpha during pulmonary ischemia-reperfusion injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L105–L113. [Google Scholar] [CrossRef]
- Shaw, J.O. Leukocytes in chemotactic-fragment-induced lung inflammation. Vascular emigration and alveolar surface migration. Am. J. Pathol. 1980, 101, 283–302. [Google Scholar] [PubMed]
- Fiser, S.M.; Tribble, C.G.; Long, S.M.; Kaza, A.K.; Kern, J.A.; Jones, D.R.; Robbins, M.K.; Kron, I.L. Ischemia-reperfusion injury after lung transplantation increases risk of late bronchiolitis obliterans syndrome. Ann. Thorac. Surg. 2002, 73, 1041–1048. [Google Scholar] [CrossRef]
- Sharma, A.K.; LaPar, D.J.; Zhao, Y.; Li, L.; Lau, C.L.; Kron, I.L.; Iwakura, Y.; Okusa, M.D.; Laubach, V.E. Natural killer T cell-derived IL-17 mediates lung ischemia-reperfusion injury. Am. J. Respir. Crit. Care Med. 2011, 183, 1539–1549. [Google Scholar] [CrossRef]
- Fiser, S.M.; Tribble, C.G.; Long, S.M.; Kaza, A.K.; Cope, J.T.; Laubach, V.E.; Kern, J.A.; Kron, I.L. Lung transplant reperfusion injury involves pulmonary macrophages and circulating leukocytes in a biphasic response. J. Thorac. Cardiovasc. Surg. 2001, 121, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Welbourn, C.R.; Goldman, G.; Paterson, I.S.; Valeri, C.R.; Shepro, D.; Hechtman, H.B. Pathophysiology of ischaemia reperfusion injury: Central role of the neutrophil. Br. J. Surg. 1991, 78, 651–655. [Google Scholar] [CrossRef]
- Zhao, M.; Fernandez, L.G.; Doctor, A.; Sharma, A.K.; Zarbock, A.; Tribble, C.G.; Kron, I.L.; Laubach, V.E. Alveolar macrophage activation is a key initiation signal for acute lung ischemia-reperfusion injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L1018–L1026. [Google Scholar] [CrossRef]
- Capucetti, A.; Albano, F.; Bonecchi, R. Multiple roles for chemokines in neutrophil biology. Front. Immunol. 2020, 11, 1259. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Collard, C.D. Vascular ischaemia and reperfusion injury. Br. Med. Bull. 2004, 70, 71–86. [Google Scholar] [CrossRef] [PubMed]
- Mortaz, E.; Alipoor, S.D.; Adcock, I.M.; Mumby, S.; Koenderman, L. Update on neutrophil function in severe inflammation. Front. Immunol. 2018, 9, 2171. [Google Scholar] [CrossRef] [PubMed]
- Short, J.D.; Downs, K.; Tavakoli, S.; Asmis, R. Protein thiol redox signaling in monocytes and macrophages. Antioxid. Redox Signal. 2016, 25, 816–835. [Google Scholar] [CrossRef]
- Iadecola, C.; Anrather, J. The immunology of stroke: From mechanisms to translation. Nat. Med. 2011, 17, 796–808. [Google Scholar] [CrossRef] [PubMed]
- Elvington, A.; Atkinson, C.; Kulik, L.; Zhu, H.; Yu, J.; Kindy, M.S.; Holers, V.M.; Tomlinson, S. Pathogenic natural antibodies propagate cerebral injury following ischemic stroke in mice. J. Immunol. 2012, 188, 1460–1468. [Google Scholar] [CrossRef]
- Kulik, L.; Fleming, S.D.; Moratz, C.; Reuter, J.W.; Novikov, A.; Chen, K.; Andrews, K.A.; Markaryan, A.; Quigg, R.J.; Silverman, G.J.; et al. Pathogenic natural antibodies recognizing annexin IV are required to develop intestinal ischemia-reperfusion injury. J. Immunol. 2009, 182, 5363–5373. [Google Scholar] [CrossRef]
- Diepenhorst, G.M.; van Gulik, T.M.; Hack, C.E. Complement- mediated ischemia-reperfusion injury: Lessons learned from animal and clinical studies. Ann. Surg. 2009, 249, 889–899. [Google Scholar] [CrossRef]
- Fisher, A.B.; Dodia, C. Lung as a model for evaluation of critical intracellular PO2 and PCO. Am. J. Physiol. 1981, 241, E47–E50. [Google Scholar] [CrossRef]
- Fukuse, T.; Hirata, T.; Nakamura, T.; Kawashima, M.; Hitomi, S.; Wada, H. Influence of deflated and anaerobic conditions during cold storage on rat lungs. Am. J. Respir. Crit. Care Med. 1999, 160, 621–627. [Google Scholar] [CrossRef]
- Yang, Q.; He, G.W.; Underwood, M.J.; Yu, C.M. Cellular and molecular mechanisms of endothelial ischemia/reperfusion injury: Perspectives and implications for postischemic myocardial protection. Am. J. Transl. Res. 2016, 8, 765–777. [Google Scholar] [PubMed]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Cell biology of ischemia/reperfusion injury. Int. Rev. Cell Mol. Biol. 2012, 298, 229–317. [Google Scholar]
- Sasaki, S.; Yasuda, K.; Mccully, J.D.; Lo Cicero, J., 3rd. Calcium channel blocker enhances lung preservation. J. Heart Lung Transplant. 1999, 18, 127–132. [Google Scholar] [CrossRef]
- Gautam, N.; Olofsson, A.M.; Herwald, H.; Iversen, L.F.; Lundgren-Akerlund, E.; Hedqvist, P.; Arfors, K.E.; Flodgaard, H.; Lindbom, L. Heparin-binding protein (HBP/CAP37): A missing link in neutrophil-evoked alteration of vascular permeability. Nat. Med. 2001, 7, 1123–1127. [Google Scholar] [CrossRef]
- Javadov, S.; Hunter, J.C.; Barreto-Torres, G.; Parodi-Rullan, R. Targeting the mitochondrial permeability transition: Cardiac ischemia-reperfusion versus carcinogenesis. Cell. Physiol. Biochem. 2011, 27, 179–190. [Google Scholar] [CrossRef]
- Haihua, C.; Wei, W.; Kun, H.; Yuanli, L.; Fei, L. Cobra venom factor-induced complement depletion protects against lung ischemia reperfusion injury through alleviating blood-air barrier damage. Sci. Rep. 2018, 8, 10346. [Google Scholar] [CrossRef] [PubMed]
- Ali, H.A.; Pavlisko, E.N.; Snyder, L.D.; Frank, M.; Palmer, S.M. Complement system in lung transplantation. Clin. Transplant. 2018, 32, e13208. [Google Scholar] [CrossRef]
- Kulkarni, H.S.; Liszewski, M.K.; Brody, S.L.; Atkinson, J.P. The complement system in the airway epithelium: An overlooked host defense mechanism and therapeutic target? J. Allergy Clin. Immunol. 2018, 141, 1582–1586.e1. [Google Scholar] [CrossRef]
- Bosmann, M.; Ward, P.A. Role of C3, C5 and anaphylatoxin receptors in acute lung injury and in sepsis. Adv. Exp. Med. Biol. 2012, 946, 147–159. [Google Scholar]
- Kulkarni, H.S.; Ramphal, K.; Ma, L.; Brown, M.; Oyster, M.; Speckhart, K.N.; Takahashi, T.; Byers, D.E.; Porteous, M.K.; Kalman, L.; et al. Local complement activation is associated with primary graft dysfunction after lung transplantation. JCI Insight 2020, 5, 138358. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.T.; Gozal, E.; Roberts, A.M. Platelet-mediated vascular dysfunction during acute lung injury. Arch. Physiol. Biochem. 2012, 118, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Schofield, Z.V.; Woodruff, T.M.; Halai, R.; Wu, M.C.; Cooper, M.A. Neutrophils—A key component of ischemia-reperfusion injury. Shock 2013, 40, 463–470. [Google Scholar] [CrossRef]
- Sato, Y.; Hogg, J.C.; English, D.; van Eeden, S.F. Endothelin-1 changes polymorphonuclear leukocytes’ deformability and CD11b expression and promotes their retention in the lung. Am. J. Respir. Cell Mol. Biol. 2000, 23, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.F. Pulmonary edema: Pathophysiology and diagnosis. Int. J. Tuberc. Lung Dis. 2011, 15, 155–160. [Google Scholar]
- Ganter, C.C.; Jakob, S.M.; Takala, J. Pulmonary capillary pressure. A review. Minerva Anestesiol. 2006, 72, 21–36. [Google Scholar]
- Lockinger, A.; Schutte, H.; Walmrath, D.; Seeger, W.; Grimminger, F. Protection against gas exchange abnormalities by pre-aerosolized PGE1, iloprost and nitroprus- side in lung ischemia-reperfusion. Transplantation 2001, 71, 185–193. [Google Scholar] [CrossRef]
- Ware, L.B.; Golden, J.A.; Finkbeiner, W.E.; Matthay, M.A. Alveolar epithelial fluid transport capacity in reperfusion lung injury after lung transplantation. Am. J. Respir. Crit. Care Med. 1999, 159, 980–988. [Google Scholar] [CrossRef]
- Nathan, S.D. The future of lung transplantation. Chest 2015, 147, 309–316. [Google Scholar] [CrossRef]
- Cantu, E.; Shah, R.J.; Lin, W.; Daye, Z.J.; Diamond, J.M.; Suzuki, Y.; Ellis, J.H.; Borders, C.F.; Andah, G.A.; Beduhn, B.; et al. Oxidant stress regulatory genetic variation in recipients and donors contributes to risk of primary graft dysfunction after lung transplantation. J. Thorac. Cardiovasc. Surg. 2015, 149, 596–602. [Google Scholar] [CrossRef]
- Cantu, E.; Suzuki, Y.; Diamond, J.M.; Ellis, J.; Tiwari, J.; Beduhn, B.; Nellen, J.R.; Shah, R.; Meyer, N.J.; Lederer, D.J.; et al. Protein quantitative trait loci analysis identifies genetic variation in the innate immune regulator TOLLIP in post-lung transplant primary graft dysfunction risk. Am. J. Transplant. 2016, 16, 833–840. [Google Scholar] [CrossRef]
- Chen, F.; Date, H. Update on ischemia-reperfusion injury in lung transplantation. Curr. Opin. Organ Transplant. 2015, 20, 515–520. [Google Scholar] [CrossRef]
- Meyers, B.F.; Lynch, J.; Trulock, E.P.; Guthrie, T.J.; Cooper, J.D.; Patterson, G.A. Lung transplantation: A decade of experience. Ann. Surg. 1999, 230, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Paradis, I. Bronchiolitis obliterans: Pathogenesis, prevention, and management. Am. J. Med. Sci. 1998, 315, 161–178. [Google Scholar]
- Heng, D.; Sharples, L.D.; McNeil, K.; Stewart, S.; Wreghitt, T.; Wallwork, J. Bronchiolitis obliterans syndrome: Incidence, natural history, prognosis, and risk factors. J. Heart Lung Transplant. 1998, 17, 1255–1263. [Google Scholar]
- Bando, K.; Paradis, I.L.; Similo, S.; Konishi, H.; Komatsu, K.; Zullo, T.G.; Yousem, S.A.; Close, J.M.; Zeevi, A.; Duquesnoy, R.J.; et al. Obliterative bronchiolitis after lung and heart–lung transplantation. An analysis of risk factors and management. J. Thorac. Cardiovasc. Surg. 1995, 110, 4–14. [Google Scholar] [CrossRef]
- Reichenspurner, H.; Girgis, R.E.; Robbins, R.C.; Yun, K.L.; Nitschke, M.; Berry, G.J.; Morris, R.E.; Theodore, J.; Reitz, B.A. Stanford experience with obliterative bronchiolitis after lung and heart–lung transplantation. Ann. Thorac. Surg. 1996, 62, 1467–1473. [Google Scholar] [CrossRef]
- Yousem, S.A.; Burke, C.M.; Billingham, M.E. Pathologic pulmonary alterations in long-term human heart–lung transplantation. Hum. Pathol. 1985, 16, 911–923. [Google Scholar] [CrossRef]
- Cooper, J.D.; Billingham, M.; Egan, T.; Hertz, M.I.; Higenbottam, T.; Lynch, J.; Mauer, J.; Paradis, I.; Patterson, G.A.; Smith, C.; et al. A working formulation for the standardization of nomenclature and for clinical staging of chronic dysfunction in lung allografts. International Society for Heart and Lung Transplantation. J. Heart Lung Transplant. 1993, 12, 713–716. [Google Scholar]
- Dhillon, G.S.; Zamora, M.R.; Roos, J.E.; Sheahan, D.; Sista, R.R.; Van der Starre, P.; Weill, D.; Nicolls, M.R. Lung transplant airway hypoxia. A diathesis to fibrosis? Am. J. Respir. Crit. Care Med. 2010, 182, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Ross, D.J.; Jordan, S.C.; Nathan, S.D.; Kass, R.M.; Koerner, S.K. Delayed development of obliterative bronchiolitis syndrome with OKT3 after unilateral lung transplantation. A plea for multicenter immunosuppressive trials. Chest 1996, 109, 870–873. [Google Scholar] [CrossRef][Green Version]
- Snell, G.I.; Esmore, D.S.; Williams, T.J. Cytolytic therapy for the bronchiolitis obliterans syndrome complicating lung transplantation. Chest 1996, 109, 874–878. [Google Scholar] [CrossRef]
- Jirsch, D.W.; Fisk, R.L.; Couves, C.M. Ex vivo evaluation of stored lungs. Ann. Thorac. Surg. 1970, 10, 163–168. [Google Scholar] [CrossRef]
- Cypel, M.; Yeung, J.C.; Hirayama, S.; Rubacha, M.; Fischer, S.; Anraku, M.; Sato, M.; Harwood, S.; Pierre, A.; Waddell, T.K.; et al. Technique for prolonged normothermic ex vivo lung perfusion. J. Heart Lung Transplant. 2008, 27, 1319–1325. [Google Scholar] [CrossRef] [PubMed]
- Steen, S.; Liao, Q.; Wierup, P.N.; Bolys, R.; Pierre, L.; Sjoberg, T. Transplantation of lungs from non-heart-beating donors after functional assessment ex vivo. Ann. Thorac. Surg. 2003, 76, 244–252. [Google Scholar] [CrossRef]
- Yeung, J.C.; Cypel, M.; Machuca, T.N.; Koike, T.; Cook, D.J.; Bonato, R.; Chen, M.; Sato, M.; Waddell, T.K.; Liu, M.; et al. Physiologic assessment of the ex vivo donor lung for transplantation. J. Heart Lung Transplant. 2012, 31, 1120–1126. [Google Scholar] [CrossRef]
- Cypel, M.; Keshavjee, S. The clinical potential of ex vivo lung perfusion. Expert Rev. Respir. Med. 2012, 6, 27–35. [Google Scholar] [CrossRef]
- Cypel, M.; Yeung, J.C.; Liu, M.; Anraku, M.; Chen, F.; Karolak, W.; Sato, M.; Laratta, J.; Azad, S.; Madonik, M.; et al. Normothermic ex vivo lung perfusion in clinical lung transplantation. N. Engl. J. Med. 2011, 364, 1431–1440. [Google Scholar] [CrossRef]
- Steen, S.; Ingemansson, R.; Eriksson, L.; Pierre, L.; Algotsson, L.; Wierup, P.; Liao, Q.; Eyjolfsson, A.; Gustafsson, R.; Sjöberg, T. First human transplantation of a nonacceptable donor lung after reconditioning ex vivo. Ann. Thorac. Surg. 2007, 83, 2191–2194. [Google Scholar] [CrossRef]
- Ingemansson, R.; Eyjolfsson, A.; Mared, L.; Pierre, L.; Algotsson, L.; Ekmehag, B.; Gustafsson, R.; Johnsson, P.; Koul, B.; Lindstedt, S.; et al. Clinical transplantation of initially rejected donor lungs after reconditioning ex vivo. Ann. Thorac. Surg. 2009, 87, 255–260. [Google Scholar] [CrossRef]
- Aboelnazar, N.S.; Himmat, S.; Hatami, S.; White, C.W.; Burhani, M.S.; Dromparis, P.; Matsumura, N.; Tian, G.; Dyck, J.R.B.; Mengel, M.; et al. Negative pressure ventilation decreases inflammation and lung edema during normothermic ex-vivo lung perfusion. J. Heart Lung Transplant. 2018, 37, 520–530. [Google Scholar] [CrossRef]
- Becker, S.; Steinmeyer, J.; Avsar, M.; Höffler, K.; Salman, J.; Haverich, A.; Warnecke, G.; Ochs, M.; Schnapper, A. Evaluating acellular versus cellular perfusate composition during prolonged ex vivo lung perfusion after initial cold ischaemia for 24 hours. Transpl. Int. 2015, 29, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Roman, M.; Gjorgjimajkoska, O.; Neil, D.; Nair, S.; Colah, S.; Parmar, J.; Tsui, S. Comparison between cellular and acellular perfusates for ex vivo lung perfusion in a porcine model. J. Heart Lung Transplant. 2015, 34, 978–987. [Google Scholar] [CrossRef]
- Van Raemdonck, D.; Neyrinck, A.; Cypel, M.; Keshavjee, S. Ex-vivo lung perfusion. Transpl. Int. 2014, 28, 643–656. [Google Scholar] [CrossRef] [PubMed]
- Warnecke, G.; Moradiellos, J.; Tudorache, I.; Kühn, C.; Avsar, M.; Wiegmann, B.; Sommer, W.; Ius, F.; Kunze, C.; Gottlieb, J.; et al. Normothermic perfusion of donor lungs for preservation and assessment with the Organ Care System Lung before bilateral transplantation: A pilot study of 12 patients. Lancet 2012, 380, 1851–1858. [Google Scholar] [CrossRef]
- Buchko, M.T.; Himmat, S.; Aboelnazar, N.S.; Stewart, C.J.; Hatami, S.; Dromparis, P.; Adam, B.; Freed, D.H.; Nagendran, J. A low-cost perfusate alternative for ex vivo lung perfusion. Transplant. Proc. 2020, 52, 2941–2946. [Google Scholar] [CrossRef] [PubMed]
- Cypel, M.; Yeung, J.C.; Donahoe, L.; Chen, M.; Zamel, R.; Hoetzenecker, K.; Yasufuku, K.; de Perrot, M.; Perre, A.F.; Waddell, T.K.; et al. Normothermic ex vivo lung perfusion: Does the indication impact organ utilization and patient out- comes after transplantation? J. Thorac. Cardiovasc. Surg. 2019, S0022-5223, 31732–31735. [Google Scholar] [CrossRef]
- Stone, J.P.; Critchley, W.R.; Major, T.; Rajan, G.; Risnes, I.; Scott, H.; Liao, Q.; Wohlfart, B.; Sjöberg, T.; Yonan, N.; et al. Altered immunogenicity of donor lungs via removal of passenger leukocytes using ex vivo lung perfusion. Am. J. Transplant. 2016, 16, 33–43. [Google Scholar] [CrossRef]
- Deffebach, M.E. Lung mechanical effects on the bronchial circulation. Eur. Respir. J. Suppl. 1990, 12, 586–590. [Google Scholar]
- Cudkowicz, L.; Armstrong, J.B. Observations on the normal anatomy of the bronchial arteries. Thorax 1951, 6, 343–358. [Google Scholar] [CrossRef] [PubMed]
- Fritts, H.W.J.; Harris, P.; Chidsey, C.A., III; Clauss, R.H.; Cournand, A. Estimation of flow through bronchial-pulmonary vascular anastomoses with use of T-1824 dye. Circulation 1961, 23, 390–398. [Google Scholar] [CrossRef]
- McCullagh, A.; Rosenthal, M.; Wanner, A.; Hurtado, A.; Padley, S.; Bush, A. The bronchial circulation—Worth a closer look: A review of the relationship between the bronchial vasculature and airway inflammation. Pediatr. Pulmonol. 2010, 45, 1–13. [Google Scholar] [CrossRef]
- Inci, I.; Weder, W. Airway complications after lung transplantation can be avoided without bronchial artery revascularization. Curr. Opin. Organ Transplant. 2010, 15, 578–581. [Google Scholar] [CrossRef]
- Nørgaard, M.A.; Olsen, P.S.; Svendsen, U.G.; Pettersson, G. Revascularization of the bronchial arteries in lung transplantation: An overview. Ann. Thorac. Surg. 1996, 62, 1215–1221. [Google Scholar] [CrossRef]
- Takao, M.; Katayama, Y.; Onoda, K.; Tanabe, H.; Hiraiwa, T.; Mizutani, T.; Yada, I.; Namikawa, S.; Yuasa, H.; Kusagawa, M. Significance of bronchial mucosal blood flow for the monitoring of acute rejection in lung transplantation. J. Heart Lung Transplant. 1991, 10, 956–967. [Google Scholar]
- Fisher, A.; Andreasson, A.; Chrysos, A.; Lally, J.; Mamasoula, C.; Exley, C.; Wilkinson, J.; Qian, J.; Watson, G.; Lewington, O.; et al. An observational study of donor ex vivo lung perfusion in UK lung transplantation: DEVELOP-UK. Health Technol. Assess. 2016, 20, 1–276. [Google Scholar] [CrossRef]
- Andreasson, A.S.; Karamanou, D.M.; Gillespie, C.S.; Özalp, F.; Butt, T.; Hill, P.; Jiwa, K.; Walden, H.R.; Green, N.J.; Borthwick, L.A.; et al. Profiling inflammation and tissue injury markers in perfusate and bronchoalveolar lavage fluid during human ex vivo lung perfusion. Eur. J. Cardiothorac. Surg. 2017, 51, 577–586. [Google Scholar] [PubMed]
- Ricard, J.D.; Dreyfuss, D.; Saumon, G. Ventilator-induced lung injury. Eur. Respir. J. 2003, 42, 2s–9s. [Google Scholar] [CrossRef] [PubMed]
- Buchko, M.T.; Boroumand, N.; Cheng, J.C.; Hirji, A.; Halloran, K.; Freed, D.H.; Nagendran, J. Clinical transplantation using negative pressure ventilation ex situ lung perfusion with extended criteria donor lungs. Nat. Commun. 2020, 11, 5765. [Google Scholar] [CrossRef] [PubMed]
- Machuca, T.N.; Cypel, M.; Bonato, R.; Yeung, J.C.; Chun, Y.M.; Juvet, S.; Guan, Z.; Hwang, D.M.; Chen, M.; Saito, T.; et al. Safety and efficacy of ex vivo donor lung adenoviral IL-10 gene therapy in a large animal lung transplant survival model. Hum. Gene Ther. 2017, 28, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Cypel, M.; Liu, M.; Rubacha, M.; Yeung, J.C.; Hirayama, S.; Anraku, M.; Sato, M.; Medin, J.; Davidson, B.L.; de Perrot, M.; et al. Functional repair of human donor lungs by IL-10 gene therapy. Sci. Transl. Med. 2009, 1, 4ra9. [Google Scholar] [CrossRef]
- Yeung, J.C.; Wagnetz, D.; Cypel, M.; Rubacha, M.; Koike, T.; Chun, Y.M.; Hu, J.; Waddell, T.K.; Hwang, D.M.; Liu, M.; et al. Ex vivo adenoviral vector gene delivery results in decreased vector-associated inflammation pre- and post-lung transplantation in the pig. Mol. Ther. 2012, 20, 1204–1211. [Google Scholar] [CrossRef] [PubMed]
- Stone, M.L.; Zhao, Y.; Smith, J.R.; Weiss, M.L.; Kron, I.L.; Laubach, V.E.; Sharma, A.K. Mesenchymal stromal cell-derived extracellular vesicles attenuate lung ischemia-reperfusion injury and enhance reconditioning of donor lungs after circulatory death. Respir. Res. 2017, 18, 212. [Google Scholar] [CrossRef] [PubMed]
- Stone, M.L.; Sharma, A.K.; Mas, V.R.; Gehrau, R.C.; Mulloy, D.P.; Zhao, Y.; Lau, C.L.; Kron, I.L.; Huerter, M.E.; Laubach, V.E. Ex vivo perfusion with adeno- sine A2A receptor agonist enhances rehabilitation of murine donor lungs after circulatory death. Transplantation 2015, 99, 2494–2503. [Google Scholar] [CrossRef]
- Wang, X.; Parapanov, R.; Francioli, C.; Perentes, J.Y.; Letovanec, I.; Gonzalez, M.; Kern, C.; Ris, H.B.; Piquilloud, L.; Marcucci, C.; et al. Experimental ex vivo lung perfusion with sevoflurane: Effects on damaged donor lung grafts. Interact. Cardiovasc. Thorac. Surg. 2018, 26, 977–984. [Google Scholar] [CrossRef]
- Kondo, T.; Chen, F.; Ohsumi, A.; Hijiya, K.; Motoyama, H.; Sowa, T.; Ohata, K.; Takahashi, M.; Yamada, T.; Sato, M.; et al. β2-Adrenoreceptor agonist inhalation during ex vivo lung perfusion attenuates lung injury. Ann. Thorac. Surg. 2015, 100, 480–486. [Google Scholar] [CrossRef]
- Hijiya, K.; Chen-Yoshikawa, T.F.; Kondo, T.; Motoyama, H.; Ohsumi, A.; Nakajima, D.; Sakamoto, J.; Ohata, K.; Takahashi, M.; Tanaka, S.; et al. Bronchodilator inhalation during ex vivo lung perfusion improves posttransplant graft function after warm ischemia. Ann. Thorac. Surg. 2017, 103, 447–453. [Google Scholar] [CrossRef]
- Hsin, M.; Au, T. Ex vivo lung perfusion: A potential platform for molecular diagnosis and ex vivo organ repair. J. Thorac. Dis. 2018, 10, S1871–S1883. [Google Scholar] [CrossRef]
- Possoz, J.; Neyrinck, A.; Van Raemdonck, D. Ex vivo lung perfusion prior to transplantation: An overview of current clinical practice worldwide. J. Thorac. Dis. 2019, 11, 1635–1650. [Google Scholar] [CrossRef]
- Tane, S.; Noda, K.; Shigemura, N. Ex vivo lung perfusion: A key tool for translational science in the lungs. Chest 2017, 151, 1220–1228. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Grade | P/F Ratio | Chest X-ray |
|---|---|---|
| 0 | >300 | Normal |
| 1 | >300 | Diffuse Allograft Infiltrates |
| 2 | 200–300 | Diffuse Allograft Infiltrates |
| 3 | <200 | Diffuse Allograft Infiltrates |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Forgie, K.A.; Fialka, N.; Freed, D.H.; Nagendran, J. Lung Transplantation, Pulmonary Endothelial Inflammation, and Ex-Situ Lung Perfusion: A Review. Cells 2021, 10, 1417. https://doi.org/10.3390/cells10061417
Forgie KA, Fialka N, Freed DH, Nagendran J. Lung Transplantation, Pulmonary Endothelial Inflammation, and Ex-Situ Lung Perfusion: A Review. Cells. 2021; 10(6):1417. https://doi.org/10.3390/cells10061417
Chicago/Turabian StyleForgie, Keir A., Nicholas Fialka, Darren H. Freed, and Jayan Nagendran. 2021. "Lung Transplantation, Pulmonary Endothelial Inflammation, and Ex-Situ Lung Perfusion: A Review" Cells 10, no. 6: 1417. https://doi.org/10.3390/cells10061417
APA StyleForgie, K. A., Fialka, N., Freed, D. H., & Nagendran, J. (2021). Lung Transplantation, Pulmonary Endothelial Inflammation, and Ex-Situ Lung Perfusion: A Review. Cells, 10(6), 1417. https://doi.org/10.3390/cells10061417