
Toll-Like Receptor Signaling in the Establishment and Function of the Immune System

{kind=link}
Abstract
1. Introduction
2. Overview of TLR Signaling
3. TLR Signaling in Immune System Development and Maintenance
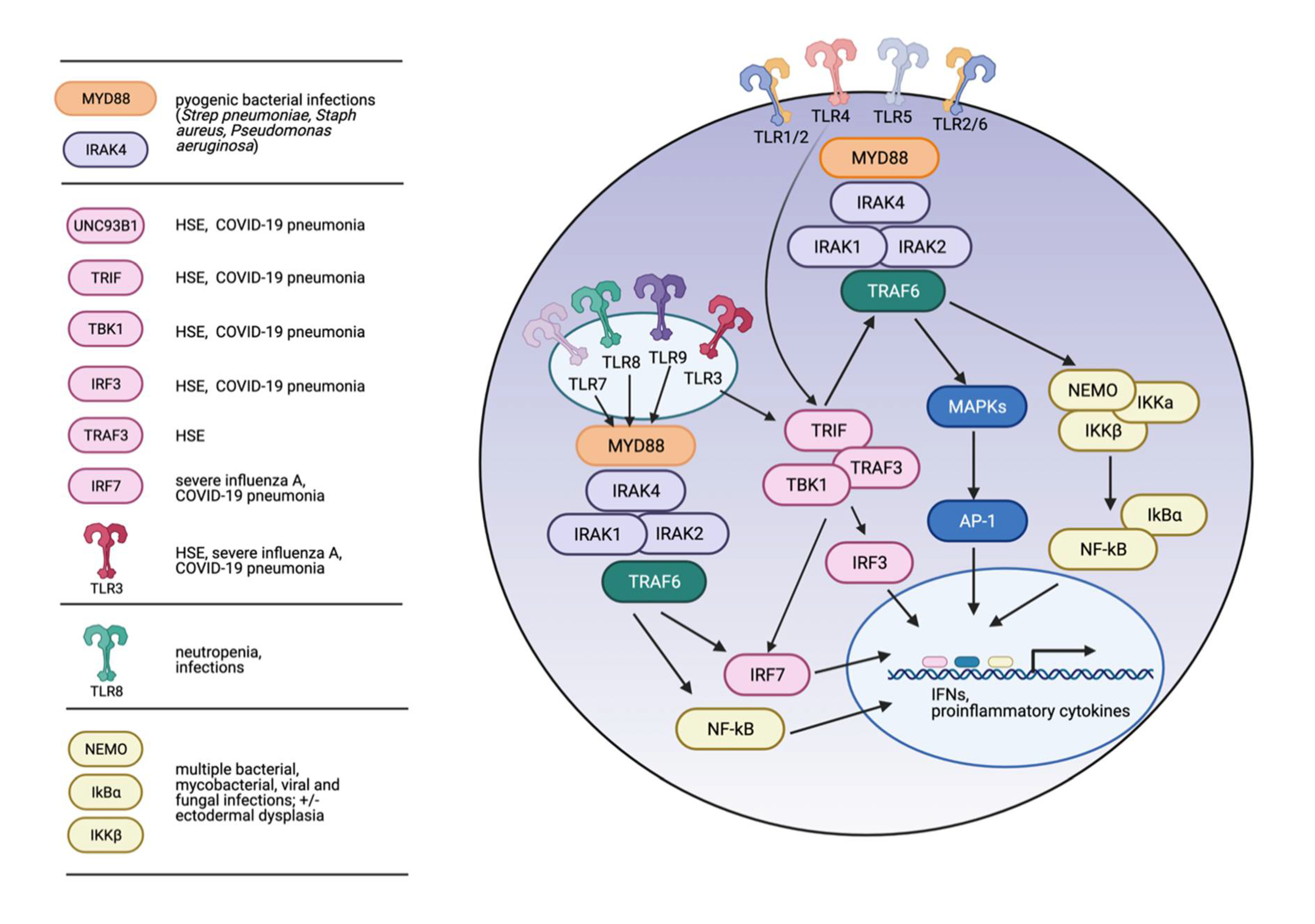
4. Inborn Errors of Immunity (IEI) Associated with Inherited Defects in TLR Signaling
5. MyD88 and IRAK-4 Deficiency and IEI
6. TLR3 Pathway Variants and IEI
7. TLR3 Pathway Variants and Herpes Simplex Encephalitis
8. TLR3 Pathway Variants and Influenza
9. TLR3 Pathway Variants and COVID-19
10. TLR8 Gain-of-Function Variants and IEI
11. IKK Complex and NFKBIA (IKBA) Variants and IEI
12. TLR Pathway Polymorphisms and Disease
13. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gay, N.J.; Symmons, M.F.; Gangloff, M.; Bryant, C.E. Assembly and localization of Toll-like receptor signalling complexes. Nat. Rev. Immunol. 2014, 14, 546–558. [Google Scholar] [CrossRef]
- O’Neill, L.A.J.; Golenbock, D.T.; Bowie, A.G. The history of Toll-like receptors—Redefining innate immunity. Nat. Rev. Immunol. 2013, 13, 453–460. [Google Scholar] [CrossRef]
- Rock, F.L.; Hardiman, G.; Timans, J.C.; Kastelein, R.A.; Bazan, J.F. A family of human receptors structurally related to Drosophila Toll. Proc. Natl. Acad. Sci. USA 1998, 95, 588–593. [Google Scholar] [CrossRef]
- Chaudhary, P.M.; Ferguson, C.; Nguyen, V.; Nguyen, O.; Massa, H.F.; Eby, M.; Jasmin, A.; Trask, B.J.; Hood, L.; Nelson, P.S. Cloning and characterization of two Toll/Interleukin-1 receptor-like genes TIL3 and TIL4: Evidence for a multi-gene receptor family in humans. Blood 1998, 91, 4020–4027. [Google Scholar] [CrossRef]
- Chuang, T.H.; Ulevitch, R.J. Cloning and characterization of a sub-family of human toll-like receptors: HTLR7, hTLR8 and hTLR9. Eur. Cytokine Netw. 2000, 11, 372–378. [Google Scholar] [PubMed]
- Du, X.; Poltorak, A.; Wei, Y.; Beutler, B. Three novel mammalian toll-like receptors: Gene structure, expression, and evolution. Eur. Cytokine Netw. 2000, 11, 362–371. [Google Scholar]
- Takeuchi, O.; Kawai, T.; Sanjo, H.; Copeland, N.; Gilbert, D.; Jenkins, N.; Takeda, K.; Akira, S. TLR6: A novel member of an expanding Toll-like receptor family. Gene 1999, 231, 59–65. [Google Scholar] [CrossRef]
- Chuang, T.-H.; Ulevitch, R.J. Identification of hTLR10: A novel human Toll-like receptor preferentially expressed in immune cells. Biochim. Biophys. Acta 2001, 1518, 157–161. [Google Scholar] [CrossRef]
- Nascimento, L.E.O.; Emassari, P.; Wetzler, L.M. The Role of TLR2 in Infection and Immunity. Front. Immunol. 2012, 3. [Google Scholar] [CrossRef]
- Takeuchi, O.; Sato, S.; Horiuchi, T.; Hoshino, K.; Takeda, K.; Dong, Z.; Modlin, R.L.; Akira, S. Cutting Edge: Role of Toll-Like Receptor 1 in Mediating Immune Response to Microbial Lipoproteins. J. Immunol. 2002, 169, 10–14. [Google Scholar] [CrossRef]
- Takeuchi, O.; Kawai, T.; Mühlradt, P.F.; Morr, M.; Radolf, J.D.; Zychlinsky, A.; Takeda, K.; Akira, S. Discrimination of bacterial lipoproteins by Toll-like receptor 6. Int. Immunol. 2001, 13, 933–940. [Google Scholar] [CrossRef] [PubMed]
- Piccinini, A.M.; Midwood, K.S. DAMPening inflammation by modulating TLR signalling. Mediat. Inflamm. 2010, 2010. [Google Scholar] [CrossRef]
- McClure, R.; Massari, P. TLR-Dependent Human Mucosal Epithelial Cell Responses to Microbial Pathogens. Front. Immunol. 2014, 5, 386. [Google Scholar] [CrossRef]
- Takizawa, H.; Boettcher, S.; Manz, M.G. Demand-adapted regulation of early hematopoiesis in infection and inflammation. Blood 2012, 119, 2991–3002. [Google Scholar] [CrossRef]
- Zarember, K.A.; Godowski, P.J. Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J. Immunol. 2002, 168, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Fitzner, N.; Clauberg, S.; Essmann, F.; Liebmann, J.; Kolb-Bachofen, V. Human Skin Endothelial Cells Can Express All 10 TLR Genes and Respond to Respective Ligands. Clin. Vaccine Immunol. 2007, 15, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Crack, P.J.; Bray, P.J. Toll-like receptors in the brain and their potential roles in neuropathology. Immunol. Cell Biol. 2007, 85, 476–480. [Google Scholar] [CrossRef]
- Schmid, M.A.; Takizawa, H.; Baumjohann, D.R.; Saito, Y.; Manz, M.G. Bone marrow dendritic cell progenitors sense pathogens via Toll-like receptors and subsequently migrate to inflamed lymph nodes. Blood 2011, 118, 4829–4840. [Google Scholar] [CrossRef] [PubMed]
- Welner, R.S.; Pelayo, R.; Nagai, Y.; Garrett, K.P.; Wuest, T.R.; Carr, D.J.; Borghesi, L.A.; Farrar, M.A.; Kincade, P.W. Lymphoid precursors are directed to produce dendritic cells as a result of TLR9 ligation during herpes infection. Blood 2008, 112, 3753–3761. [Google Scholar] [CrossRef] [PubMed]
- Nagai, Y.; Garrett, K.P.; Ohta, S.; Bahrun, U.; Kouro, T.; Akira, S.; Takatsu, K.; Kincade, P.W. Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity 2006, 24, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Kawai, T. Toll-Like Receptor Signaling Pathways. Front. Immunol. 2014, 5. [Google Scholar] [CrossRef]
- Medzhitov, R.; Preston-Hurlburt, P.; Kopp, E.; Stadlen, A.; Chen, C.; Ghosh, S.; A Janeway, C. MyD88 Is an Adaptor Protein in the hToll/IL-1 Receptor Family Signaling Pathways. Mol. Cell 1998, 2, 253–258. [Google Scholar] [CrossRef]
- Burns, K.; Janssens, S.; Brissoni, B.; Olivos, N.; Beyaert, R.; Tschopp, J. Inhibition of Interleukin 1 Receptor/Toll-like Receptor Signaling through the Alternatively Spliced, Short Form of MyD88 Is Due to Its Failure to Recruit IRAK-4. J. Exp. Med. 2003, 197, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.C.; Lo, Y.C.; Wu, H. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature 2010, 465, 885–890. [Google Scholar] [CrossRef]
- Motshwene, P.G.; Moncrieffe, M.C.; Grossmann, J.G.; Kao, C.; Ayaluru, M.; Sandercock, A.M.; Robinson, C.V.; Latz, E.; Gay, N.J. An Oligomeric Signaling Platform Formed by the Toll-like Receptor Signal Transducers MyD88 and IRAK-4. J. Biol. Chem. 2009, 284, 25404–25411. [Google Scholar] [CrossRef]
- Landstrom, M. The TAK1-TRAF6 signalling pathway. Int. J. Biochem. Cell Biol. 2010, 42, 585–589. [Google Scholar] [CrossRef]
- Oshiumi, H.; Matsumoto, M.; Funami, K.; Akazawa, T.; Seya, T. TICAM-1, an adaptor molecule that participates in Toll-like receptor 3-mediated interferon-beta induction. Nat. Immunol. 2003, 4, 161–167. [Google Scholar] [CrossRef]
- Hoebe, K.; Du, X.; Georgel, P.; Janssen, E.; Tabeta, K.; Kim, S.O.; Goode, J.; Lin, P.; Mann, N.; Mudd, S.; et al. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nat. Cell Biol. 2003, 424, 743–748. [Google Scholar] [CrossRef]
- Maniatis, T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 2003, 4, 491–496. [Google Scholar]
- Sato, S.; Sugiyama, M.; Yamamoto, M.; Watanabe, Y.; Kawai, T.; Takeda, K.; Akira, S. Toll/IL-1 receptor domain-containing adaptor inducing IFN-beta (TRIF) associates with TNF receptor-associated factor 6 and TANK-binding kinase 1, and activates two distinct transcription factors, NF-κ B and IFN-regulatory factor-3, in the Toll-like receptor signaling. J. Immunol. 2003, 171, 4304–4310. [Google Scholar] [PubMed]
- He, Q.; Zhang, C.; Wang, L.; Zhang, P.; Ma, N.; Lv, J.; Liu, F. Inflammatory signaling regulates hematopoietic stem and progenitor cell emergence in vertebrates. Blood 2015, 125, 1098–1106. [Google Scholar] [CrossRef] [PubMed]
- Espín-Palazón, R.; Stachura, D.L.; Campbell, C.A.; García-Moreno, D.; Del Cid, N.; Kim, A.D.; Candel, S.; Meseguer, J.; Mulero, V.; Traver, D. Proinflammatory Signaling Regulates Hematopoietic Stem Cell Emergence. Cell 2014, 159, 1070–1085. [Google Scholar] [CrossRef]
- Megías, J.; Yáñez, A.; Moriano, S.; O’Connor, J.-E.; Gozalbo, D.; Gil, M.-L. Direct Toll-Like Receptor-Mediated Stimulation of Hematopoietic Stem and Progenitor Cells Occurs In Vivo and Promotes Differentiation Toward Macrophages. Stem Cells 2012, 30, 1486–1495. [Google Scholar] [CrossRef]
- Esplin, B.L.; Shimazu, T.; Welner, R.S.; Garrett, K.P.; Nie, L.; Zhang, Q.; Humphrey, M.B.; Yang, Q.; Borghesi, L.A.; Kincade, P.W. Chronic Exposure to a TLR Ligand Injures Hematopoietic Stem Cells. J. Immunol. 2011, 186, 5367–5375. [Google Scholar] [CrossRef] [PubMed]
- Herman, A.C.; Monlish, D.A.; Romine, M.P.; Bhatt, S.T.; Zippel, S.; Schuettpelz, L.G. Systemic TLR2 agonist exposure regulates hematopoietic stem cells via cell-autonomous and cell-non-autonomous mechanisms. Blood Cancer J. 2016, 6, e437. [Google Scholar] [CrossRef]
- Zhang, H.; Rodriguez, S.; Wang, L.; Wang, S.; Serezani, C.H.; Kapur, R.; Cardoso, A.A.; Carlesso, N. Sepsis Induces Hematopoietic Stem Cell Exhaustion and Myelosuppression through Distinct Contributions of TRIF and MYD88. Stem Cell Rep. 2016, 6, 940–956. [Google Scholar] [CrossRef]
- Takizawa, H.; Fritsch, K.; Kovtonyuk, L.V.; Saito, Y.; Yakkala, C.; Jacobs, K.; Ahuja, A.K.; Lopes, M.; Hausmann, A.; Hardt, W.D.; et al. Pathogen-Induced TLR4-TRIF Innate Immune Signaling in Hematopoietic Stem Cells Promotes Proliferation but Reduces Competitive Fitness. Cell Stem Cell 2017, 21, 225–240.e225. [Google Scholar] [CrossRef] [PubMed]
- Thoma-Uszynski, S.; Stenger, S.; Takeuchi, O.; Ochoa, M.T.; Engele, M.; Sieling, P.A.; Barnes, P.F.; Röllinghoff, M.; Bölcskei, P.L.; Wagner, M.; et al. Induction of Direct Antimicrobial Activity Through Mammalian Toll-Like Receptors. Science 2001, 291, 1544–1547. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Takeda, K.; Kaisho, T. Toll-like receptors: Critical proteins linking innate and acquired immunity. Nat. Immunol. 2001, 2, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Marshak-Rothstein, A. Toll-like receptors in systemic autoimmune disease. Nat. Rev. Immunol. 2006, 6, 823–835. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Xiong, Y.; Li, Q.; Yang, H. Inhibition of Toll-Like Receptor Signaling as a Promising Therapy for Inflammatory Diseases: A Journey from Molecular to Nano Therapeutics. Front. Physiol. 2017, 8, 508. [Google Scholar] [CrossRef] [PubMed]
- Ospelt, C.; Gay, S. TLRs and chronic inflammation. Int. J. Biochem. Cell Biol. 2010, 42, 495–505. [Google Scholar] [CrossRef]
- Rakoff-Nahoum, S.; Medzhitov, R. Toll-like receptors and cancer. Nat. Rev. Cancer 2008, 9, 57–63. [Google Scholar] [CrossRef]
- Mokhtari, Y.; Pourbagheri-Sigaroodi, A.; Zafari, P.; Bagheri, N.; Ghaffari, S.H.; Bashash, D. Toll-like receptors (TLRs): An old family of immune receptors with a new face in cancer pathogenesis. J. Cell. Mol. Med. 2021, 25, 639–651. [Google Scholar] [CrossRef] [PubMed]
- Arend, W.P.; Palmer, G.; Gabay, C. IL-1, IL-18, and IL-33 families of cytokines. Immunol. Rev. 2008, 223, 20–38. [Google Scholar] [CrossRef]
- Dinarello, C.A. Interleukin-1beta and the autoinflammatory diseases. N. Engl. J. Med. 2009, 360, 2467–2470. [Google Scholar] [CrossRef] [PubMed]
- Von Bernuth, H.; Picard, C.; Jin, Z.; Pankla, R.; Xiao, H.; Ku, C.L. Pyogenic bacterial infections in humans with MyD88 deficiency. Science 2008, 321. [Google Scholar] [CrossRef]
- Picard, C.; Puel, A.; Bonnet, M.; Ku, C.-L.; Bustamante, J.; Yang, K.; Soudais, C.; Boisson-Dupuis, S.; Feinberg, J.; Fieschi, C.; et al. Pyogenic Bacterial Infections in Humans with IRAK-4 Deficiency. Science 2003, 299, 2076–2079. [Google Scholar] [CrossRef] [PubMed]
- Picard, C.; von Bernuth, H.; Ghandil, P.; Chrabieh, M.; Levy, O.; Arkwright, P.D.; McDonald, D.; Geha, R.S.; Takada, H.; Krause, J.C.; et al. Clinical Features and Outcome of Patients With IRAK-4 and MyD88 Deficiency. Medicine 2010, 89, 403–425. [Google Scholar] [CrossRef] [PubMed]
- Ku, C.-L.; Von Bernuth, H.; Picard, C.; Zhang, S.-Y.; Chang, H.-H.; Yang, K.; Chrabieh, M.; Issekutz, A.C.; Cunningham, C.K.; Gallin, J.; et al. Selective predisposition to bacterial infections in IRAK-4–deficient children: IRAK-4–dependent TLRs are otherwise redundant in protective immunity. J. Exp. Med. 2007, 204, 2407–2422. [Google Scholar] [CrossRef]
- von Bernuth, H.; Picard, C.; Puel, A.; Casanova, J.L. Experimental and natural infections in MyD88- and IRAK-4-deficient mice and humans. Eur. J. Immunol. 2012, 42, 3126–3135. [Google Scholar] [CrossRef] [PubMed]
- Mestas, J.; Hughes, C.C.W. Of mice and not men: Differences between mouse and human immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef] [PubMed]
- Farina, C.; Krumbholz, M.; Giese, T.; Hartmann, G.; Aloisi, F.; Meinl, E. Preferential expression and function of Toll-like receptor 3 in human astrocytes. J. Neuroimmunol. 2005, 159, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Town, T.; Jeng, D.; Alexopoulou, L.; Tan, J.; Flavell, R.A. Microglia Recognize Double-Stranded RNA via TLR3. J. Immunol. 2006, 176, 3804–3812. [Google Scholar] [CrossRef] [PubMed]
- Muzio, M.; Bosisio, D.; Polentarutti, N.; D’Amico, G.; Stoppacciaro, A.; Mancinelli, R.; Veer, C.V.; Penton-Rol, G.; Ruco, L.P.; Allavena, P.; et al. Differential Expression and Regulation of Toll-Like Receptors (TLR) in Human Leukocytes: Selective Expression of TLR3 in Dendritic Cells. J. Immunol. 2000, 164, 5998–6004. [Google Scholar] [CrossRef]
- Visintin, A.; Mazzoni, A.; Spitzer, J.H.; Wyllie, D.H.; Dower, S.K.; Segal, D.M. Regulation of Toll-Like Receptors in Human Monocytes and Dendritic Cells. J. Immunol. 2001, 166, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, N.; Ho, S.; Antonenko, S.; Malefyt, R.D.W.; Kastelein, R.A.; Bazan, F.; Liu, Y.-J. Subsets of Human Dendritic Cell Precursors Express Different Toll-like Receptors and Respond to Different Microbial Antigens. J. Exp. Med. 2001, 194, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Rothenfusser, S.; Britsch, S.; Krug, A.; Jahrsdörfer, B.; Giese, T.; Endres, S.; Hartmann, G. Quantitative Expression of Toll-Like Receptor 1–10 mRNA in Cellular Subsets of Human Peripheral Blood Mononuclear Cells and Sensitivity to CpG Oligodeoxynucleotides. J. Immunol. 2002, 168, 4531–4537. [Google Scholar] [CrossRef]
- Matsumoto, M.; Funami, K.; Tanabe, M.; Oshiumi, H.; Shingai, M.; Seto, Y.; Yamamoto, A.; Seya, T. Subcellular localization of Toll-like receptor 3 in human dendritic cells. J. Immunol. 2003, 171, 3154–3162. [Google Scholar] [CrossRef]
- Jongbloed, S.L.; Kassianos, A.J.; McDonald, K.J.; Clark, G.J.; Ju, X.; Angel, C.E.; Chen, C.-J.J.; Dunbar, P.R.; Wadley, R.B.; Jeet, V.; et al. Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J. Exp. Med. 2010, 207, 1247–1260. [Google Scholar] [CrossRef]
- Poulin, L.F.; Salio, M.; Griessinger, E.; Anjos-Afonso, F.; Craciun, L.; Chen, J.-L.; Keller, A.M.; Joffre, O.; Zelenay, S.; Nye, E.; et al. Characterization of human DNGR-1+ BDCA3+ leukocytes as putative equivalents of mouse CD8α+ dendritic cells. J. Exp. Med. 2010, 207, 1261–1271. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Seya, T. TLR3: Interferon induction by double-stranded RNA including poly(I:C). Adv. Drug Deliv. Rev. 2008, 60, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-Y.; Jouanguy, E.; Ugolini, S.; Smahi, A.; Elain, G.; Romero, P.; Segal, D.; Sancho-Shimizu, V.; Lorenzo, L.; Puel, A.; et al. TLR3 Deficiency in Patients with Herpes Simplex Encephalitis. Science 2007, 317, 1522–1527. [Google Scholar] [CrossRef]
- Guo, Y.; Audry, M.; Ciancanelli, M.; Alsina, L.; Azevedo, J.; Herman, M.; Anguiano, E.; Sancho-Shimizu, V.; Lorenzo, L.; Pauwels, E.; et al. Herpes simplex virus encephalitis in a patient with complete TLR3 deficiency: TLR3 is otherwise redundant in protective immunity. J. Exp. Med. 2011, 208, 2083–2098. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.K.; Seppänen, M.; Hautala, T.; Ciancanelli, M.J.; Itan, Y.; Lafaille, F.G.; Dell, W.; Lorenzo, L.; Byun, M.; Pauwels, E.; et al. TLR3 deficiency in herpes simplex encephalitis: High allelic heterogeneity and recurrence risk. Neurology 2014, 83, 1888–1897. [Google Scholar] [CrossRef]
- Mork, N.J.; Kofodolsen, E.; Sorensen, K.; Bach, E.; Orntoft, T.F.; Ostergaard, L.; Paludan, S.R.; Christiansen, M.H.; Mogensen, T.H. Mutations in the TLR3 signaling pathway and beyond in adult patients with herpes simplex encephalitis. Genes Immun. 2015, 16, 552–566. [Google Scholar] [CrossRef] [PubMed]
- Sironi, M.; Peri, A.M.; Cagliani, R.; Forni, D.; Riva, S.; Biasin, M.; Clerici, M.; Gori, A. TLR3 Mutations in Adult Patients with Herpes Simplex Virus and Varicella-Zoster Virus Encephalitis. J. Infect. Dis. 2017, 215, 1430–1434. [Google Scholar] [CrossRef]
- Casrouge, A.; Zhang, S.-Y.; Eidenschenk, C.; Jouanguy, E.; Puel, A.; Yang, K.; Alcais, A.; Picard, C.; Mahfoufi, N.; Nicolas, N.; et al. Herpes Simplex Virus Encephalitis in Human UNC-93B Deficiency. Science 2006, 314, 308–312. [Google Scholar] [CrossRef]
- Sancho-Shimizu, V.; De Diego, R.P.; Lorenzo, L.; Halwani, R.; Alangari, A.; Israelsson, E.; Fabrega, S.; Cardon, A.; Maluenda, J.; Tatematsu, M.; et al. Herpes simplex encephalitis in children with autosomal recessive and dominant TRIF deficiency. J. Clin. Investig. 2011, 121, 4889–4902. [Google Scholar] [CrossRef]
- de Diego, R.P.; Sancho-Shimizu, V.; Lorenzo, L.; Puel, A.; Plancoulaine, S.; Picard, C.; Herman, M.; Cardon, A.; Durandy, A.; Bustamante, J.; et al. Human TRAF3 adaptor molecule deficiency leads to impaired Toll-like receptor 3 response and susceptibility to herpes simplex encephalitis. Immunity 2010, 33, 400–411. [Google Scholar] [CrossRef]
- Herman, M.; Ciancanelli, M.; Ou, Y.-H.; Lorenzo, L.; Klaudel-Dreszler, M.; Pauwels, E.; Sancho-Shimizu, V.; De Diego, R.P.; Abhyankar, A.; Israelsson, E.; et al. Heterozygous TBK1 mutations impair TLR3 immunity and underlie herpes simplex encephalitis of childhood. J. Exp. Med. 2012, 209, 1567–1582. [Google Scholar] [CrossRef]
- Andersen, L.L.; Mørk, N.; Reinert, L.S.; Kofod-Olsen, E.; Narita, R.; Jørgensen, S.E.; Skipper, K.; Höning, K.; Gad, H.H.; Østergaard, L.; et al. Functional IRF3 deficiency in a patient with herpes simplex encephalitis. J. Exp. Med. 2015, 212, 1371–1379. [Google Scholar] [CrossRef]
- Bsibsi, M.; Persoon-Deen, C.; Verwer, R.W.; Meeuwsen, S.; Ravid, R.; Van Noort, J.M. Toll-like receptor 3 on adult human astrocytes triggers production of neuroprotective mediators. Glia 2006, 53, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Bsibsi, M.; Ravid, R.; Gveric, D.; Van Noort, J.M. Broad Expression of Toll-Like Receptors in the Human Central Nervous System. J. Neuropathol. Exp. Neurol. 2002, 61, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Peltier, D.C.; Simms, A.; Farmer, J.R.; Miller, D.J. Human Neuronal Cells Possess Functional Cytoplasmic and TLR-Mediated Innate Immune Pathways Influenced by Phosphatidylinositol-3 Kinase Signaling. J. Immunol. 2010, 184, 7010–7021. [Google Scholar] [CrossRef] [PubMed]
- Gazquez, I.; Jover-Saenz, A.; Puig, T.; De Vera, C.V.; Rubio, M. Familial herpes encephalitis. Lancet 1996, 347, 910. [Google Scholar] [CrossRef]
- Jackson, A.; Melanson, M.; Mb, J.P.R. Familial herpes simplex encephalitis. Ann. Neurol. 2002, 51, 406–407. [Google Scholar] [CrossRef]
- Koskiniemi, M.; Saarinen, A.; Klapper, P.; Sarna, S.; Cleator, G.; Vaheri, A. Familial herpes encephalitis. Lancet 1995, 346, 1553. [Google Scholar] [CrossRef]
- Lerner, A.M.; Levine, D.P.; Reyes, M.P. Two cases of herpes simplex virus encephalitis in the same family. N. Engl. J. Med. 1983, 308, 1481. [Google Scholar] [PubMed]
- Abel, L.; Plancoulaine, S.; Jouanguy, E.; Zhang, S.-Y.; Mahfoufi, N.; Nicolas, N.; Sancho-Shimizu, V.; Alcaïs, A.; Guo, Y.; Cardon, A.; et al. Age-Dependent Mendelian Predisposition to Herpes Simplex Virus Type 1 Encephalitis in Childhood. J. Pediatr. 2010, 157, 623–629. [Google Scholar] [CrossRef]
- Brinkmann, M.M.; Spooner, E.; Hoebe, K.; Beutler, B.; Ploegh, H.L.; Kim, Y.M. The interaction between the ER membrane protein UNC93B and TLR3, 7, and 9 is crucial for TLR signaling. J. Cell Biol. 2007, 177, 265–275. [Google Scholar] [CrossRef]
- Tabeta, K.; Hoebe, K.; Janssen, E.M.; Du, X.; Georgel, P.; Crozat, K.; Mudd, S.; Mann, N.; Sovath, S.; Goode, J.R.; et al. The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nat. Immunol. 2006, 7, 156–164. [Google Scholar] [CrossRef]
- Bishop, G.A.; Xie, P. Multiple roles of TRAF3 signaling in lymphocyte function. Immunol. Res. 2007, 39, 22–32. [Google Scholar] [CrossRef]
- He, J.Q.; Oganesyan, G.; Saha, S.K.; Zarnegar, B.; Cheng, G. TRAF3 and its biological function. Adv. Exp. Med. Biol. 2007, 597, 48–59. [Google Scholar]
- Annunziata, C.M.; Davis, R.E.; Demchenko, Y.; Bellamy, W.; Gabrea, A.; Zhan, F.; Lenz, G.; Hanamura, I.; Wright, G.; Xiao, W.; et al. Frequent engagement of the classical and alternative NF-κB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell 2007, 12, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Keats, J.J.; Fonseca, R.; Chesi, M.; Schop, R.; Baker, A.; Chng, W.J.; Van Wier, S.; Tiedemann, R.; Shi, X.-C.; Sebag, M.; et al. Promiscuous mutations activate the noncanonical NF-κB pathway in multiple myeloma. Cancer Cell 2007, 12, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q. Human genetics of life-threatening influenza pneumonitis. Qual. Life Res. 2020, 139, 941–948. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.K.; Huang, S.X.; Chen, J.; Kerner, G.; Gilliaux, O.; Bastard, P.; Dobbs, K.; Hernandez, N.; Goudin, N.; Hasek, M.L.; et al. Severe influenza pneumonitis in children with inherited TLR3 deficiency. J. Exp. Med. 2019, 216, 2038–2056. [Google Scholar] [CrossRef]
- Ciancanelli, M.J.; Huang, S.X.L.; Luthra, P.; Garner, H.; Itan, Y.; Volpi, S.; Lafaille, F.G.; Trouillet, C.; Schmolke, M.; Albrecht, R.; et al. Life-threatening influenza and impaired interferon amplification in human IRF7 deficiency. Science 2015, 348, 448–453. [Google Scholar] [CrossRef]
- Hernandez, N.; Melki, I.; Jing, H.; Habib, T.; Huang, S.S.; Danielson, J.; Kula, T.; Drutman, S.; Belkaya, S.; Rattina, V.; et al. Life-threatening influenza pneumonitis in a child with inherited IRF9 deficiency. J. Exp. Med. 2018, 215, 2567–2585. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Botos, I.; Wang, Y.; Leonard, J.; Shiloach, J.; Segal, D.M.; Davies, D.R. Structural Basis of Toll-Like Receptor 3 Signaling with Double-Stranded RNA. Science 2008, 320, 379–381. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, L.; Davies, D.R.; Segal, D.M. Dimerization of Toll-like Receptor 3 (TLR3) Is Required for Ligand Binding. J. Biol. Chem. 2010, 285, 36836–36841. [Google Scholar] [CrossRef]
- Zhang, Q.; Bastard, P.; Liu, Z.; Le Pen, J.; Moncada-Velez, M.; Chen, J.; Ogishi, M.; Sabli, I.K.D.; Hodeib, S.; Korol, C.; et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 2020, 370, 4570. [Google Scholar] [CrossRef]
- Levy, R.; Bastard, P.; Lanternier, F.; Lecuit, M.; Zhang, S.Y.; Casanova, J.L. IFN-α2a Therapy in Two Patients with Inborn Errors of TLR3 and IRF3 Infected with SARS-CoV-2. J. Clin. Immunol. 2021, 41, 26–27. [Google Scholar] [CrossRef] [PubMed]
- de Marcken, M.; Dhaliwal, K.; Danielsen, A.C.; Gautron, A.S.; Dominguez-Villar, M. TLR7 and TLR8 activate distinct pathways in monocytes during RNA virus infection. Sci. Signal. 2019, 12, 605. [Google Scholar] [CrossRef]
- Melchjorsen, J.; Jensen, S.B.; Malmgaard, L.; Rasmussen, S.B.; Weber, F.; Bowie, A.G.; Matikainen, S.; Paludan, S.R. Activation of Innate Defense against a Paramyxovirus Is Mediated by RIG-I and TLR7 and TLR8 in a Cell-Type-Specific Manner. J. Virol. 2005, 79, 12944–12951. [Google Scholar] [CrossRef] [PubMed]
- Triantafilou, K.; Orthopoulos, G.; Vakakis, E.; Ahmed, M.A.E.; Golenbock, D.T.; Lepper, P.M.; Triantafilou, M. Human cardiac inflammatory responses triggered by Coxsackie B viruses are mainly Toll-like receptor (TLR) 8-dependent. Cell. Microbiol. 2005, 7, 1117–1126. [Google Scholar] [CrossRef]
- Davila, S.; Hibberd, M.L.; Dass, R.H.; Wong, H.E.E.; Sahiratmadja, E.; Bonnard, C.; Alisjahbana, B.; Szeszko, J.S.; Balabanova, Y.; Drobniewski, F.; et al. Genetic Association and Expression Studies Indicate a Role of Toll-Like Receptor 8 in Pulmonary Tuberculosis. PLoS Genet. 2008, 4, e1000218. [Google Scholar] [CrossRef]
- Gantier, M.P.; Irving, A.T.; Kaparakis-Liaskos, M.; Xu, D.; Evans, V.A.; Cameron, P.U.; Bourne, J.A.; Ferrero, R.; John, M.; Behlke, M.A.; et al. Genetic modulation of TLR8 response following bacterial phagocytosis. Hum. Mutat. 2010, 31, 1069–1079. [Google Scholar] [CrossRef]
- Patinote, C.; Karroum, N.B.; Moarbess, G.; Cirnat, N.; Kassab, I.; Bonnet, P.-A.; Deleuze-Masquéfa, C. Agonist and antagonist ligands of toll-like receptors 7 and 8: Ingenious tools for therapeutic purposes. Eur. J. Med. Chem. 2020, 193, 112238. [Google Scholar] [CrossRef]
- Itoh, H.; Tatematsu, M.; Watanabe, A.; Iwano, K.; Funami, K.; Seya, T.; Matsumoto, M. UNC93B1 Physically Associates with Human TLR8 and Regulates TLR8-Mediated Signaling. PLoS ONE 2011, 6, e28500. [Google Scholar] [CrossRef]
- Tanji, H.; Ohto, U.; Shibata, T.; Taoka, M.; Yamauchi, Y.; Isobe, T.; Miyake, K.; Shimizu, T. Toll-like receptor 8 senses degradation products of single-stranded RNA. Nat. Struct. Mol. Biol. 2015, 22, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xu, C.; Hsu, L.C.; Luo, Y.; Xiang, R.; Chuang, T.H. A five-amino-acid motif in the undefined region of the TLR8 ectodomain is required for species-specific ligand recognition. Mol. Immunol. 2010, 47, 1083–1090. [Google Scholar] [CrossRef] [PubMed]
- Guiducci, C.; Gong, M.; Cepika, A.-M.; Xu, Z.; Tripodo, C.; Bennett, L.; Crain, C.; Quartier, P.; Cush, J.J.; Pascual, V.; et al. RNA recognition by human TLR8 can lead to autoimmune inflammation. J. Exp. Med. 2013, 210, 2903–2919. [Google Scholar] [CrossRef] [PubMed]
- Aluri, J.; Bach, A.; Kaviany, S.; Paracatu, L.C.; Kitcharoensakkul, M.; Walkiewicz, M.A.; Putnam, C.D.; Shinawi, M.; Saucier, N.; Rizzi, E.M.; et al. Immunodeficiency and bone marrow failure with mosaic and germline TLR8 gain of function. Blood 2021, 137, 2450–2462. [Google Scholar] [CrossRef]
- Zonana, J.; Elder, M.E.; Schneider, L.C.; Orlow, S.J.; Moss, C.; Golabi, M.; Shapira, S.K.; Farndon, P.A.; Wara, D.W.; Emmal, S.A.; et al. A Novel X-Linked Disorder of Immune Deficiency and Hypohidrotic Ectodermal Dysplasia Is Allelic to Incontinentia Pigmenti and Due to Mutations in IKK-gamma (NEMO). Am. J. Hum. Genet. 2000, 67, 1555–1562. [Google Scholar] [CrossRef]
- Döffinger, R.; Smahi, A.; Bessia, C.; Geissmann, F.; Feinberg, J.; Durandy, A.; Bodemer, C.; Kenwrick, C.; Dupuis-Girod, S.; Blanche, S.; et al. X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-κB signaling. Nat. Genet. 2001, 27, 277–285. [Google Scholar] [CrossRef]
- Picard, C.; Casanova, J.-L.; Puel, A. Infectious Diseases in Patients with IRAK-4, MyD88, NEMO, or I B Deficiency. Clin. Microbiol. Rev. 2011, 24, 490–497. [Google Scholar] [CrossRef]
- Hanson, E.P.; Monaco-Shawver, L.; Solt, L.A.; Madge, L.A.; Banerjee, P.P.; May, M.J.; Orange, J.S. Hypomorphic nuclear factor-κB essential modulator mutation database and reconstitution system identifies phenotypic and immunologic diversity. J. Allergy Clin. Immunol. 2008, 122, 1169–1177.e1116. [Google Scholar] [CrossRef]
- Courtois, G.; Smahi, A.; Reichenbach, J.; Döffinger, R.; Cancrini, C.; Bonnet, M.; Puel, A.; Chable-Bessia, C.; Yamaoka, S.; Feinberg, J.; et al. A hypermorphic IκBα mutation is associated with autosomal dominant anhidrotic ectodermal dysplasia and T cell immunodeficiency. J. Clin. Investig. 2003, 112, 1108–1115. [Google Scholar] [CrossRef]
- Boisson, B.; Puel, A.; Picard, C.; Casanova, J.L. Human IκBα Gain of Function: A Severe and Syndromic Immunodeficiency. J. Clin. Immunol. 2017, 37, 397–412. [Google Scholar] [CrossRef] [PubMed]
- Janssen, R.; Van Wengen, A.; Hoeve, M.A.; Ten Dam, M.; Van Der Burg, M.; Van Dongen, J.; Van de Vosse, E.; Van Tol, M.; Bredius, R.; Ottenhoff, T.H.; et al. The same IκBα mutation in two related individuals leads to completely different clinical syndromes. J. Exp. Med. 2004, 200, 559–568. [Google Scholar] [CrossRef] [PubMed]
- McDonald, D.R.; Mooster, J.L.; Reddy, M.; Bawle, E.; Secord, E.; Geha, R.S. Heterozygous N-terminal deletion of IκBα results in functional nuclear factor κB haploinsufficiency, ectodermal dysplasia, and immune deficiency. J. Allergy Clin. Immunol. 2007, 120, 900–907. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Granados, E.; Keenan, J.E.; Kinney, M.C.; Leo, H.; Jain, N.; Ma, C.A.; Quinones, R.; Gelfand, E.W.; Jain, A. A novel mutation in NFKBIA/IKBA results in a degradation-resistant N-truncated protein and is associated with ectodermal dysplasia with immunodeficiency. Hum. Mutat. 2008, 29, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, H.; Miyata, R.; Suzuki, T.; Nose, T.; Kubota, K.; Kato, Z.; Kaneko, H.; Kondo, N. A rapid screening method to detect autosomal-dominant ectodermal dysplasia with immune deficiency syndrome. J. Allergy Clin. Immunol. 2012, 129, 578–580. [Google Scholar] [CrossRef] [PubMed]
- Giancane, G.; Ferrari, S.; Carsetti, R.; Papoff, P.; Iacobini, M.; Duse, M. Anhidrotic ectodermal dysplasia: A new mutation. J. Allergy Clin. Immunol. 2013, 132, 1451–1453. [Google Scholar] [CrossRef]
- Schimke, L.F.; Rieber, N.; Rylaarsdam, S.; Marques, O.C.; Hubbard, N.; Puel, A.; Kallmann, L.; Sombke, S.A.; Notheis, G.; Schwarz, H.-P.; et al. A Novel Gain-of-Function IKBA Mutation Underlies Ectodermal Dysplasia with Immunodeficiency and Polyendocrinopathy. J. Clin. Immunol. 2013, 33, 1088–1099. [Google Scholar] [CrossRef]
- Yoshioka, T.; Nishikomori, R.; Hara, J.; Okada, K.; Hashii, Y.; Okafuji, I.; Nodomi, S.; Kawai, T.; Izawa, K.; Ohnishi, H.; et al. Autosomal Dominant Anhidrotic Ectodermal Dysplasia with Immunodeficiency Caused by a Novel NFKBIA Mutation, p.Ser36Tyr, Presents with Mild Ectodermal Dysplasia and Non-Infectious Systemic Inflammation. J. Clin. Immunol. 2013, 33, 1165–1174. [Google Scholar] [CrossRef]
- Lee, A.J.; Moncada-Vélez, M.; Picard, C.; Llanora, G.; Huang, C.H.; Abel, L. Severe Mycobacterial Diseases in a Patient with GOF IκBα Mutation without EDA. J. Clin. Immunol. 2016, 36, 12–15. [Google Scholar] [CrossRef]
- Staples, E.; Morillo-Gutierrez, B.; Davies, J.; Petersheim, D.; Massaad, M.; Slatter, M.; Dimou, D.; Doffinger, R.; Hackett, S.; Kumararatne, D.; et al. Disseminated Mycobacterium malmoense and Salmonella Infections Associated with a Novel Variant in NFKBIA. J. Clin. Immunol. 2017, 37, 415–418. [Google Scholar] [CrossRef]
- Petersheim, D.; Massaad, M.J.; Lee, S.; Scarselli, A.; Cancrini, C.; Moriya, K.; Sasahara, Y.; Lankester, A.C.; Dorsey, M.; Di Giovanni, D.; et al. Mechanisms of genotype-phenotype correlation in autosomal dominant anhidrotic ectodermal dysplasia with immune deficiency. J. Allergy Clin. Immunol. 2018, 141, 1060–1073.e3. [Google Scholar] [CrossRef]
- Pannicke, U.; Baumann, B.; Fuchs, S.; Henneke, P.; Rensing-Ehl, A.; Rizzi, M.; Janda, A.; Hese, K.; Schlesier, M.; Holzmann, K.; et al. Deficiency of Innate and Acquired Immunity Caused by an IKBKB Mutation. N. Engl. J. Med. 2013, 369, 2504–2514. [Google Scholar] [CrossRef]
- Cuvelier, G.D.; Rubin, T.S.; Junker, A.; Sinha, R.; Rosenberg, A.M.; Wall, D.A.; Schroeder, M.L. Clinical presentation, immunologic features, and hematopoietic stem cell transplant outcomes for IKBKB immune deficiency. Clin. Immunol. 2019, 205, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Medvedev, A.E. Toll-Like Receptor Polymorphisms, Inflammatory and Infectious Diseases, Allergies, and Cancer. J. Interf. Cytokine Res. 2013, 33, 467–484. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Wijmenga, C.; O’Neill, L.A.J. Genetic variation in Toll-like receptors and disease susceptibility. Nat. Immunol. 2012, 13, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Maglione, P.J.; Simchoni, N.; Cunningham-Rundles, C. Toll-like receptor signaling in primary immune deficiencies. Ann. N. Y. Acad. Sci. 2015, 1356, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Plantinga, T.; Johnson, M.D.; Scott, W.K.; Van De Vosse, E.; Edwards, D.R.V.; Smith, P.B.; Alexander, B.D.; Yang, J.C.; Kremer, D.; Laird, G.M.; et al. Toll-like Receptor 1 Polymorphisms Increase Susceptibility to Candidemia. J. Infect. Dis. 2012, 205, 934–943. [Google Scholar] [CrossRef]
- Bochud, P.-Y.; Chien, J.W.; Marr, K.A.; Leisenring, W.M.; Upton, A.; Janer, M.; Rodrigues, S.D.; Li, S.; Hansen, J.A.; Zhao, L.P.; et al. Toll-like Receptor 4 Polymorphisms and Aspergillosis in Stem-Cell Transplantation. N. Engl. J. Med. 2008, 359, 1766–1777. [Google Scholar] [CrossRef]
- Kesh, S.; Mensah, N.Y.; Peterlongo, P.; Jaffe, D.; Hsu, K.; Brink, M.; O’Reilly, R.; Pamer, E.; Satagopan, J.; Papanicolaou, G. TLR1 and TLR6 Polymorphisms Are Associated with Susceptibility to Invasive Aspergillosis after Allogeneic Stem Cell Transplantation. Ann. N. Y. Acad. Sci. 2005, 1062, 95–103. [Google Scholar] [CrossRef]
- Van der Graaf, C.A.; Netea, M.G.; Morré, S.A.; Den Heijer, M.; Verweij, P.E.; Van der Meer, J.W.; Kullberg, B.J. Toll-like receptor 4 Asp299Gly/Thr399Ile polymorphisms are a risk factor for Candida bloodstream infection. Eur. Cytokine Netw. 2006, 17, 29–34. [Google Scholar]
- Woehrle, T.; Du, W.; Goetz, A.; Hsu, H.-Y.; Joos, T.O.; Weiss, M.; Bauer, U.; Brueckner, U.B.; Schneider, E.M. Pathogen specific cytokine release reveals an effect of TLR2 Arg753Gln during Candida sepsis in humans. Cytokine 2008, 41, 322–329. [Google Scholar] [CrossRef]
- Johnson, C.M.; Lyle, E.A.; Omueti, K.O.; Stepensky, V.A.; Yegin, O.; Alpsoy, E.; Hamann, L.; Schumann, R.R.; Tapping, R.I. Cutting Edge: A Common Polymorphism Impairs Cell Surface Trafficking and Functional Responses of TLR1 but Protects against Leprosy. J. Immunol. 2007, 178, 7520–7524. [Google Scholar] [CrossRef] [PubMed]
- Sironi, M.; Biasin, M.; Cagliani, R.; Forni, D.; De Luca, M.; Saulle, I.; Caputo, S.L.; Mazzotta, F.; Macías, J.; Pineda, J.A.; et al. A Common Polymorphism in TLR3 Confers Natural Resistance to HIV-1 Infection. J. Immunol. 2011, 188, 818–823. [Google Scholar] [CrossRef] [PubMed]
- Hawn, T.R.; Verbon, A.; Lettinga, K.D.; Zhao, L.P.; Li, S.S.; Laws, R.J.; Skerrett, S.J.; Beutler, B.; Schroeder, L.; Nachman, A.; et al. A Common Dominant TLR5 Stop Codon Polymorphism Abolishes Flagellin Signaling and Is Associated with Susceptibility to Legionnaires’ Disease. J. Exp. Med. 2003, 198, 1563–1572. [Google Scholar] [CrossRef]
- Schott, E.; Witt, H.; Neumann, K.; Bergk, A.; Halangk, J.; Weich, V.; Muller, T.; Puhl, G.; Wiedenmann, B.; Berg, T. Association of TLR7 single nucleotide polymorphisms with chronic HCV-infection and response to interferon-a-based therapy. J. Viral Hepat. 2007, 15, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-H.; Eng, H.-L.; Lin, K.-H.; Chang, C.-H.; Hsieh, C.-A.; Lin, Y.-L.; Lin, T.-M. TLR7 and TLR8 Gene Variations and Susceptibility to Hepatitis C Virus Infection. PLoS ONE 2011, 6, e26235. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aluri, J.; Cooper, M.A.; Schuettpelz, L.G. Toll-Like Receptor Signaling in the Establishment and Function of the Immune System. Cells 2021, 10, 1374. https://doi.org/10.3390/cells10061374
Aluri J, Cooper MA, Schuettpelz LG. Toll-Like Receptor Signaling in the Establishment and Function of the Immune System. Cells. 2021; 10(6):1374. https://doi.org/10.3390/cells10061374
Chicago/Turabian StyleAluri, Jahnavi, Megan A. Cooper, and Laura G. Schuettpelz. 2021. "Toll-Like Receptor Signaling in the Establishment and Function of the Immune System" Cells 10, no. 6: 1374. https://doi.org/10.3390/cells10061374
APA StyleAluri, J., Cooper, M. A., & Schuettpelz, L. G. (2021). Toll-Like Receptor Signaling in the Establishment and Function of the Immune System. Cells, 10(6), 1374. https://doi.org/10.3390/cells10061374