1. Introduction
Glycosylation, the posttranslational modification of proteins and lipids with sugars, plays an important role in regulating protein–protein interactions, protein–ligand signaling, and cellular behaviors, particularly during development [
1,
2,
3]. The biological importance of glycosylation can be most appreciated in the number of developmental disorders that result from defects in genes encoding for glycosyltransferases or other proteins with integral roles in the various glycosylation pathways (e.g., nucleotide sugar transporters). Growing evidence has highlighted active roles that alterations in glycosylation in post-developmental disease states (e.g., cancer) can play in disease development and/or progression. In either context, interventions designed to modify or target defects or alterations in glycosylation in disease states have shown promise as therapeutic strategies.
Fucosylation, the posttranslational modification of proteins or lipids with the sugar L-fucose, has recently been shown to be altered during the progression of melanoma and pancreatic cancer [
4,
5,
6]. In melanoma, global fucosylation is altered during disease progression, suggesting that alterations in the fucosylation pathway play a role in disease progression. Indeed, we and others have reported that fucosylation can regulate melanoma motility, although the precise underlying molecular mechanisms remain unclear. Characterizing the substrates that are subject to fucosylation and understanding how fucosylation influences their function, downstream signaling, and ultimately cellular behavior, are expected to highlight how and in which context(s) fucosylation might be exploited as a viable therapeutic target or to identify the key proteins and cellular signaling pathways that respond to alterations in fucosylation and may themselves be targeted for therapeutic intervention.
Here, we characterized melanoma proteins that are recognized by UEAI lectin, a sugar-binding protein that recognizes fucose-containing glycans. We found that UEAI lectin recognizes almost exclusively intracellular proteins, an unexpected observation suggesting that fucosylation can occur on intracellular proteins independent of the canonical fucosylation pathway. We identified and validated ribosomal protein S3 (RPS3) as a fucosylated protein in human cancer cells as well as in normal mouse tissues. We determined that the fucosylated species is ribosome-independent, instead interacting with a group of cytosolic proteins that posttranscriptionally regulate RNAs. This report suggests the existence of an as yet uncharacterized fucosylation pathway that is altered in disease states and that plays a role in regulating intracellular protein function.
2. Materials and Methods
2.1. Cell Lines and Reagents
All cell lines used in this manuscript were validated for identity by the H. Lee Moffitt Cancer Center Molecular Genomics Core within 2 years of testing and were confirmed to be mycoplasma-free using the Plasmo Test kit (InvivoGen, San Diego, CA, USA). Cell lines were cultured in DMEM (VWR, Radnor, PA, USA) supplemented with heat inactivated fetal bovine serum 1:10 (Peak Serum, Denver, CO, USA). Antibodies: RPS3 (Bethyl, Montgomery, TX, USA), HNRNPU (ThermoFisher, Waltham, MA, USA), BCLAF1 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), EIF3K (Bethyl), FLAG (Sigma, St. Louis, MO, USA), β-actin (Sigma), β-tubulin (Developmental Studies Hybridoma Bank, DSHB, Iowa City, IA, USA), LaminA/C (DSHB). UEAI, AAL, UEAI-488, AAL-488, UEAI-biotin, AAL-biotin were purchased from Vector Laboratories (Burlingame, CA, USA). Secondary antibodies: anti-rabbit-HRP (Santa Cruz Biotechnology, Inc.), m-IgGκ-BP-HRP (Santa Cruz Biotechnology, Inc.), streptavidin-800 (Life Technologies, Carlsbad, CA, USA), IRDye 680RD anti-mouse (LI-COR, Lincoln, NE, USA).
2.2. Plasmids and Cloning
RPS3 coding sequence was cloned into pLENTI-C-MYC-DDK-IRES-puro plasmid (Origene, Rockville, MD, USA), transformed into Stbl3 bacterial cells, and selected on chloramphenicol plates (50 µg/mL) following standard protocols. Colonies were picked, grown, and miniprepped (Qiagen, Hilden, Germany) for sequence verification by Eton Bioscience. Lentiviral particles were produced in 293T cells with VSVG and Δ8.9 plasmids following standard protocols. Cells were transduced and selected on puromycin (1 µg/mL) for stable line expression. pcDNA3.1 MCS-BirA (R118G)-HA plasmid for RPS3 BioID was purchased from Addgene (Watertown, MA, USA) (#36047). MYC-DDK (mDDK; DDK is also known as FLAG) tagged RPS3 was cloned into pCDNA3.1 plasmid for mutagenesis. Mutagenesis was carried out using the Q5 Site-Directed Mutagenesis Kit following the manufacturer’s instructions (NEB, Ipswich, MA, USA).
Primers for constructing RPS3 Δ129-160:
5-GGAGACCCTGTTAACTAC-3
5-CTCCATGATGAACCGCAG-3
2.3. Western Blot
Western blotting was carried out following standard protocols. Briefly, cells were washed in cold PBS and harvested for lysate in IP Lysis Buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 2 mM EDTA, 1% Triton X-100) or RIPA Lysis Buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS) supplemented with protease and phosphatase inhibitor tablets (ThermoFisher, Waltham, MA, USA) where appropriate. For cell fractions, cells were harvested in 0.5% Triton X-100 PBS supplemented with protease and phosphatase inhibitor on ice for 10 min (m). Lysates were then spun down at 12,000× g for 5 m at 4 °C. Supernatant was removed (cytoplasmic fraction) and Triton-X insoluble material was then lysed/solubilized in RIPA buffer (nuclear fraction). Lysates were then subjected to sonication using a probe sonicator or DNA was sheared using a Dounce homogenizer. Mouse tissue was ground in RIPA Lysis Buffer using a mortar and pestle and further processed using a Dounce homogenizer. Lysates were then cleared of insoluble material by spinning at 12,000× g for 5 m at 4 °C. Cell fractionation was carried out by incubating cells on ice for 20 m in 0.5% Triton X-100 PBS supplemented with protease and phosphatase inhibitor tablets (ThermoFisher). Cells were then vortexed for 10 s, and Triton-insoluble material was pelleted by centrifugation at 12,000× g for 5 m at 4 °C. Supernatant was then removed and RIPA Lysis Buffered applied to the pellet. Protein concentration was determined using DC Protein Assay (Bio-Rad Laboratories, Hercules, CA, USA). Equal amounts of protein then prepared in 4× Laemmli Buffer (62.5 mM Tris-HCl pH 6.8, 10% glycerol, 0.005% Bromophenol blue, 10% beta-mercaptoethanol) and heated for 10 m at 95 °C prior to SDS-PAGE gel loading. Proteins were blotted onto PVDF membrane (Bio-Rad Laboratories) and blocked in 5% BSA PBST, 5% non-fat dried milk PBST, or Carbo-Free blocking solution (Vector Laboratories) where appropriate. Membranes were then probed for protein of interest overnight at 4 °C. Membranes were then washed in PBST and secondary reagent applied for 1–2 h at room temperature (RT). Membranes were then developed by film or imaged on a LI-COR Odyssey Fc Imaging System where appropriate.
2.4. PCR and qRT-PCR
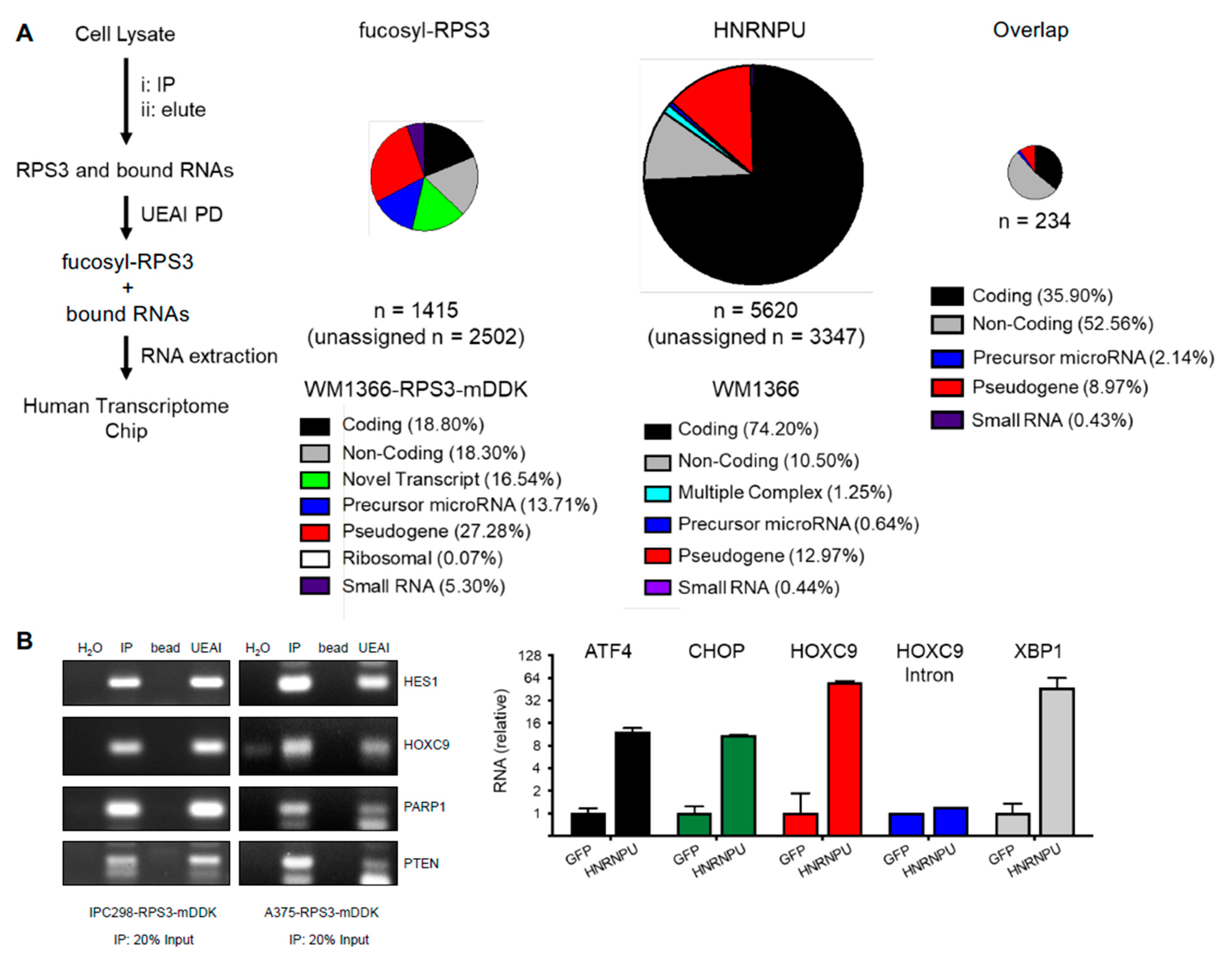
Cells were washed in cold PBS and then RNA was harvested using the GeneElute Mammalian Total RNA Kit (Sigma) or using TRIzol (ThermoFisher) following the manufacturer’s instructions. RNA was quantitated on a NanoDrop and reverse transcribed to cDNA using High-Capacity cDNA Reverse Transcription Kit (ThermoFisher). cDNA was analyzed by PCR using Phusion High-Fidelity DNA Polymerase (NEB) or by qRT-PCR using FastStart SYBR Green Master Mix (Roche, Basel, Switzerland) and quantitated on a CFX96 Touch Real-Time PCR Detection System (Bio-Rad Laboratories).
qRT-PCR primers:
18S rRNA
5-GTAACCCGTTGAACCCCATT-3
5-CCATCCAATCGGTAGTAGCG-3
HES1
5-AAACCAAAGACAGCATCTGAGC-3
5-TTCCCCAGCACACTTGGGTC-3
HOXC9
5-GGGAGGGTTCAGTGTTGAGA-3
5-GGGATGACCTGGACCAAATA-3
HOXC9 Intron
5-TGGGCATCTCCCCAGATTAGA-3
5-GTATAATTAGGCCCTGGCCCC-3
PARP1
5-GGTACCATCAGGCTGCTTT-3
5-TTCGCCACTTCATCCACTCC-3
PTEN
5-AGCTGGAAAGGGACGAACTG-3
5-CCTTTAGCTGGCAGACCACA-3
Histone H3
5-AAGCAGACTGCCCGCAAAT-3
5-GGCCTGTAACGATGAGGTTTC-3
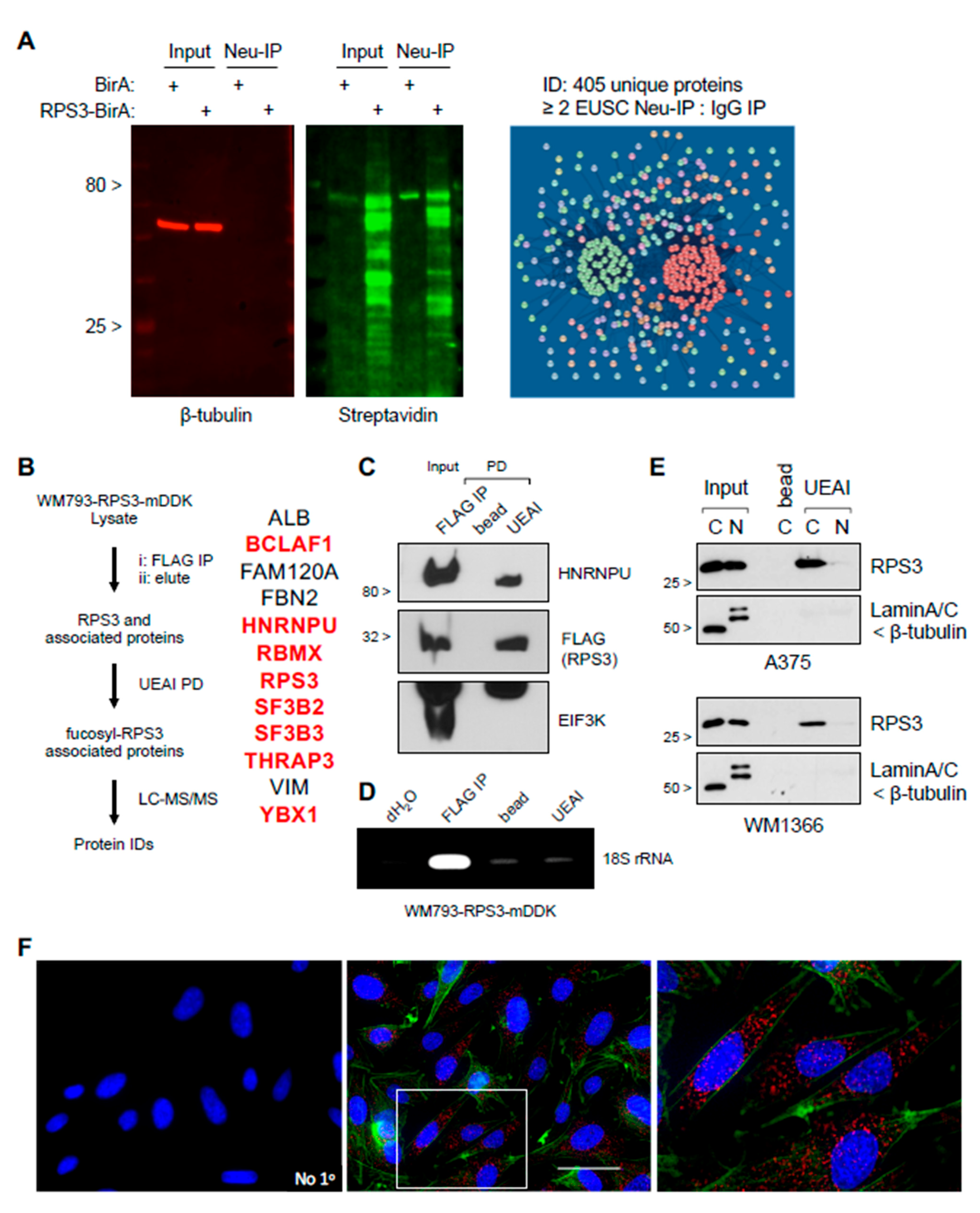
2.5. Immunoprecipitation for Protein, RNA, and Mass Spectrometry Analysis
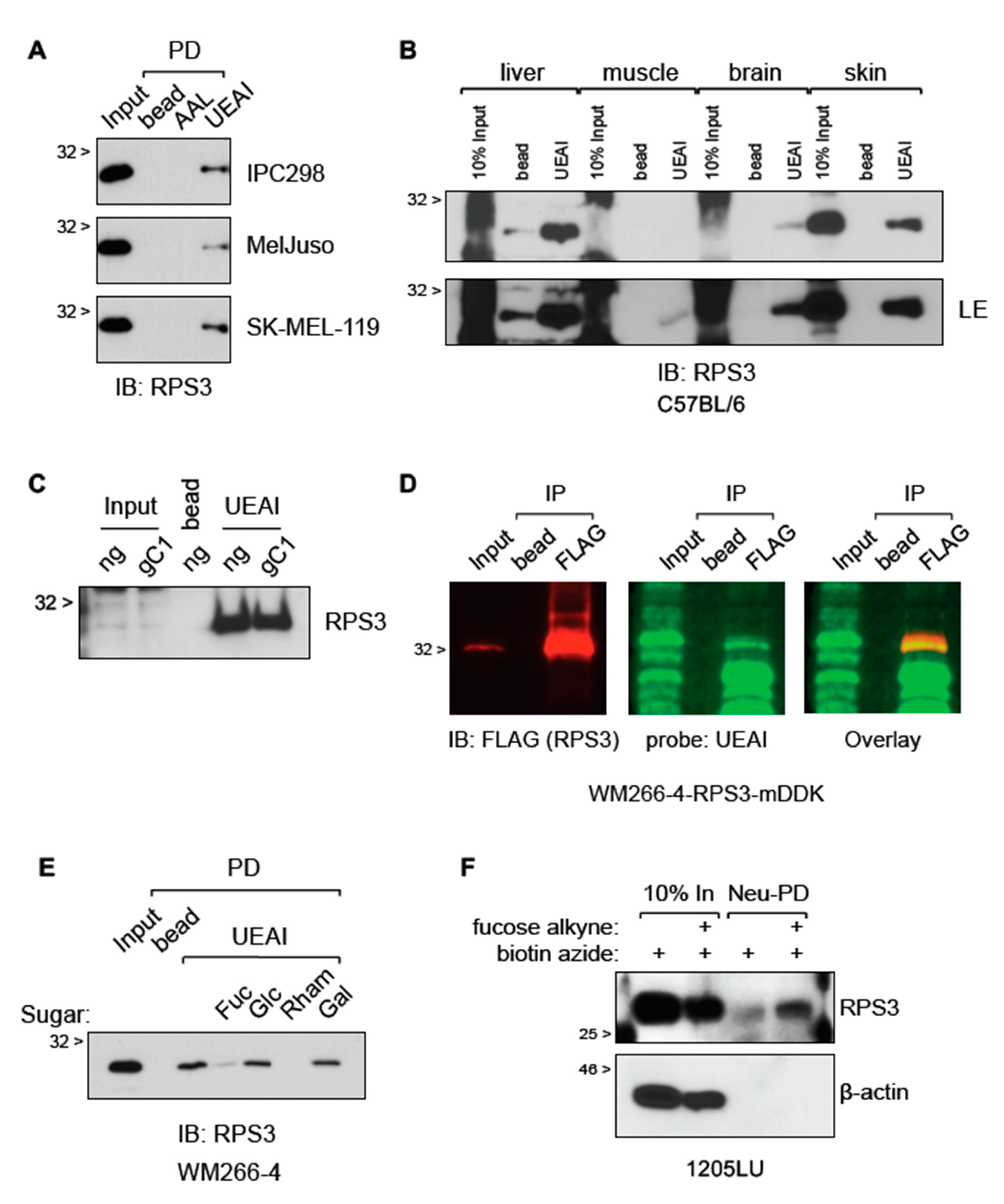
Cells were washed in PBS and then lysed in ice cold IP Lysis Buffer or RIPA Lysis Buffer using a Dounce homogenizer. Lysates were then cleared of insoluble material by spinning at 12,000× g for 5 m at 4 °C. Protein concentration was determined using DC Protein Assay (Bio-Rad Laboratories). Lysate concentrations were equalized using IP Lysis Buffer. Equal amounts of protein were incubated overnight at 4 °C with end-over-end rotation with anti-FLAG-agarose (Sigma), lectin-agarose conjugates (Vector Labs), or indicated control (normal mouse IgG or agarose bead). The next day, immunoprecipitations were washed in ice cold 0.1% Triton X-100 PBS. For protein, the immunoprecipitations were reconstituted in Western sample buffer (for Western blotting). For RNA, the samples were reconstituted in TRIzol following the manufacturer’s instructions (for PCR, qRT-PCR, or for analysis by Chip (RIP-Chip). For RIP-Chip, unbiased identification of RNAs bound by RPS3 was carried out by the Molecular Genetics Core Facility at Moffitt Cancer Center on a Clariom D Array (Applied Biosystems, Foster City, CA, USA) following the manufacturer’s instructions. Data were analyzed on the Transcriptome Analysis Console (TAC) following the manufacturer’s instructions. Cutoff for RNAs bound by RPS3 was set at ≤ 2-fold enriched in RPS3 IP relative to control IP. For mass spectrometry, samples were reconstituted in PBS and submitted to the Proteomics Core Facility at Moffitt Cancer Center for processing and analysis. For IP elution and subsequent lectin PD, washed FLAG IP protein complexes were eluted with 3× FLAG Peptide following the manufacturer’s instructions (Sigma). FLAG agarose was then removed by centrifugation, and eluted complexes were then incubated with lectin-agarose conjugates or control beads as described above.
2.6. Analysis by LC-MS/MS Mass Spectrometry
Gel bands between ~10 and 50 kDa and from ~50 to 250 were excised and treated with Tris (2-carboxy-ethyl) phosphine hydrochloride (TCEP) and iodoacetamide. In-gel digestion using trypsin was performed overnight at 37 °C. Tryptic peptides were analyzed using nanoflow liquid chromatography (U3000, Dionex, Sunnyvale, CA, USA) coupled to an electrospray bench top orbitrap mass spectrometer (Q-Exactive plus, ThermoFisher) in a data-dependent manner for tandem mass spectrometry peptide sequencing. The peptide mixtures were separated on a 75 µm ID × 25 cm, 2 µm, 100 Å, C18 analytical column (New Objective, Woburn, MA, USA) using a 90 m program at a flow rate of 300 nL/m of 5% solvent B for 8 m, 5% to 38.5% solvent B over 60 m, then 50% to 90% solvent B over 7 m and held at 90% for 5 m, followed by 90% to 5% solvent B in 1 m and re-equilibrated for 10 m. Solvent A was composed of 98% ddH2O and 2% acetonitrile containing 0.1% FA. Solvent B was 90% acetonitrile and 10% ddH2O containing 0.1% FA. Sixteen tandem mass spectra were collected in a data-dependent manner following each survey scan. Data analysis was performed using MASCOT and SEQUEST against the Human UniProt database (downloaded April 2016) to identify proteins from IP samples. The raw files were processed using select parameters, including at least 7 amino acids per peptide, as many as 3 missed cleavages, and a false discovery rate of 0.01 was selected for peptides and proteins. Methionine oxidation and peptide N-terminal acetylation were selected as variable modifications. Both MASCOT [
7] and SEQUEST [
8] search results were summarized in Scaffold 4.7.3 (Proteome Software,
http://www.proteomesoftware.com; accessed date: 30 January 2017).
2.7. Immunofluorescence
Cells were cultured and treated on glass coverslips in a 6-well tissue culture dish. Cells were washed in cold PBS and fixed in 4% formaldehyde PBS at RT for 10 m or in ice cold methanol at −20 °C for 15 m. Fixed cells were washed in PBS. Fixed cells were blocked/permeabilized in 5% BSA 0.3% Triton X-100 PBS for 1 h at RT. Primary antibody (as indicated) diluted in blocking buffer was applied ON at 4 °C. The next day, cells were washed in PBS, and fluorophore-conjugated secondary antibody (Invitrogen, Carlsbad, CA, USA) diluted in blocking buffer was applied for 1 h at RT. Cells were washed in PBS and then mounted on glass sides using Prolong Gold Antifade Mountant (ThermoFisher). Cells were analyzed and imaged on a Keyence BZ-700 fluorescent microscope.
2.8. Tissue Microarray Staining
Melanoma tissue microarray (TMA) was purchased from US Biomax (Derwood, MD, USA) (Cat. No. ME1004f). Paraffin was melted away by heating at ~70 °C for 30 m before rehydrating the tissue through xylene and ethanol dilution rinses. Antigen retrieval was conducted using DAKO Target Retrieval Solution (Agilent, Santa Clara, CA, USA) following the manufacturer’s instructions. Tissue was blocked using DAKO Protein Block, Serum Free reagent (Agilent). The TMA was probed for RPS3 and Phalloidin-FITC (Invitrogen) overnight at 4 °C. The next day, the TMA was washed and incubated with secondary antibody for 2 h at RT. The slide was then washed 1 time in PBS containing DAPI (ThermoFisher), followed by another 3 washes in PBS. A coverslip was then mounted over the tissue using ProLong Gold Antifade Reagent (ThermoFisher). The tissue slide was imaged at the Moffitt Cancer Center Clinical Testing Department Core Facility, and the signal was quantitated in an unbiased manner using the HALO platform.
2.9. Flow Cytometry
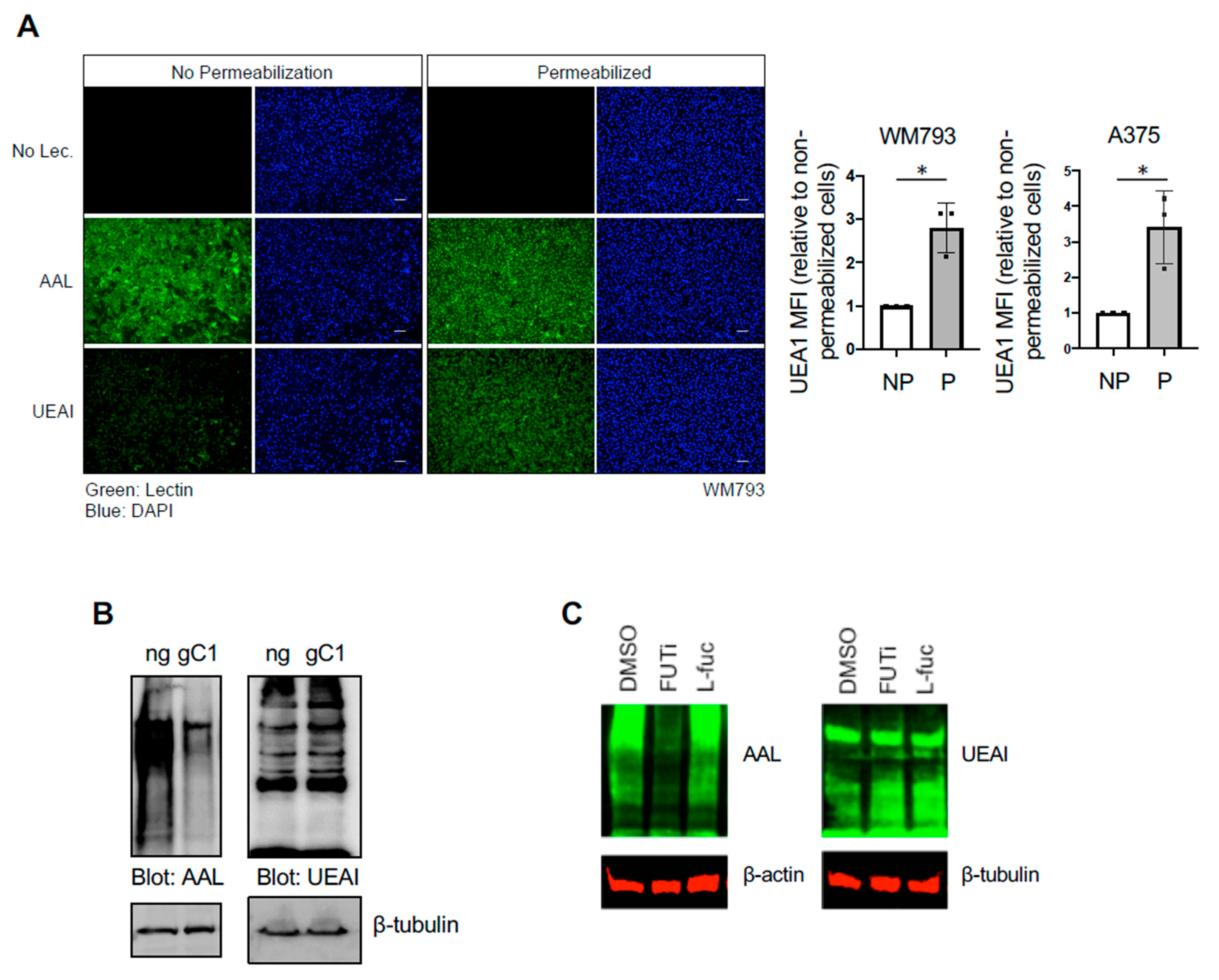
Subconfluent cells were harvested in trypsin (Corning Inc., Corning, NY, USA) and washed in cold PBS. Cells were fixed in 4% formaldehyde PBS for 10 m at RT and then washed in PBS. Non-permeabilized cells were then blocked in 5% BSA PBS and incubated with 0.2 µg/mL fluorophore-conjugated lectin (Vector Laboratories) for 1 h at 4 °C. For permeabilization and staining, cells were incubated in 5% BSA 0.3% Triton X-100 PBS for 30–60 m at RT and then incubated with 0.2 µg/mL fluorophore-conjugated lectin (Vector Laboratories) for another 1 h at RT. Stained cells were then washed in PBS twice before being subjected to flow cytometry. Samples were analyzed using FlowJo software (BD Biosciences, East Rutherford, NJ, USA), and statistical analyses were performed on Prism (GraphPad, San Diego, CA, USA).
2.10. Biotinylation Identification (BioID)
RPS3 coding sequence was cloned into pCDNA3.1-MCS-BirA (R118G)-HA plasmid (Addgene #36047). The 293T cells were transfected for RPS3-BioID using Lipofectamine 2000 reagent following the manufacturer’s protocol (ThermoFisher), and cells were then cultured for 2 days in media supplemented with biotin prior to lysis. Cells were washed in cold PBS and lysed in IP Lysis Buffer using a Dounce homogenizer. Lysates were cleared on insoluble material by centrifugation at 12,000×
g for 5 m at 4 °C. Cell lysates were then incubated with neutravidin-conjugated agarose (ThermoFisher) or control agarose beads ON at 4 °C with end-over-end rotation. The next day, IPs were washed in 0.1% Triton X-100 PBS, and the pelleted agarose-IP conjugates were submitted to the Proteomics Core Facility at Moffitt Cancer Center for processing and analysis. Proteins identified with 2 or more exclusive unique spectrum counts were considered as a positive ID. Proteins identified were analyzed and visualized by String (
http://string-db.org; accessed date: 4 April 2019) using default parameters.
2.11. Click-Chemistry Identification of RPS3 Fucosylation
Click-IT Protein Reaction Buffer Kit and Click-IT Fucose Alkyne were purchased from ThermoFisher and used in accordance with the manufacturer’s instructions. Briefly, 1205LU cells were cultured in media supplemented with or without 50 µM Fucose Alkyne for two days. Cells were subsequently washed in cold PBS and lysed in IP Lysis Buffer using a Dounce homogenizer. Lysates were cleared of insoluble material by centrifugation at 12,000× g for 5 m at 4 °C. Lysates were then incubated with biotin azide and the click chemistry reaction was performed following the kit instructions. Proteins were then precipitated in methanol/chloroform. Precipitated proteins were washed thoroughly in methanol and resuspended in 1% SDS with heat. Solubilized protein samples were then diluted in IP Lysis Buffer, followed by removal of a small aliquot to hold in reserve as “Input”. Neutravidin-agarose (ThermoFisher) was then applied to capture proteins that integrated the labeled fucose by IP. Samples were incubated ON at 4 °C with end-over-end rotation. The next day, neutravidin-captured proteins were washed in 0.1% Triton X-100 and run for analysis by Western blot.
4. Discussion
Glycosylation is known to play an important role in influencing protein–protein interactions, particularly in how cells interact with proteins in the extracellular environment. Glycosylation of extracellular receptors, secreted proteins, or exogenous ligands can influence downstream cellular signaling pathways and in turn impact cellular behavior (e.g., adhesion/migration). Due to the spatial restriction of fucosyltransferase enzymes to the secretory network, fucosylation has generally been thought to only influence proteins and signaling pathways, and thus cellular behavior, through modification of extracellular receptors and/or secreted proteins. Aside from posttranslational N-acetylglucosylation (GlcNAC), a reversible posttranslational modification on intracellular proteins with a GlcNAC moiety, there is no clearly recognized role for other intracellular glycosylation events that can influence protein function and/or cellular signaling pathways and downstream cellular behavior. In this report, we find that a large number of intracellular proteins are potentially posttranslationally modified with L-fucose. An unbiased UEAI lectin screen for fucosylated proteins identified only intracellular proteins, and we subsequently validated RPS3 as fucosylated using an approach for labelling fucosylated proteins with a fucose analog that allows for click chemistry-mediated identification of proteins that are posttranslationally fucosylated.
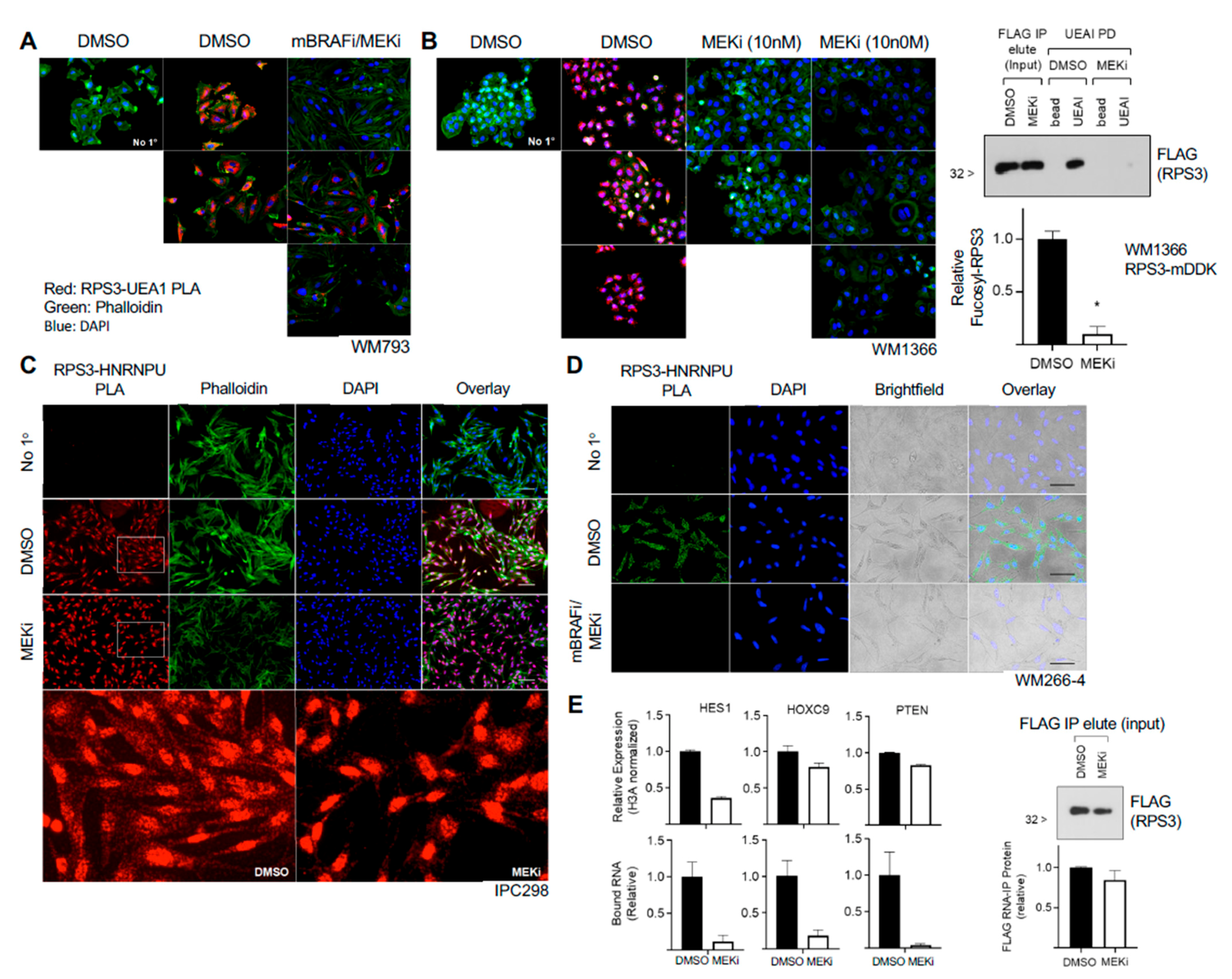
We find that fucosylation of RPS3 appears to be independent of the canonical fucosylation pathway. Knockout of the Golgi GDP-L-fucose transporter SLC35C1 had no effect on fucosylation of RPS3 or global UEAI-recognized fucosylation, and inhibition of global GDP-L-fucose synthesis with 2-FF also did not affect UEAI-recognized fucosylation. These data suggest that fucosylation outside of the ER–Golgi network and de novo GDP-L-fucose synthesis pathway can and does occur and that there are uncharacterized pathway(s) that control fucosylation on intracellular proteins that may be independent of the canonical fucosylation pathway. It is possible that SLC35C1 knockout or 2-FF are insufficient to entirely block fucosylation (i.e., compensation by SLC35C2), and retrograde trafficking/transport represents one mechanism that could result in cytoplasmic localization of fucosylated RPS3. However, as a majority of our in vitro studies have been performed in melanoma cell lines, it is also possible that the aberrant expression of splice variants of fucosyltransferases that are no longer restricted in localization to within the ER–Golgi network mediate RPS3 fucosylation. Further studies are required to test this possibility.
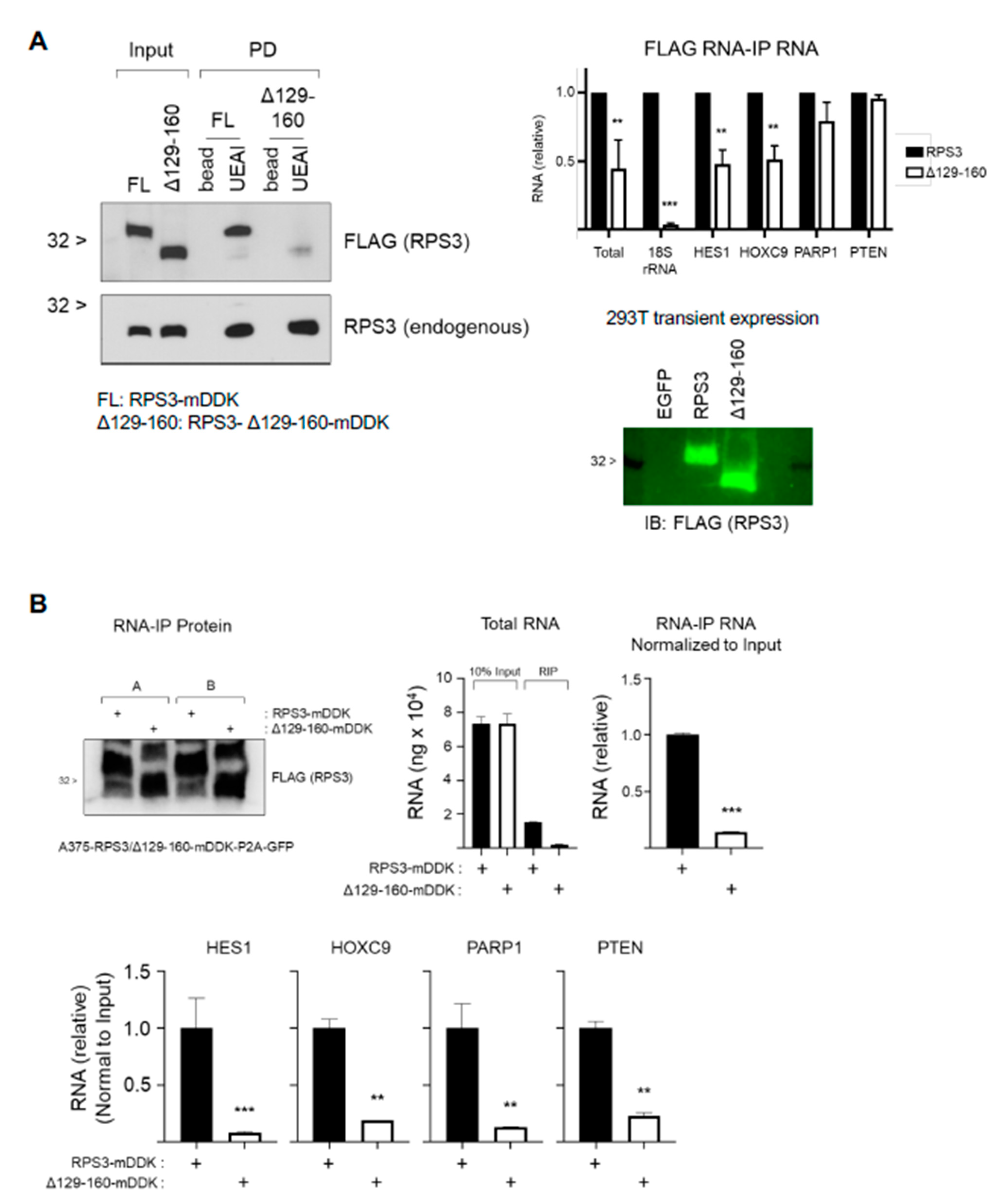
Fucosylation of RPS3 is associated with a species of RPS3 that is independent of the ribosome, and we also found that fucosylation level can respond to stimulus (MAPKi). This suggests that fucosylation of RPS3 is regulated and influences protein function, either tagging a pool of ribosome-independent RPS3 for interaction with a protein complex that posttranscriptionally regulates RNAs or excluding a pool of RPS3 from the ribosome and/or nucleus. The stress-/therapeutic agent-responsive nature of this mechanism in melanoma suggests potential pathogenic contributions that will require further study. We were unable to identify an individual specific site where RPS3 is fucosylated (or glycosylated), and a deletion mutant that abolished fucosylation on RPS3 could not be stably expressed, as it compromised protein stability. The finding that several deletion mutants of RPS3 can be expressed and are glycosylated, whereas the single deletion mutant identified that cannot be fucosylated is rapidly degraded, suggests that this region is both important for protein stability and glycosylation. These findings support the notion that the fucosylation or glycosylation of RPS3 is not necessarily site-specific but is instead spread across multiple sites within a region. This might be indicative of association with a specific protein complex and glycosylation by close proximity to glycosyltransferase enzymes. Further work is needed to clarify the mechanism by which RPS3 is fucosylated, how it influences its function, and how fucosylation can occur outside of the canonical fucosylation pathway. Subsequent elucidation of such non-canonical fucosylation events is anticipated to provide important insight into how fucosylation, and defects therein, might significantly contribute to cancer pathology and therapeutic responsiveness.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





