
Characterization of PARP6 Function in Knockout Mice and Patients with Developmental Delay

 , , , and
, , , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Housing
2.2. Cell Cultures: Human (HEK293T Cell Line), Rat Primary Cortical and Hippocampal Neurons
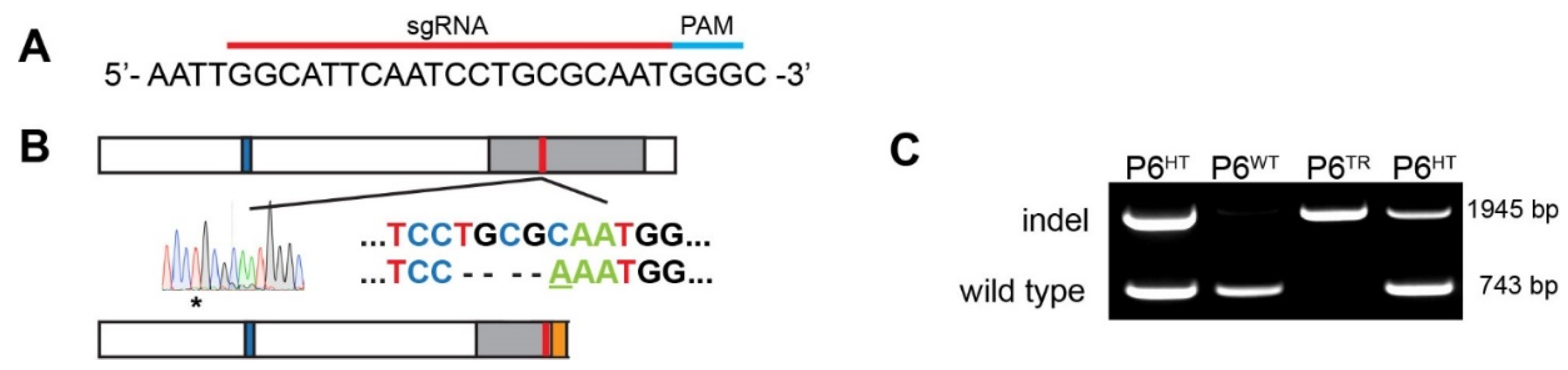
2.3. CRISPR: sgRNA Design and Generation of Mouse Line Using Cas9
2.4. Genotyping: gDNA Extraction and PCR
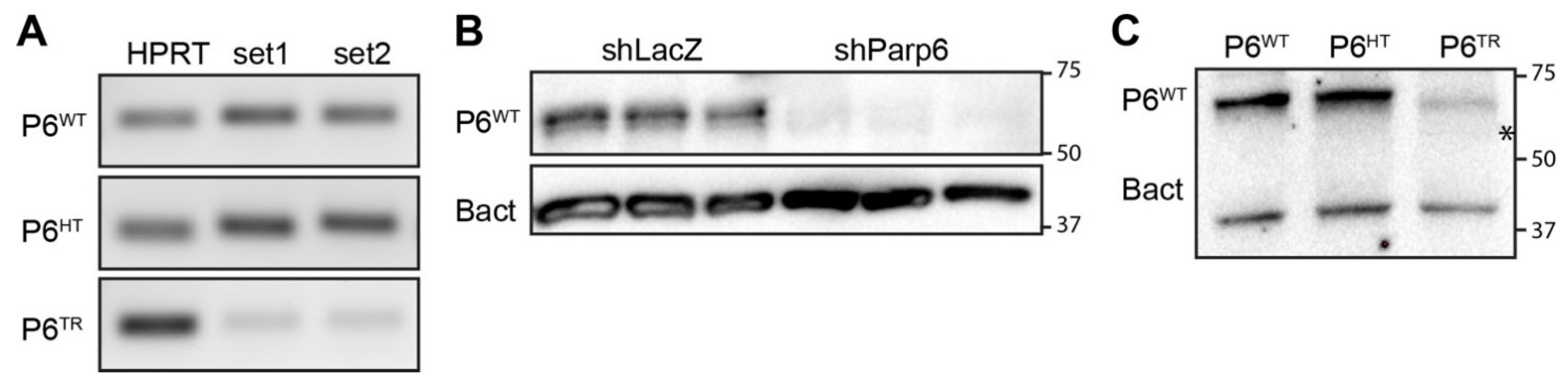
2.5. Total RNA Extraction from Parp6WT and Parp6TR Brain for cDNA, RT-PCR and RNAseq
2.6. Immunoblots
2.7. Whole Mount Immunohistochemistry and Imaging
2.8. Whole Mount E12.5 Embryo Immunolabeling, Clearing, Imaging, and Quantification
2.9. Cloning: Parp6 RNA Interference and Expression Constructs
2.10. Transduction of Cortical Neurons, BioID, NeutrAvidin Enrichment, and LC-MS/MS Analysis
2.11. PARP6 Immunoprecipitation Auto-MARylation Activity Assay
2.12. Transfection of Rat Hippocampal Cultures, Immunostaining, Imaging, and Sholl Analysis
2.13. Whole Exome Sequencing (WES), Sequence Alignment, and Analysis
2.14. Statistical Analysis
3. Results
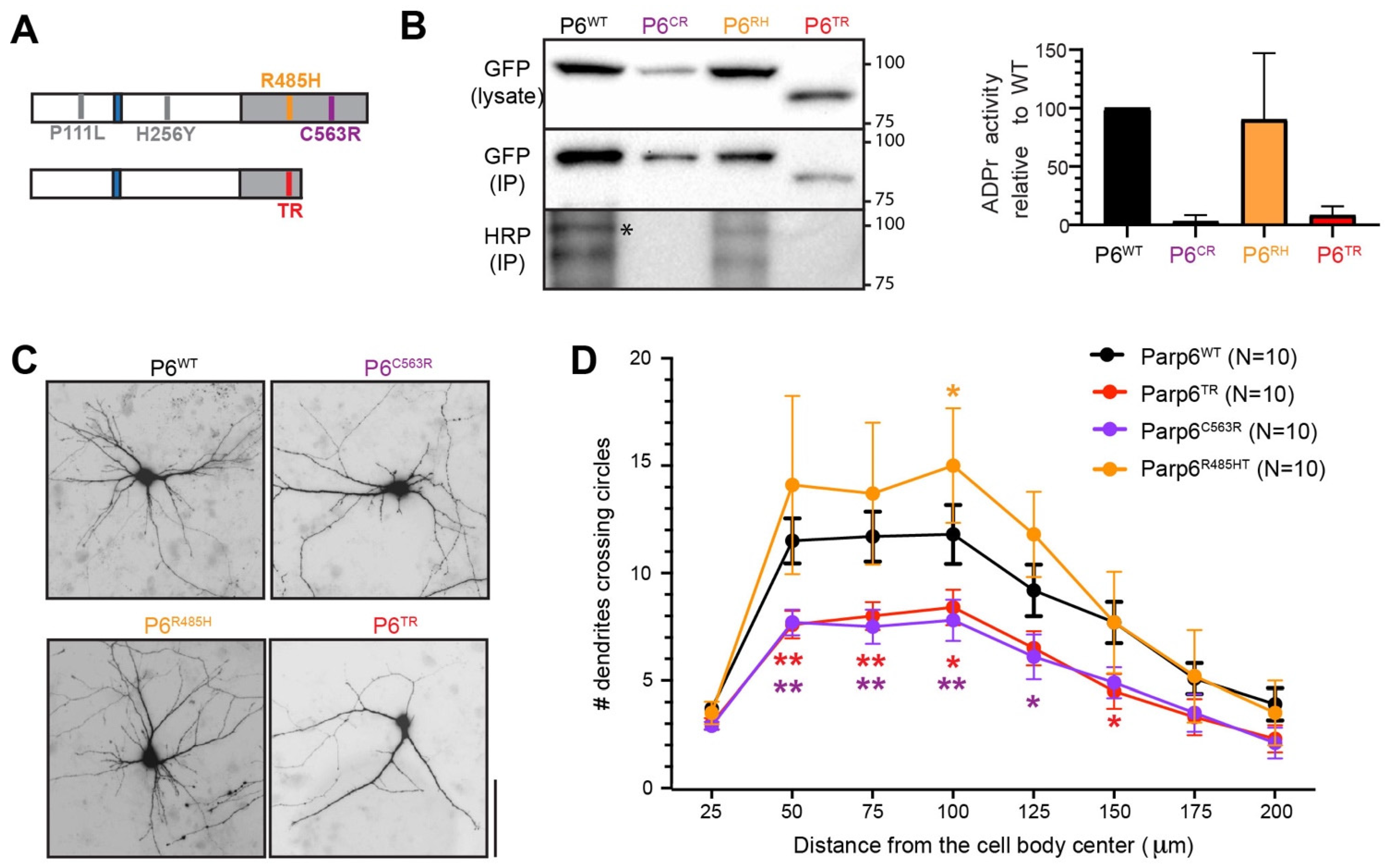
3.1. Generation of a Parp6 Knockout Mouse Line Using CRISPR-Cas9 Mutagenesis
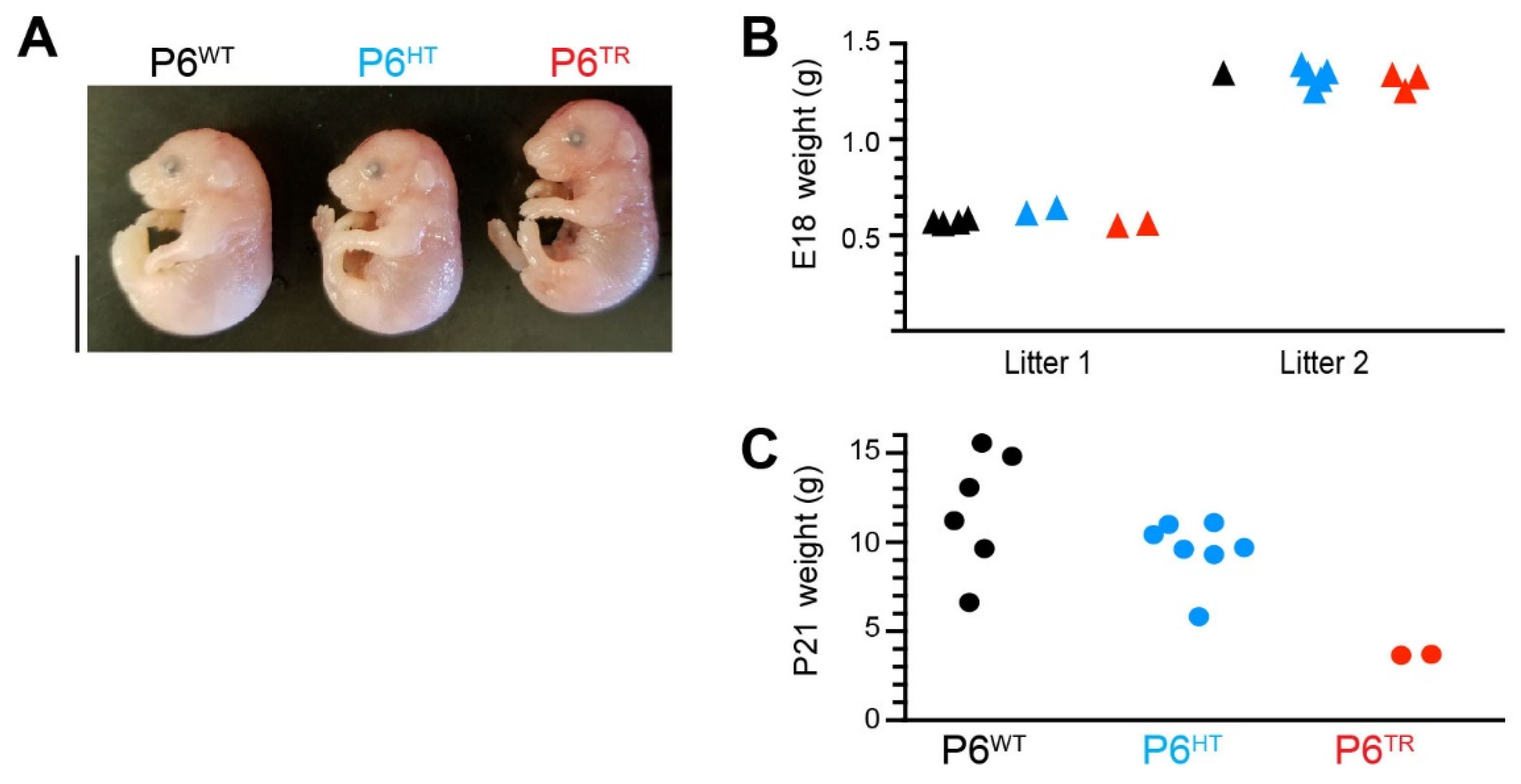
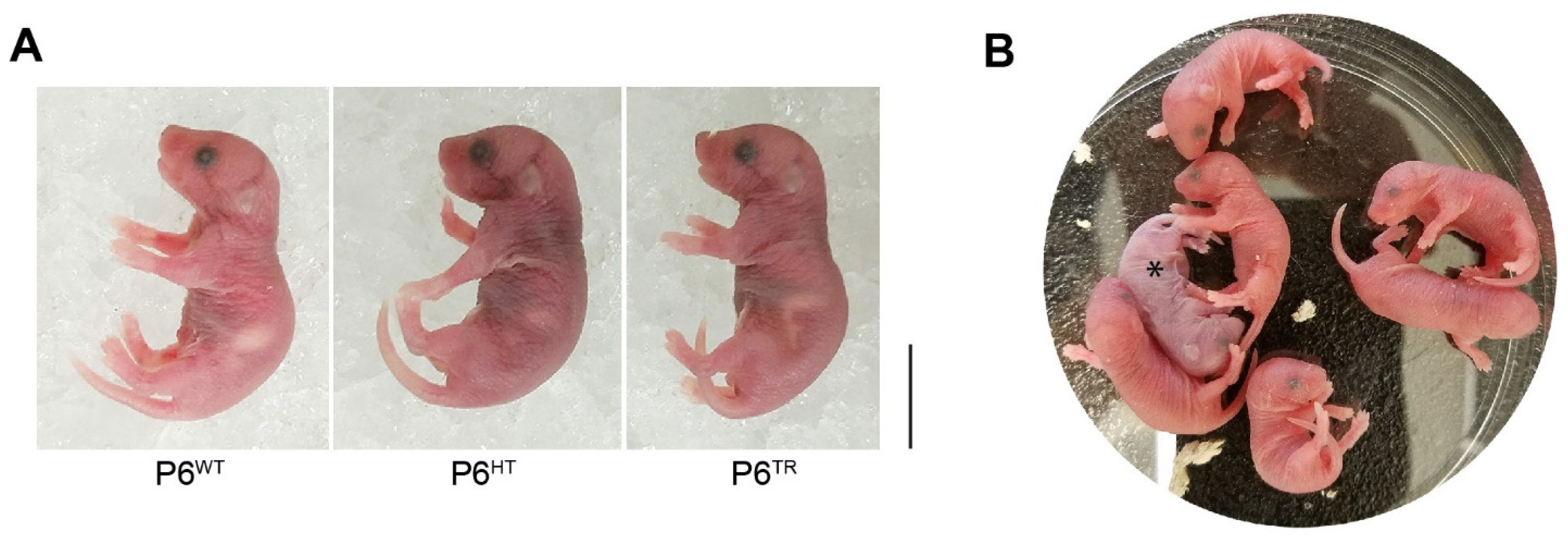
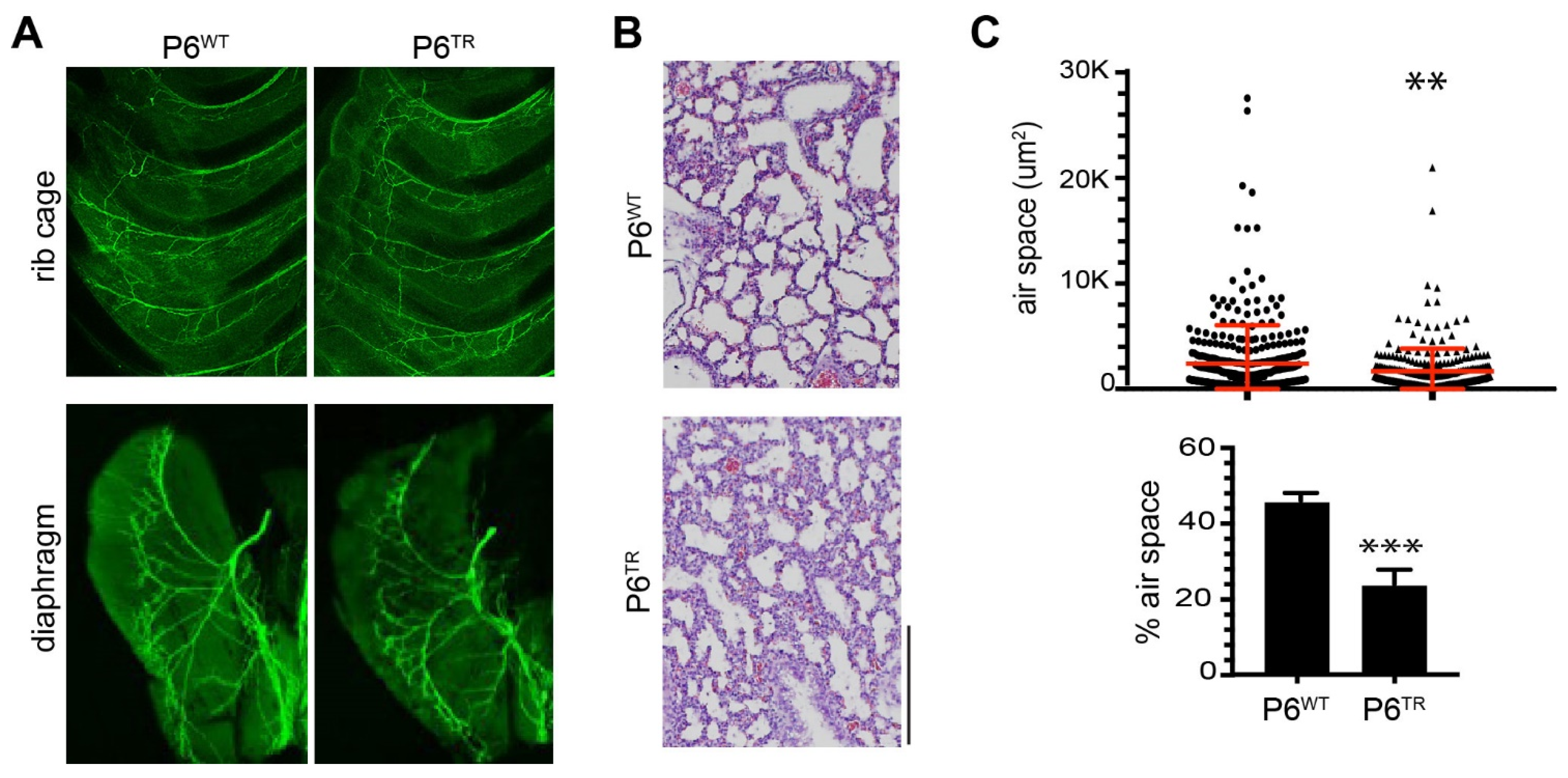
3.2. Characterization of Parp6TR Embryos and Few Surviving Adults
3.3. Loss of Full-Length Parp6 Leads to Perinatal Lethality
3.4. Human PARP6 Mutations: Clinical Findings
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jubin, T.; Kadam, A.; Jariwala, M.; Bhatt, S.; Sutariya, S.; Gani, A.R.; Gautam, S.; Begum, R. The PARP family: Insights into functional aspects of poly (ADP-ribose) polymerase-1 in cell growth and survival. Cell Prolif. 2016, 49, 421–437. [Google Scholar] [CrossRef] [PubMed]
- Krishnakumar, R.; Kraus, W.L. The PARP side of the nucleus: Molecular actions, physiological outcomes, and clinical targets. Mol. Cell 2010, 39, 8–24. [Google Scholar] [CrossRef]
- Vyas, S.; Chesarone-Cataldo, M.; Todorova, T.; Huang, Y.H.; Chang, P. A systematic analysis of the PARP protein family identifies new functions critical for cell physiology. Nat. Commun. 2013, 4, 2240. [Google Scholar] [CrossRef] [PubMed]
- Vyas, S.; Matic, I.; Uchima, L.; Rood, J.; Zaja, R.; Hay, R.T.; Ahel, I.; Chang, P. Family-wide analysis of poly(ADP-ribose) polymerase activity. Nat. Commun. 2014, 5, 4426. [Google Scholar] [CrossRef] [PubMed]
- Challa, S.; Stokes, M.S.; Kraus, W.L. MARTs and MARylation in the cytosol: Biological functions, mechanisms of action, and therapeutic potential. Cells 2021, 10, 313. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, D.J.; Cohen, M.S. Mechanisms governing PARP expression, localization, and activity in cells. Crit. Rev. Biochem. Mol. Biol. 2020, 55, 541–554. [Google Scholar] [CrossRef]
- Huang, J.Y.; Wang, K.; Vermehren-Schmaedick, A.; Adelman, J.P.; Cohen, M.S. PARP6 is a regulator of hippocampal dendritic morphogenesis. Sci. Rep. 2016, 6, 18512. [Google Scholar] [CrossRef]
- Roux, K.J.; Kim, D.I.; Burke, B. BioID: A screen for protein-protein interactions. Curr. Protoc. Protein. Sci. 2013, 74, 19–23. [Google Scholar] [CrossRef]
- Carter-O’Connell, I.; Jin, H.; Morgan, R.K.; Zaja, R.; David, L.L.; Ahel, I.; Cohen, M.S. Identifying family-member-specific targets of mono-ARTDs by using a chemical genetics approach. Cell Rep. 2016, 14, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Carter-O’Connell, I.; Vermehren-Schmaedick, A.; Jin, H.; Morgan, R.K.; David, L.L.; Cohen, M.S. Combining chemical genetics with proximity-dependent labeling reveals cellular targets of Poly(ADP-ribose) polymerase 14 (PARP14). ACS Chem. Biol. 2018, 13, 2841–2848. [Google Scholar] [CrossRef]
- You, K.T.; Li, L.S.; Kim, N.G.; Kang, H.J.; Koh, K.H.; Chwae, Y.J.; Kim, K.M.; Kim, Y.K.; Park, S.M.; Jang, S.K.; et al. Selective translational repression of truncated proteins from frameshift mutation-derived mRNAs in tumors. PLoS Biol. 2007, 5, e109. [Google Scholar] [CrossRef]
- Jayachandran, P.; Olmo, V.N.; Sanchez, S.P.; McFarland, R.J.; Vital, E.; Werner, J.M.; Hong, E.; Sanchez-Alberola, N.; Molodstov, A.; Brewster, R.M. Microtubule-associated protein 1b is required for shaping the neural tube. Neural Dev. 2016, 11, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Dehmelt, L.; Halpain, S. The MAP2/Tau family of microtubule-associated proteins. Genome Biol. 2005, 6, 204. [Google Scholar] [CrossRef] [PubMed]
- Matamoros, A.J.; Baas, P.W. Microtubules in health and degenerative disease of the nervous system. Brain Res. Bull. 2016, 126, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Chakraborti, S.; Natarajan, K.; Curiel, J.; Janke, C.; Liu, J. The emerging role of the tubulin code: From the tubulin molecule to neuronal function and disease. Cytoskeleton 2016, 73, 521–550. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.K.; Schwartz, C.E. Intellectual disability and autism spectrum disorders: Causal genes and molecular mechanisms. Neurosci. Biobehav. Rev. 2014, 46, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Lasser, M.; Tiber, J.; Lowery, L.A. The role of the microtubule cytoskeleton in neurodevelopmental disorders. Front. Cell. Neurosci. 2018, 12, 165. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, D.; Rivenson, H. Accounting principles and measuring health care quality. JAMA 2008, 299, 764–765. [Google Scholar] [CrossRef] [PubMed]
- Barkovich, A.J.; Guerrini, R.; Kuzniecky, R.I.; Jackson, G.D.; Dobyns, W.B. A developmental and genetic classification for malformations of cortical development: Update 2012. Brain 2012, 135, 1348–1369. [Google Scholar] [CrossRef]
- Gilmore, E.C.; Walsh, C.A. Genetic causes of microcephaly and lessons for neuronal development. Wiley Interdiscip. Rev. Dev. Biol. 2013, 2, 461–478. [Google Scholar] [CrossRef]
- Duerinckx, S.; Abramowicz, M. The genetics of congenitally small brains. Semin. Cell Dev. Biol. 2018, 76, 76–85. [Google Scholar] [CrossRef]
- Wang, Z.; Grosskurth, S.E.; Cheung, T.; Petteruti, P.; Zhang, J.; Wang, X.; Wang, W.; Gharahdaghi, F.; Wu, J.; Su, N.; et al. Pharmacological inhibition of PARP6 triggers multipolar spindle formation and elicits therapeutic effects in breast cancer. Cancer Res. 2018, 78, 6691–6702. [Google Scholar] [CrossRef] [PubMed]
- Johannes, J.W.; Almeida, L.; Daly, K.; Ferguson, A.D.; Grosskurth, S.E.; Guan, H.; Howard, T.; Ioannidis, S.; Kazmirski, S.; Lamb, M.L.; et al. Discovery of AZ0108, an orally bioavailable phthalazinone PARP inhibitor that blocks centrosome clustering. Bioorg. Med. Chem. Lett. 2015, 25, 5743–5747. [Google Scholar] [CrossRef] [PubMed]
- Maiato, H.; Logarinho, E. Mitotic spindle multipolarity without centrosome amplification. Nat. Cell Biol. 2014, 16, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Higginbotham, H.R.; Gleeson, J.G. The centrosome in neuronal development. Trends Neurosci. 2007, 30, 276–283. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age (y) | 28/28 | 3/5 | 7/8 | 10/13 | 12/17 | 4/9 |
|---|---|---|---|---|---|---|
| Sex | male | female | female | male | male | female |
| Gene | c.332 C>T | c. 766 C>T | c. 1453 C>T | c. 1454 G>A | c. 1687 T>C | c. 1687 T>C |
| Protein | p. P111L | p. H256Y | p. R485C | p. R485H | p. C563R | p. C563R |
| Zygosity status | heterozygous (de novo) | heterozygous (de novo) | heterozygous (de novo) | heterozygous (de novo) | homozygous | homozygous |
| Other relatives | sporadic | sporadic | Father: no Mother: ND | Sporadic Parents: ND | Parents heterozygous carriers | Parents heterozygous carriers |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vermehren-Schmaedick, A.; Huang, J.Y.; Levinson, M.; Pomaville, M.B.; Reed, S.; Bellus, G.A.; Gilbert, F.; Keren, B.; Heron, D.; Haye, D.; et al. Characterization of PARP6 Function in Knockout Mice and Patients with Developmental Delay. Cells 2021, 10, 1289. https://doi.org/10.3390/cells10061289
Vermehren-Schmaedick A, Huang JY, Levinson M, Pomaville MB, Reed S, Bellus GA, Gilbert F, Keren B, Heron D, Haye D, et al. Characterization of PARP6 Function in Knockout Mice and Patients with Developmental Delay. Cells. 2021; 10(6):1289. https://doi.org/10.3390/cells10061289
Chicago/Turabian StyleVermehren-Schmaedick, Anke, Jeffrey Y. Huang, Madison Levinson, Matthew B. Pomaville, Sarah Reed, Gary A. Bellus, Fred Gilbert, Boris Keren, Delphine Heron, Damien Haye, and et al. 2021. "Characterization of PARP6 Function in Knockout Mice and Patients with Developmental Delay" Cells 10, no. 6: 1289. https://doi.org/10.3390/cells10061289
APA StyleVermehren-Schmaedick, A., Huang, J. Y., Levinson, M., Pomaville, M. B., Reed, S., Bellus, G. A., Gilbert, F., Keren, B., Heron, D., Haye, D., Janello, C., Makowski, C., Danhauser, K., Fedorov, L. M., Haack, T. B., Wright, K. M., & Cohen, M. S. (2021). Characterization of PARP6 Function in Knockout Mice and Patients with Developmental Delay. Cells, 10(6), 1289. https://doi.org/10.3390/cells10061289