
Traumatic Optic Neuropathy Is Associated with Visual Impairment, Neurodegeneration, and Endoplasmic Reticulum Stress in Adolescent Mice

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Traumatic Brain Injury
2.3. Behavioral Testing
2.3.1. Circadian Rhythm and Locomotion
2.3.2. Optomotor/Optokinetic Response (OKR) and Visual Acuity Testing
Optomotor Machine (OMM)
Sine-Wave Gratings
Testing
Scoring
2.4. Retina Dissection
2.5. Western Blots
2.6. Histology
2.7. Immunofluorescence
2.8. Image Analysis
2.9. Statistical Analysis
3. Results
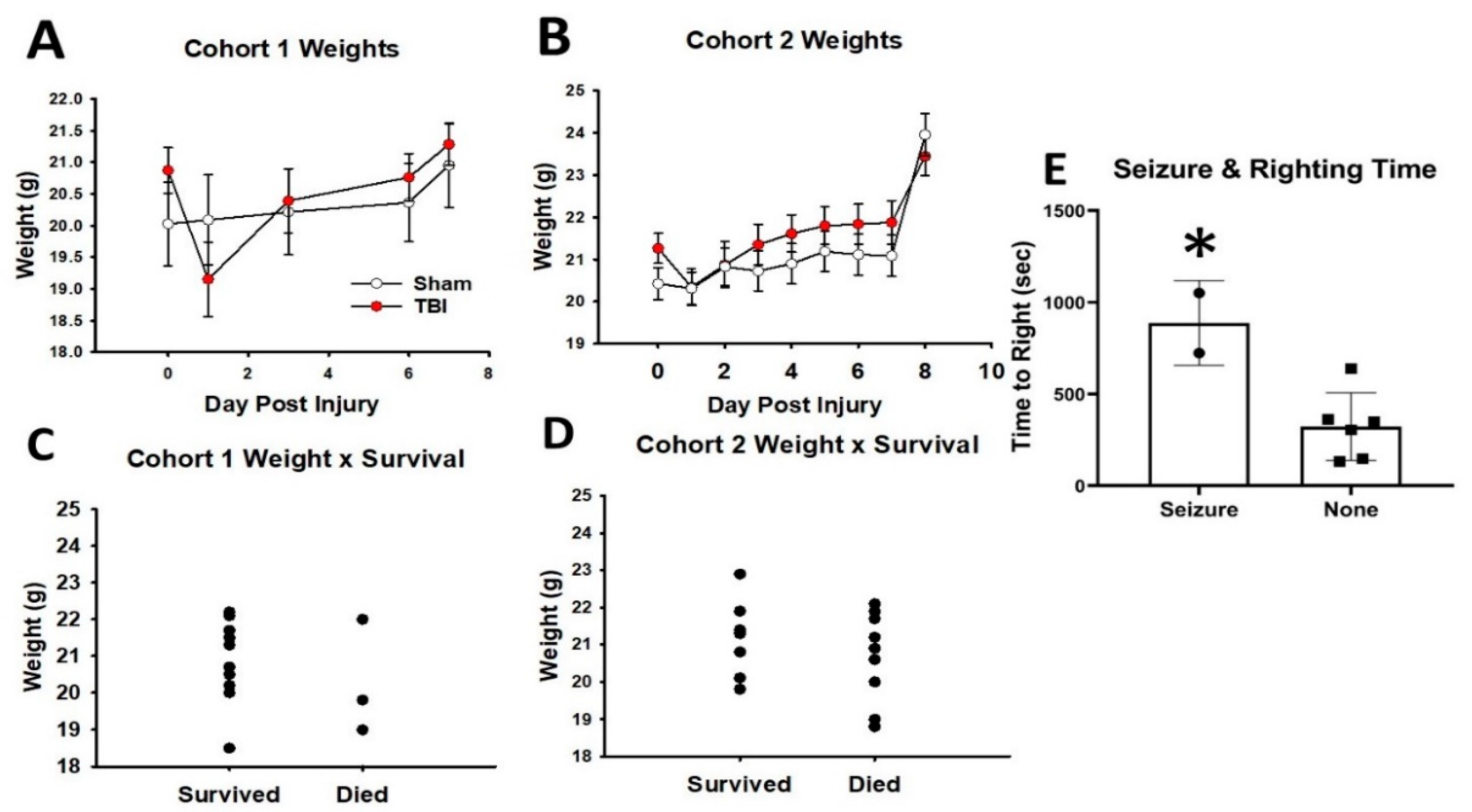
3.1. Weight, Mortality, and Morbidity Following Mild TBI in Adolescent Mice
3.2. iTON Injured Mice Have a Blunted Optokinetic Response
3.3. Activity But Not Circadian Rhythm Is Affected in This Model of Traumatic Optic Neuropathy
3.4. There Is Significant Retinal Ganglion Cell Death in TON Mice Throughout the Retina
3.5. Injured Mice Have Significantly Increased Axonal Degeneration Throughout the Primary Visual System
3.6. There Is Microglial Activation in Some Downstream Optic Subcortical Thalamic Targets
3.7. There Is Astrogliosis after TBI Throughout the Visual System Indicative of Traumatic Optic Neuropathy
3.8. ER Stress Markers Are Elevated after Experimental Head Trauma
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ventura, R.E.; Balcer, L.J.; Galetta, S.L. The neuro-ophthalmology of head trauma. Lancet Neurol. 2014, 13, 1006–1016. [Google Scholar] [CrossRef]
- Chan, J.W.; Hills, N.K.; Bakall, B.; Fernandez, B. Indirect Traumatic Optic Neuropathy in Mild Chronic Traumatic Brain Injury. Investig. Ophthalmol. Vis. Sci. 2019, 60, 2005–2011. [Google Scholar] [CrossRef]
- Zelinsky, D.G. Brain injury rehabilitation: Cortical and subcortical interfacing via retinal pathways. PM R 2010, 2, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Swanson, M.W.; Weise, K.K.; Dreer, L.E.; Johnston, J.; Davis, R.D.; Ferguson, D.; Hale, M.H.; Gould, S.J.; Christy, J.B.; Busettini, C.; et al. Academic Difficulty and Vision Symptoms in Children with Concussion. Optom. Vis. Sci. 2017, 94, 60–67. [Google Scholar] [CrossRef]
- Cook, M.W.; Levin, L.A.; Joseph, M.P.; Pinczower, E.F. Traumatic optic neuropathy. A meta-analysis. Arch. Otolaryngol. Head Neck Surg. 1996, 122, 389–392. [Google Scholar] [CrossRef]
- Steinsapir, K.D.; Goldberg, R.A. Traumatic optic neuropathy. Surv. Ophthalmol. 1994, 38, 487–518. [Google Scholar] [CrossRef]
- Ford, R.L.; Lee, V.; Xing, W.; Bunce, C. A 2-year prospective surveillance of pediatric traumatic optic neuropathy in the United Kingdom. J. Aapos Off. Publ. Am. Assoc. Pediatr. Ophthalmol. Strabismus 2012, 16, 413–417. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P. Traumatic optic neuropathy-Clinical features and management issues. Taiwan J. Ophthalmol. 2015, 5, 3–8. [Google Scholar] [CrossRef]
- Sen, N. An insight into the vision impairment following traumatic brain injury. Neurochem. Int. 2017, 11, 103–107. [Google Scholar] [CrossRef]
- Alhilali, L.M.; Yaeger, K.; Collins, M.; Fakhran, S. Detection of central white matter injury underlying vestibulopathy after mild traumatic brain injury. Radiology 2014, 272, 224–232. [Google Scholar] [CrossRef]
- Jang, S.H.; Seo, J.P. Damage to the Optic Radiation in Patients With Mild Traumatic Brain Injury. J. Neuroophthalmol. 2015, 35, 270–273. [Google Scholar] [CrossRef]
- Athanasiou, D.; Aguila, M.; Bevilacqua, D.; Novoselov, S.S.; Parfitt, D.A.; Cheetham, M.E. The cell stress machinery and retinal degeneration. FEBS Lett. 2013, 587, 2008–2017. [Google Scholar] [CrossRef]
- Cansler, S.M.; Evanson, N.K. Connecting endoplasmic reticulum and oxidative stress to retinal degeneration, TBI, and traumatic optic neuropathy. J. Neurosci. Res. 2020, 98, 571–574. [Google Scholar] [CrossRef] [PubMed]
- Kroeger, H.; Chiang, W.C.; Felden, J.; Nguyen, A.; Lin, J.H. ER stress and unfolded protein response in ocular health and disease. FEBS J. 2019, 286, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Bashir, A.; Abebe, Z.A.; McInnes, K.A.; Button, E.B.; Tatarnikov, I.; Cheng, W.H.; Haber, M.; Wilkinson, A.; Barron, C.; Diaz-Arrastia, R.; et al. Increased severity of the CHIMERA model induces acute vascular injury, sub-acute deficits in memory recall, and chronic white matter gliosis. Exp. Neurol. 2020, 324, 113116. [Google Scholar] [CrossRef] [PubMed]
- Heldt, S.A.; Elberger, A.J.; Deng, Y.; Guley, N.H.; Del Mar, N.; Rogers, J.; Choi, G.W.; Ferrell, J.; Rex, T.S.; Honig, M.G.; et al. A novel closed-head model of mild traumatic brain injury caused by primary overpressure blast to the cranium produces sustained emotional deficits in mice. Front. Neurol. 2014, 5, 2. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Fox, M.A.; Povlishock, J.T. Diffuse traumatic axonal injury in the optic nerve does not elicit retinal ganglion cell loss. J. Neuropathol. Exp. Neurol. 2013, 72, 768–781. [Google Scholar] [CrossRef]
- Chen, H.; Desai, A.; Kim, H.Y. Repetitive Closed-Head Impact Model of Engineered Rotational Acceleration Induces Long-Term Cognitive Impairments with Persistent Astrogliosis and Microgliosis in Mice. J. Neurotrauma 2017, 34, 2291–2302. [Google Scholar] [CrossRef] [PubMed]
- Desai, A.; Chen, H.; Kim, H.Y. Multiple Mild Traumatic Brain Injuries Lead to Visual Dysfunction in a Mouse Model. J Neurotrauma 2020, 37, 286–294. [Google Scholar] [CrossRef]
- Keating, C.E.; Browne, K.D.; Duda, J.E.; Cullen, D.K. Neurons in Subcortical Oculomotor Regions Are Vulnerable to Plasma Membrane Damage after Repetitive Diffuse Traumatic Brain Injury in Swine. J. Neurotrauma 2020, 37, 1918–1932. [Google Scholar] [CrossRef]
- Tzekov, R.; Quezada, A.; Gautier, M.; Biggins, D.; Frances, C.; Mouzon, B.; Jamison, J.; Mullan, M.; Crawford, F. Repetitive mild traumatic brain injury causes optic nerve and retinal damage in a mouse model. J. Neuropathol. Exp. Neurol. 2014, 73, 345–361. [Google Scholar] [CrossRef]
- Bernardo-Colon, A.; Vest, V.; Cooper, M.L.; Naguib, S.A.; Calkins, D.J.; Rex, T.S. Progression and Pathology of Traumatic Optic Neuropathy From Repeated Primary Blast Exposure. Front. Neurosci. 2019, 13, 719. [Google Scholar] [CrossRef]
- Evanson, N.K.; Guilhaume-Correa, F.; Herman, J.P.; Goodman, M.D. Optic tract injury after closed head traumatic brain injury in mice: A model of indirect traumatic optic neuropathy. PLoS ONE 2018, 13, e0197346. [Google Scholar] [CrossRef] [PubMed]
- Tao, W.; Dvoriantchikova, G.; Tse, B.C.; Pappas, S.; Chou, T.H.; Tapia, M.; Porciatti, V.; Ivanov, D.; Tse, D.T.; Pelaez, D. A Novel Mouse Model of Traumatic Optic Neuropathy Using External Ultrasound Energy to Achieve Focal, Indirect Optic Nerve Injury. Sci. Rep. 2017, 7, 11779. [Google Scholar] [CrossRef]
- Guilhaume-Correa, F.; Cansler, S.M.; Shalosky, E.M.; Goodman, M.D.; Evanson, N.K. Greater neurodegeneration and behavioral deficits after single closed head traumatic brain injury in adolescent versus adult male mice. J. Neurosci. Res. 2020, 98, 557–570. [Google Scholar] [CrossRef] [PubMed]
- Fan, P.; Yamauchi, T.; Noble, L.J.; Ferriero, D.M. Age-dependent differences in glutathione peroxidase activity after traumatic brain injury. J. Neurotrauma 2003, 20, 437–445. [Google Scholar] [CrossRef] [PubMed]
- McColl, T.J.; Brady, R.D.; Shultz, S.R.; Lovick, L.; Webster, K.M.; Sun, M.; McDonald, S.J.; O’Brien, T.J.; Semple, B.D. Mild Traumatic Brain Injury in Adolescent Mice Alters Skull Bone Properties to Influence a Subsequent Brain Impact at Adulthood: A Pilot Study. Front. Neurol. 2018, 9, 372. [Google Scholar] [CrossRef]
- Prins, M.L.; Alexander, D.; Giza, C.C.; Hovda, D.A. Repeated mild traumatic brain injury: Mechanisms of cerebral vulnerability. J. Neurotrauma 2013, 30, 30–38. [Google Scholar] [CrossRef]
- Prins, M.L.; Lee, S.M.; Cheng, C.L.; Becker, D.P.; Hovda, D.A. Fluid percussion brain injury in the developing and adult rat: A comparative study of mortality, morphology, intracranial pressure and mean arterial blood pressure. Brain Res. Dev. Brain Res. 1996, 95, 272–282. [Google Scholar] [CrossRef]
- Sullivan, P.G.; Keller, J.N.; Mattson, M.P.; Scheff, S.W. Traumatic brain injury alters synaptic homeostasis: Implications for impaired mitochondrial and transport function. J. Neurotrauma 1998, 15, 789–798. [Google Scholar] [CrossRef]
- Thomas, S.; Prins, M.L.; Samii, M.; Hovda, D.A. Cerebral metabolic response to traumatic brain injury sustained early in development: A 2-deoxy-D-glucose autoradiographic study. J. Neurotrauma 2000, 17, 649–665. [Google Scholar] [CrossRef]
- Xu, L.; Ryu, J.; Nguyen, J.V.; Arena, J.; Rha, E.; Vranis, P.; Hitt, D.; Marsh-Armstrong, N.; Koliatsos, V.E. Evidence for accelerated tauopathy in the retina of transgenic P301S tau mice exposed to repetitive mild traumatic brain injury. Exp. Neurol. 2015, 273, 168–176. [Google Scholar] [CrossRef]
- Xu, L.; Nguyen, J.V.; Lehar, M.; Menon, A.; Rha, E.; Arena, J.; Ryu, J.; Marsh-Armstrong, N.; Marmarou, C.R.; Koliatsos, V.E. Repetitive mild traumatic brain injury with impact acceleration in the mouse: Multifocal axonopathy, neuroinflammation, and neurodegeneration in the visual system. Exp. Neurol. 2016, 275, 436–449. [Google Scholar] [CrossRef] [PubMed]
- Singman, E.L.; Daphalapurkar, N.; White, H.; Nguyen, T.D.; Panghat, L.; Chang, J.; McCulley, T. Indirect traumatic optic neuropathy. Mil. Med. Res. 2016, 3, 2. [Google Scholar] [CrossRef]
- Hu, Y.; Park, K.K.; Yang, L.; Wei, X.; Yang, Q.; Cho, K.S.; Thielen, P.; Lee, A.H.; Cartoni, R.; Glimcher, L.H.; et al. Differential effects of unfolded protein response pathways on axon injury-induced death of retinal ganglion cells. Neuron 2012, 73, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Semple, B.D.; Blomgren, K.; Gimlin, K.; Ferriero, D.M.; Noble-Haeusslein, L.J. Brain development in rodents and humans: Identifying benchmarks of maturation and vulnerability to injury across species. Prog. Neurobiol. 2013, 106, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.H.; Gustafson, J.; Gangidine, M.; Stepien, D.; Schuster, R.; Pritts, T.A.; Goodman, M.D.; Remick, D.G.; Lentsch, A.B. A murine model of mild traumatic brain injury exhibiting cognitive and motor deficits. J. Surg. Res. 2013, 184, 981–988. [Google Scholar] [CrossRef]
- Thaung, C.; Arnold, K.; Jackson, I.J.; Coffey, P.J. Presence of visual head tracking differentiates normal sighted from retinal degenerate mice. Neurosci. Lett. 2002, 325, 21–24. [Google Scholar] [CrossRef]
- Peirce, J.W. PsychoPy-Psychophysics software in Python. J. Neurosci. Methods 2007, 162, 8–13. [Google Scholar] [CrossRef]
- Heitz, F.D.; Erb, M.; Anklin, C.; Robay, D.; Pernet, V.; Gueven, N. Idebenone protects against retinal damage and loss of vision in a mouse model of Leber’s hereditary optic neuropathy. PLoS ONE 2012, 7, e45182. [Google Scholar] [CrossRef]
- Abdeljalil, J.; Hamid, M.; Abdel-Mouttalib, O.; Stephane, R.; Raymond, R.; Johan, A.; Jose, S.; Pierre, C.; Serge, P. The optomotor response: A robust first-line visual screening method for mice. Vis. Res. 2005, 45, 1439–1446. [Google Scholar] [CrossRef] [PubMed]
- Ullmann, J.F.; Moore, B.A.; Temple, S.E.; Fernandez-Juricic, E.; Collin, S.P. The retinal wholemount technique: A window to understanding the brain and behaviour. Brain Behav. Evol. 2012, 79, 26–44. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Schmued, L.C.; Hopkins, K.J. Fluoro-Jade B: A high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 2000, 874, 123–130. [Google Scholar] [CrossRef]
- Namdar, I.; Feldman, R.; Glazer, S.; Meningher, I.; Shlobin, N.A.; Rubovitch, V.; Bikovski, L.; Been, E.; Pick, C.G. Motor Effects of Minimal Traumatic Brain Injury in Mice. J. Mol. Neurosci. 2020, 70, 365–377. [Google Scholar] [CrossRef]
- Willie, J.T.; Lim, M.M.; Bennett, R.E.; Azarion, A.A.; Schwetye, K.E.; Brody, D.L. Controlled cortical impact traumatic brain injury acutely disrupts wakefulness and extracellular orexin dynamics as determined by intracerebral microdialysis in mice. J. Neurotrauma 2012, 29, 1908–1921. [Google Scholar] [CrossRef]
- Nadal-Nicolas, F.M.; Jimenez-Lopez, M.; Sobrado-Calvo, P.; Nieto-Lopez, L.; Canovas-Martinez, I.; Salinas-Navarro, M.; Vidal-Sanz, M.; Agudo, M. Brn3a as a marker of retinal ganglion cells: Qualitative and quantitative time course studies in naive and optic nerve-injured retinas. Investig. Ophthalmol. Vis. Sci. 2009, 50, 3860–3868. [Google Scholar] [CrossRef]
- Mead, B.; Thompson, A.; Scheven, B.A.; Logan, A.; Berry, M.; Leadbeater, W. Comparative evaluation of methods for estimating retinal ganglion cell loss in retinal sections and wholemounts. PLoS ONE 2014, 9, e110612. [Google Scholar] [CrossRef]
- Quina, L.A.; Pak, W.; Lanier, J.; Banwait, P.; Gratwick, K.; Liu, Y.; Velasquez, T.; O’Leary, D.D.; Goulding, M.; Turner, E.E. Brn3a-expressing retinal ganglion cells project specifically to thalamocortical and collicular visual pathways. J. Neurosci. 2005, 25, 11595–11604. [Google Scholar] [CrossRef]
- Kim, Y.S.; Joh, T.H. Microglia, major player in the brain inflammation: Their roles in the pathogenesis of Parkinson’s disease. Exp. Mol. Med. 2006, 38, 333–347. [Google Scholar] [CrossRef]
- Kumar, A.; Loane, D.J. Neuroinflammation after traumatic brain injury: Opportunities for therapeutic intervention. Brain Behav. Immun. 2012, 26, 1191–1201. [Google Scholar] [CrossRef]
- Glimcher, P.W. Eye Movement, Control of (Oculomotor Control). In International Encyclopedia of the Social & Behavioral Sciences; Smelser, N.J., Baltes, P.B., Eds.; Pergamon: Oxford, UK, 2001; pp. 5205–5208. [Google Scholar]
- Perri, E.R.; Thomas, C.J.; Parakh, S.; Spencer, D.M.; Atkin, J.D. The Unfolded Protein Response and the Role of Protein Disulfide Isomerase in Neurodegeneration. Front. Cell Dev. Biol. 2015, 3, 80. [Google Scholar] [CrossRef] [PubMed]
- Kranz, P.; Neumann, F.; Wolf, A.; Classen, F.; Pompsch, M.; Ocklenburg, T.; Baumann, J.; Janke, K.; Baumann, M.; Goepelt, K.; et al. PDI is an essential redox-sensitive activator of PERK during the unfolded protein response (UPR). Cell Death Dis. 2017, 8, e2986. [Google Scholar] [CrossRef]
- Zhang, S.X.; Sanders, E.; Fliesler, S.J.; Wang, J.J. Endoplasmic reticulum stress and the unfolded protein responses in retinal degeneration. Exp. Eye Res. 2014, 125, 30–40. [Google Scholar] [CrossRef]
- Tavender, T.J.; Bulleid, N.J. Molecular mechanisms regulating oxidative activity of the Ero1 family in the endoplasmic reticulum. Antioxid. Redox Signal. 2010, 13, 1177–1187. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.S.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef]
- Eletto, D.; Chevet, E.; Argon, Y.; Appenzeller-Herzog, C. Redox controls UPR to control redox. J. Cell Sci. 2014, 127, 3649–3658. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, L.; Zhou, L.; Lei, Y.; Zhang, Y.; Huang, C. Redox signaling and unfolded protein response coordinate cell fate decisions under ER stress. Redox Biol. 2019, 25, 101047. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hamm, R.J.; Povlishock, J.T. Traumatic axonal injury in the optic nerve: Evidence for axonal swelling, disconnection, dieback, and reorganization. J. Neurotrauma 2011, 28, 1185–1198. [Google Scholar] [CrossRef]
- Ohlsson, M.; Bellander, B.M.; Langmoen, I.A.; Svensson, M. Complement activation following optic nerve crush in the adult rat. J. Neurotrauma 2003, 20, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Fitch, M.T.; Silver, J. Glial cells, inflammation, and CNS trauma: Modulation of the inflammatory environment after injury can lead to long-distance regeneration beyond the glial scar. In CNS Regeneration, 2nd ed.; Kordower, J.H., Tuszynski, M.H., Eds.; Academic Press: San Diego, CA, USA, 2008; pp. 59–94. [Google Scholar]
- Gentleman, S.M.; Leclercq, P.D.; Moyes, L.; Graham, D.I.; Smith, C.; Griffin, W.S.; Nicoll, J.A. Long-term intracerebral inflammatory response after traumatic brain injury. Forensic Sci. Int. 2004, 146, 97–104. [Google Scholar] [CrossRef]
- Crunelli, V.; Kelly, J.S.; Leresche, N.; Pirchio, M. The ventral and dorsal lateral geniculate nucleus of the rat: Intracellular recordings in vitro. J. Physiol. 1987, 384, 587–601. [Google Scholar] [CrossRef] [PubMed]
- Reid, R.C.; Usrey, W.M. Chapter 26—Vision. In Fundamental Neuroscience, 4th ed.; Squire, L.R., Berg, D., Bloom, F.E., du Lac, S., Ghosh, A., Spitzer, N.C., Eds.; Academic Press: San Diego, CA, USA, 2013; pp. 577–595. [Google Scholar]
- Cui, Q.; Ren, C.; Sollars, P.J.; Pickard, G.E.; So, K.F. The injury resistant ability of melanopsin-expressing intrinsically photosensitive retinal ganglion cells. Neuroscience 2015, 284, 845–853. [Google Scholar] [CrossRef] [PubMed]
- VanderWall, K.B.; Lu, B.; Wang, S.; Meyer, J.S. Differential Susceptibility of Rat Retinal Ganglion Cells Following Optic Nerve Crush. bioRxiv 2018, 429282. [Google Scholar] [CrossRef]
- Karve, I.P.; Taylor, J.M.; Crack, P.J. The contribution of astrocytes and microglia to traumatic brain injury. Br. J. Pharm. 2016, 173, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Cekanaviciute, E.; Fathali, N.; Doyle, K.P.; Williams, A.M.; Han, J.; Buckwalter, M.S. Astrocytic transforming growth factor-beta signaling reduces subacute neuroinflammation after stroke in mice. Glia 2014, 62, 1227–1240. [Google Scholar] [CrossRef]
- Lööv, C.; Hillered, L.; Ebendal, T.; Erlandsson, A. Engulfing astrocytes protect neurons from contact-induced apoptosis following injury. PLoS ONE 2012, 7, e33090. [Google Scholar] [CrossRef]
- Myer, D.J.; Gurkoff, G.G.; Lee, S.M.; Hovda, D.A.; Sofroniew, M.V. Essential protective roles of reactive astrocytes in traumatic brain injury. Brain 2006, 129, 2761–2772. [Google Scholar] [CrossRef]
- Harrington, M.E. The ventral lateral geniculate nucleus and the intergeniculate leaflet: Interrelated structures in the visual and circadian systems. Neurosci. Biobehav. Rev. 1997, 21, 705–727. [Google Scholar] [CrossRef]
- Llompart-Pou, J.A.; Pérez, G.; Raurich, J.M.; Riesco, M.; Brell, M.; Ibáñez, J.; Pérez-Bárcena, J.; Abadal, J.M.; Homar, J.; Burguera, B. Loss of cortisol circadian rhythm in patients with traumatic brain injury: A microdialysis evaluation. Neurocrit. Care 2010, 13, 211–216. [Google Scholar] [CrossRef]
- Shekleton, J.A.; Parcell, D.L.; Redman, J.R.; Phipps-Nelson, J.; Ponsford, J.L.; Rajaratnam, S.M. Sleep disturbance and melatonin levels following traumatic brain injury. Neurology 2010, 74, 1732–1738. [Google Scholar] [CrossRef] [PubMed]
- Hattar, S.; Kumar, M.; Park, A.; Tong, P.; Tung, J.; Yau, K.W.; Berson, D.M. Central projections of melanopsin-expressing retinal ganglion cells in the mouse. J. Comp. Neurol. 2006, 497, 326–349. [Google Scholar] [CrossRef] [PubMed]
- Bunt, S.M.; Horder, T.J. Evidence for an orderly arrangement of optic axons within the optic nerves of the major nonmammalian vertebrate classes. J. Comp. Neurol. 1983, 213, 94–114. [Google Scholar] [CrossRef]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hetzer, S.M.; Guilhaume-Correa, F.; Day, D.; Bedolla, A.; Evanson, N.K. Traumatic Optic Neuropathy Is Associated with Visual Impairment, Neurodegeneration, and Endoplasmic Reticulum Stress in Adolescent Mice. Cells 2021, 10, 996. https://doi.org/10.3390/cells10050996
Hetzer SM, Guilhaume-Correa F, Day D, Bedolla A, Evanson NK. Traumatic Optic Neuropathy Is Associated with Visual Impairment, Neurodegeneration, and Endoplasmic Reticulum Stress in Adolescent Mice. Cells. 2021; 10(5):996. https://doi.org/10.3390/cells10050996
Chicago/Turabian StyleHetzer, Shelby M., Fernanda Guilhaume-Correa, Dylan Day, Alicia Bedolla, and Nathan K. Evanson. 2021. "Traumatic Optic Neuropathy Is Associated with Visual Impairment, Neurodegeneration, and Endoplasmic Reticulum Stress in Adolescent Mice" Cells 10, no. 5: 996. https://doi.org/10.3390/cells10050996
APA StyleHetzer, S. M., Guilhaume-Correa, F., Day, D., Bedolla, A., & Evanson, N. K. (2021). Traumatic Optic Neuropathy Is Associated with Visual Impairment, Neurodegeneration, and Endoplasmic Reticulum Stress in Adolescent Mice. Cells, 10(5), 996. https://doi.org/10.3390/cells10050996