
Selection, Expansion, and Unique Pretreatment of Allogeneic Human Natural Killer Cells with Anti-CD38 Monoclonal Antibody for Efficient Multiple Myeloma Treatment

 , , , , ,
, , , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Isolation of NK Cells from Peripheral Blood and Abnormal Plasmatic Cells from Bone Marrow Aspirates
2.3. Cell Culture, Pretreatment with Daratumumab, and Induction of Interferon Gamma (IFN-γ) Expression by Increasing Concentrations of Platelet-Derived Growth Factor (PDGF-DD)
2.4. Phenotypic and Functional Analyses of Expanded PBNK Cells
2.5. Lentiviral Vectors
2.6. Cytotoxic Activity of PBNK Cells and Daratumumab-Mediated Cytotoxicity
2.7. Quantification of IFN- γ and PDGF-DD
2.8. Migration Assay
2.9. In Vivo Experiments
2.10. Statistical Analysis
3. Results
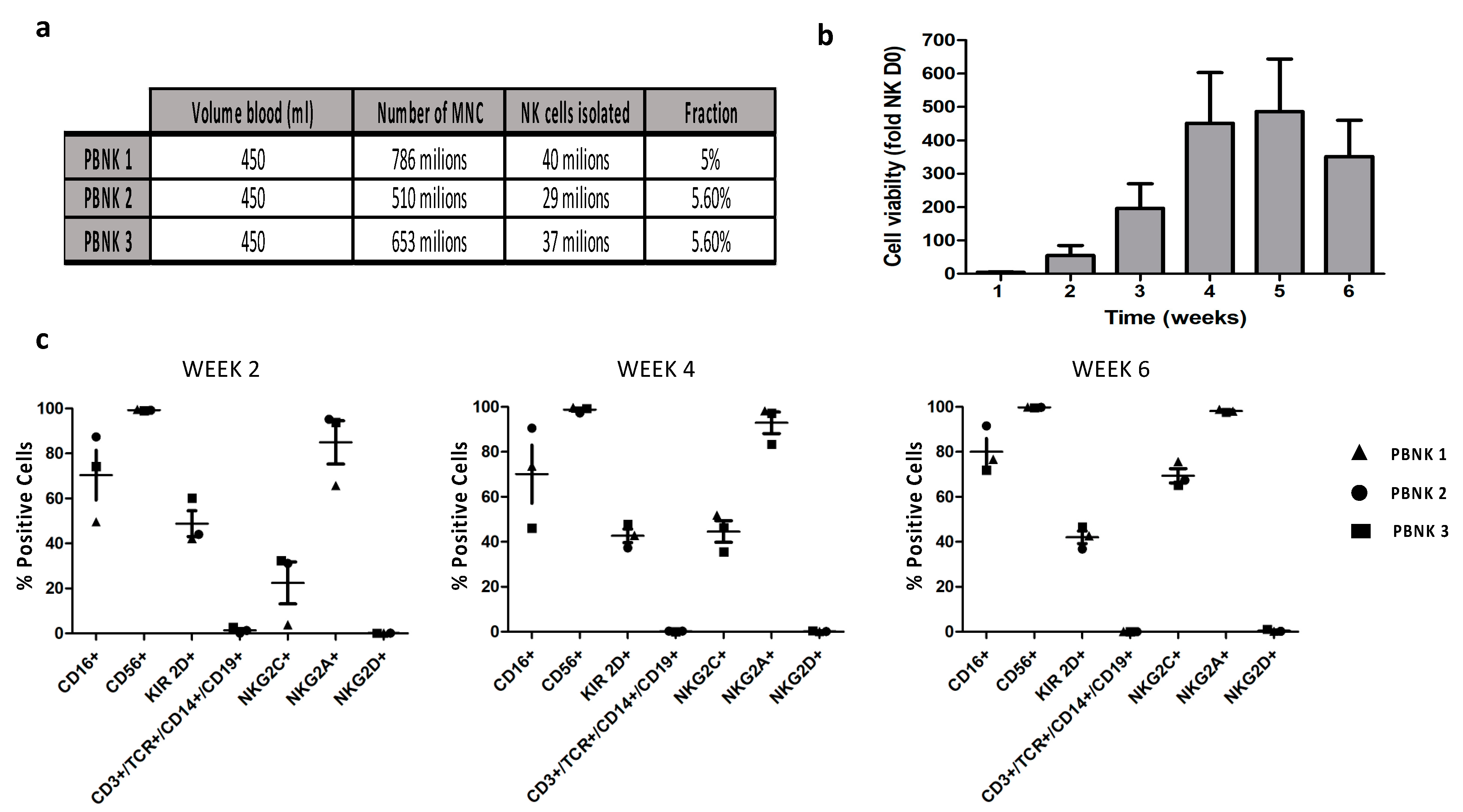
3.1. Isolation and Phenotypic Characterization of Expanded PBNK Cells
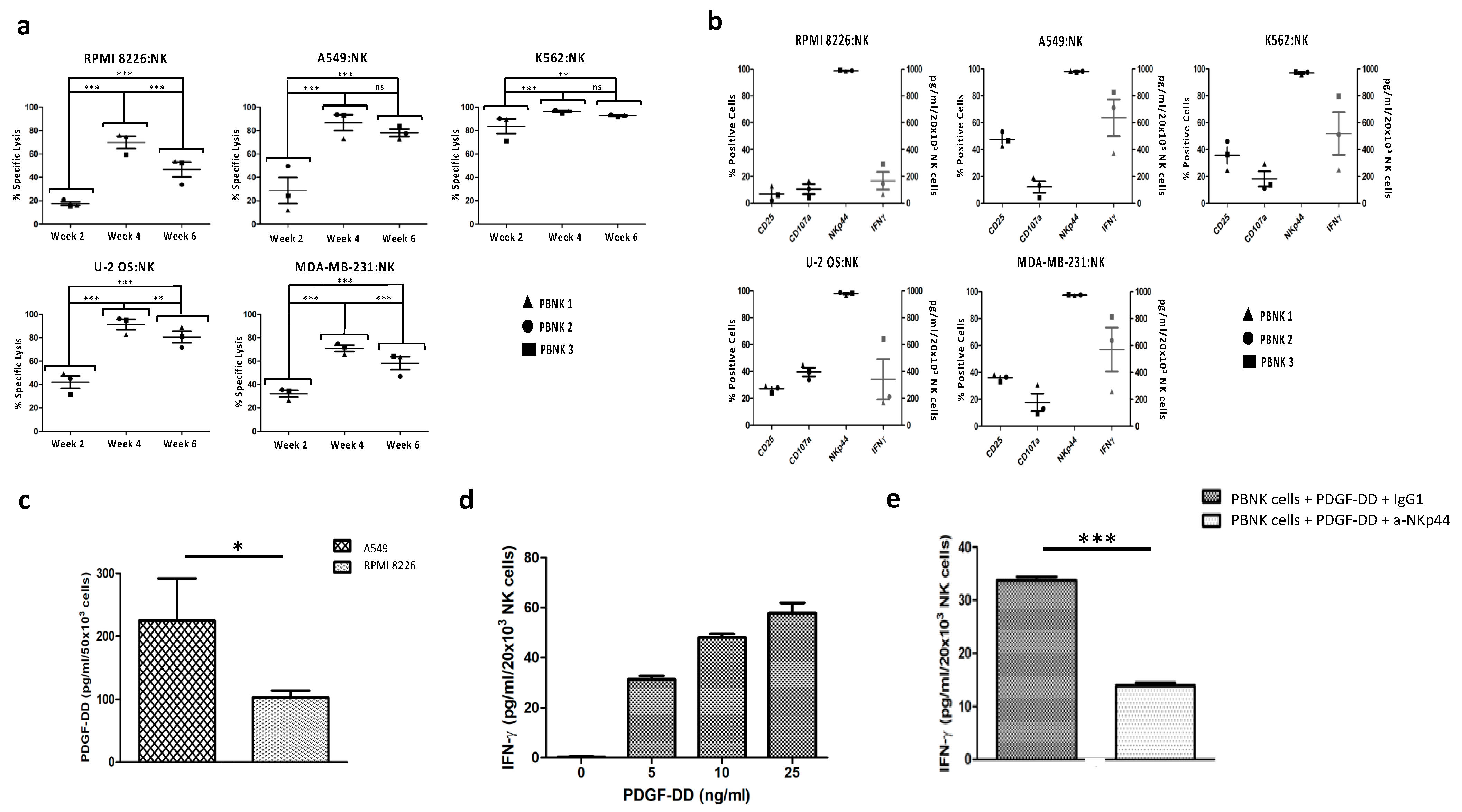
3.2. Functional Characterization of Expanded PBNK Cells
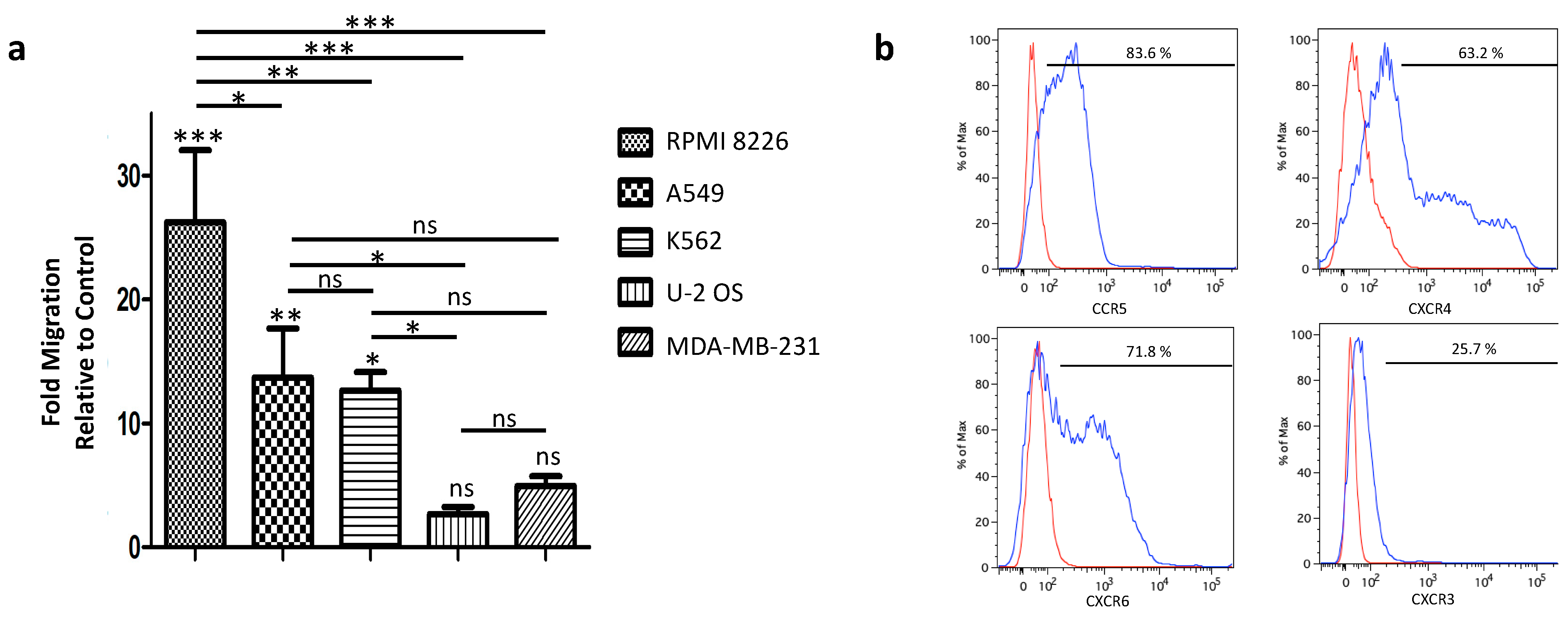
3.3. Tumor-Homing of Expanded PBNK Cells
3.4. Combinatorial Treatment of Expanded PBNK Cells with Monoclonal Antibodies
3.5. Efficacious Combinatorial Therapy with Expanded PBNK Cells and Monoclonal Antibody in Primary MM Cells and an MM Mouse Model
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric Antigen Receptor-Modified T Cells in Chronic Lymphoid Leukemia. N. Engl. J. Med. 2016, 374, 725–733. [Google Scholar] [CrossRef]
- Porter, D.L.; Hwang, W.-T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V.; et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015, 7, 303ra139. [Google Scholar] [CrossRef]
- Davila, M.L.; Riviere, I.; Wang, X.; Bartido, S.; Park, J.; Curran, K.; Chung, S.S.; Stefanski, J.; Borquez-Ojeda, O.; Olszewska, M.; et al. Efficacy and Toxicity Management of 19-28z CAR T Cell Therapy in B Cell Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2014, 6, 224ra25. [Google Scholar] [CrossRef]
- Richman, S.A.; Nunez-Cruz, S.; Moghimi, B.; Li, L.Z.; Gershenson, Z.T.; Mourelatos, Z.; Barrett, D.M.; Grupp, S.A.; Milone, M.C. High-Affinity GD2-Specific CAR T Cells Induce Fatal Encephalitis in a Preclinical Neuroblastoma Model. Cancer Immunol. Res. 2018, 6, 36–46. [Google Scholar] [CrossRef]
- Sotillo, E.; Barrett, D.M.; Black, K.L.; Bagashev, A.A.; Oldridge, D.; Wu, G.; Sussman, R.T.; LaNauze, C.; Ruella, M.; Gazzara, M.R.; et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov. 2015, 5, 1282–1295. [Google Scholar] [CrossRef]
- Ichiki, Y.; Bowlus, C.L.; Shimoda, S.; Ishibashi, H.; Vierling, J.M.; Gershwin, M.E. T cell immunity and graft-versus-host disease (GVHD). Autoimmun. Rev. 2006, 5, 1–9. [Google Scholar] [CrossRef]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Kerbauy, L.N.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef]
- Morrissey, M.A.; Williamson, A.P.; Steinbach, A.M.; Roberts, E.W.; Kern, N.; Headley, M.B.; Vale, R.D. Chimeric antigen receptors that trigger phagocytosis. eLife 2018, 7, e36688. [Google Scholar] [CrossRef] [PubMed]
- Tel, J.; Anguille, S.; Waterborg, C.E.; Smits, E.L.; Figdor, C.G.; De Vries, I.J.M. Tumoricidal activity of human dendritic cells. Trends Immunol. 2014, 35, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Pegram, H.J.; Andrews, D.M.; Smyth, M.J.; Darcy, P.K.; Kershaw, M.H. Activating and inhibitory receptors of natural killer cells. Immunol. Cell Biol. 2010, 89, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Motais, B.; Charvátová, S.; Hrdinka, M.; Šimíček, M.; Jelínek, T.; Ševčíková, T.; Kořístek, Z.; Hájek, R.; Bagó, J.R. A Bird’s-Eye View of Cell Sources for Cell-Based Therapies in Blood Cancers. Cancers 2020, 12, 1333. [Google Scholar] [CrossRef] [PubMed]
- Barrow, A.D.; Edeling, M.A.; Trifonov, V.; Luo, J.; Goyal, P.; Bohl, B.; Bando, J.K.; Kim, A.H.; Walker, J.; Andahazy, M.; et al. Natural Killer Cells Control Tumor Growth by Sensing a Growth Factor. Cell 2018, 172, 534–548.e19. [Google Scholar] [CrossRef] [PubMed]
- Rubnitz, J.E.; Inaba, H.; Ribeiro, R.C.; Pounds, S.; Rooney, B.; Bell, T.; Pui, C.-H.; Leung, W. NKAML: A Pilot Study to Determine the Safety and Feasibility of Haploidentical Natural Killer Cell Transplantation in Childhood Acute Myeloid Leukemia. J. Clin. Oncol. 2010, 28, 955–959. [Google Scholar] [CrossRef]
- Williams, B.A.; Law, A.D.; Routy, B.; Denhollander, N.; Gupta, V.; Wang, X.-H.; Chaboureau, A.; Viswanathan, S.; Keating, A. A phase I trial of NK-92 cells for refractory hematological malignancies relapsing after autologous hematopoietic cell transplantation shows safety and evidence of efficacy. Oncotarget 2017, 8, 89256–89268. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.; Li, L.; Mccarty, J.; Kaur, I.; Yvon, E.; Shaim, H.; Muftuoglu, M.; Liu, E.; Orlowski, R.Z.; Cooper, L.; et al. Phase I study of cord blood-derived natural killer cells combined with autologous stem cell transplantation in multiple myeloma. Br. J. Haematol. 2017, 177, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Bachanova, V.; McKenna, D.H.; Luo, X.; Defor, T.E.; Cooley, S.; Warlick, E.; Weisdorf, D.J.; Brachya, G.; Peled, T.; Miller, J.S.; et al. First-in-Human Phase I Study of Nicotinamide-Expanded Related Donor Natural Killer Cells for the Treatment of Relapsed/Refractory Non-Hodgkin Lymphoma and Multiple Myeloma. Biol. Blood Marrow Transplant. 2019, 25, S175–S176. [Google Scholar] [CrossRef]
- Shah, N.; Mehta, R.; Li, L.; Mccarty, J.; Kaur, I.; Orlowski, R.Z.; Cooper, L.; Lee, D.A.; Cao, K.; Parmar, S.; et al. Phase II study of ex vivo expanded cord blood natural killer cells for multiple myeloma. J. Clin. Oncol. 2018, 36, 8006. [Google Scholar] [CrossRef]
- Minarik, J.; Pour, L.; Maisnar, V.; Spicka, I.; Jungova, A.; Jelinek, T.; Brozova, L.; Krhovska, P.; Scudla, V.; Hajek, R. Single agent daratumumab in advanced multiple myeloma possesses significant efficacy even in an unselected “real-world” population. Biomed. Pap. 2019, 163, 279–283. [Google Scholar] [CrossRef]
- Alderson, K.L.; Sondel, P.M. Clinical Cancer Therapy by NK Cells via Antibody-Dependent Cell-Mediated Cytotoxicity. J. Biomed. Biotechnol. 2011, 2011, 379123. [Google Scholar] [CrossRef]
- Hatjiharissi, E.; Xu, L.; Santos, D.D.; Hunter, Z.R.; Ciccarelli, B.T.; Verselis, S.; Modica, M.; Cao, Y.; Manning, R.J.; Leleu, X.; et al. Increased natural killer cell expression of CD16, augmented binding and ADCC activity to rituximab among individuals expressing the Fc{gamma}RIIIa-158 V/V and V/F polymorphism. Blood 2007, 110, 2561–2564. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, M.; Canfield, R.; Robbins, M.; Ritchie, D.S.; Trapani, J.; Neeson, P. Targeting Mechanisms for Natural Killer Cell Dysfunction in Patients with Multiple Myeloma. Blood 2015, 126, 4237. [Google Scholar] [CrossRef]
- Veluchamy, J.P.; Delso-Vallejo, M.; Kok, N.; Bohme, F.; Seggewiss-Bernhardt, R.; Van Der Vliet, H.J.; De Gruijl, T.D.; Huppert, V.; Spanholtz, J. Standardized and flexible eight colour flow cytometry panels harmonized between different laboratories to study human NK cell phenotype and function. Sci. Rep. 2017, 7, srep43873. [Google Scholar] [CrossRef] [PubMed]
- Bottcher, S.; Ritgen, M.; Bruggemann, M.; Raff, T.; Luschen, S.; Humpe, A.; Kneba, M.; Pott, C. Flow cytometric assay for determination of FcgammaRIIIA-158 V/F polymorphism. J. Immunol. Methods 2005, 306, 128–136. [Google Scholar] [CrossRef]
- Karimi, M.A.; Lee, E.; Bachmann, M.H.; Salicioni, A.M.; Behrens, E.M.; Kambayashi, T.; Baldwin, C.L. Measuring Cytotoxicity by Bioluminescence Imaging Outperforms the Standard Chromium-51 Release Assay. PLoS ONE 2014, 9, e89357. [Google Scholar] [CrossRef]
- Paul, S.; Lal, G. The Molecular Mechanism of Natural Killer Cells Function and Its Importance in Cancer Immunotherapy. Front. Immunol. 2017, 8, 1124. [Google Scholar] [CrossRef]
- Benevolo, M.; Mottolese, M.; Tremante, E.; Rollo, F.; Diodoro, M.G.; Ercolani, C.; Sperduti, I.; Monaco, E.L.; Cosimelli, M.; Giacomini, P. High expression of HLA-E in colorectal carcinoma is associated with a favorable prognosis. J. Transl. Med. 2011, 9, 184. [Google Scholar] [CrossRef]
- Marín, R.; Ruiz-Cabello, F.; Pedrinaci, S.; Méndez, R.; Jiménez, P.; Geraghty, D.E.; Garrido, F. Analysis of HLA-E expression in human tumors. Immunogenetics 2003, 54, 767–775. [Google Scholar] [CrossRef]
- Felices, M.; Lenvik, A.J.; McElmurry, R.; Chu, S.; Hinderlie, P.; Bendzick, L.; Geller, M.A.; Tolar, J.; Blazar, B.R.; Miller, J.S. Continuous treatment with IL-15 exhausts human NK cells via a metabolic defect. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Brandt, C.S.; Baratin, M.; Yi, E.C.; Kennedy, J.; Gao, Z.; Fox, B.; Haldeman, B.; Ostrander, C.D.; Kaifu, T.; Chabannon, C.; et al. The B7 family member B7-H6 is a tumor cell ligand for the activating natural killer cell receptor NKp30 in humans. J. Exp. Med. 2009, 206, 1495–1503. [Google Scholar] [CrossRef]
- Ishigami, S.; Natsugoe, S.; Tokuda, K.; Nakajo, A.; Che, X.; Iwashige, H.; Aridome, K.; Hokita, S.; Aikou, T. Prognostic value of intratumoral natural killer cells in gastric carcinoma. Cancer 2000, 88, 577–583. [Google Scholar] [CrossRef]
- Coca, S.; Perez-Piqueras, J.; Martinez, D.; Colmenarejo, A.; Saez, M.A.; Vallejo, C.; Martos, J.A.; Moreno, M. The prognostic significance of intratumoral natural killer cells in patients with colorectal carcinoma. Cancer 1997, 79, 2320–2328. [Google Scholar] [CrossRef]
- Villegas, F.R.; Coca, S.; Villarrubia, V.G.; Jiménez, R.; Chillón, M.J.; Jareño, J.; Zuil, M.; Callol, L. Prognostic significance of tumor infiltrating natural killer cells subset CD57 in patients with squamous cell lung cancer. Lung Cancer 2002, 35, 23–28. [Google Scholar] [CrossRef]
- Wendel, M.; Galani, I.E.; Suri-Payer, E.; Cerwenka, A. Natural killer cell accumulation in tumors is dependent on IFN-gamma and CXCR3 ligands. Cancer Res. 2008, 68, 8437–8445. [Google Scholar] [CrossRef] [PubMed]
- Lima, M.; Leander, M.; Santos, M.; Santos, A.H.; Lau, C.; Queirós, M.L.; Gonçalves, M.; Fonseca, S.; Moura, J.; Teixeira, M.D.A.; et al. Chemokine Receptor Expression on Normal Blood CD56+NK-Cells Elucidates Cell Partners That Comigrate during the Innate and Adaptive Immune Responses and Identifies a Transitional NK-Cell Population. J. Immunol. Res. 2015, 2015, 839684. [Google Scholar] [CrossRef]
- Freire-De-Lima, L.; Nardy, A.F.F.R.; Ramos-Junior, E.S.; Conde, L.; Lemos, J.S.; Da Fonseca, L.M.; Lima, J.E.; Maiolino, A.; Morrot, A. Multiple Myeloma Cells Express Key Immunoregulatory Cytokines and Modulate the Monocyte Migratory Response. Front. Med. 2017, 4, 92. [Google Scholar] [CrossRef]
- Zahavi, D.; Weiner, L. Monoclonal Antibodies in Cancer Therapy. Antibodies 2020, 9, 34. [Google Scholar] [CrossRef]
- Hubert, P.; Amigorena, S. Antibody-dependent cell cytotoxicity in monoclonal antibody-mediated tumor immunotherapy. OncoImmunology 2012, 1, 103–105. [Google Scholar] [CrossRef] [PubMed]
- Koene, H.R.; Kleijer, M.; Algra, J.; Roos, D.; Borne, A.E.V.D.; De Haas, M. Fc gammaRIIIa-158V/F polymorphism influences the binding of IgG by natural killer cell Fc gammaRIIIa, independently of the Fc gammaRIIIa-48L/R/H phenotype. Blood 1997, 90, 1109–1114. [Google Scholar] [CrossRef] [PubMed]
- Niwa, R.; Hatanaka, S.; Shoji-Hosaka, E.; Sakurada, M.; Kobayashi, Y.; Uehara, A.; Yokoi, H.; Nakamura, K.; Shitara, K. Enhancement of the antibody-dependent cellular cytotoxicity of low-fucose IgG1 Is independent of FcgammaRIIIa functional polymorphism. Clin. Cancer Res. 2004, 10, 6248–6255. [Google Scholar] [CrossRef]
- De Weers, M.; Tai, Y.-T.; Van Der Veer, M.S.; Bakker, J.M.; Vink, T.; Jacobs, D.C.H.; Oomen, L.A.; Peipp, M.; Valerius, T.; Slootstra, J.W.; et al. Daratumumab, a Novel Therapeutic Human CD38 Monoclonal Antibody, Induces Killing of Multiple Myeloma and Other Hematological Tumors. J. Immunol. 2010, 186, 1840–1848. [Google Scholar] [CrossRef]
- Van Der Veer, M.S.; De Weers, M.; Van Kessel, B.; Bakker, J.M.; Wittebol, S.I.; Parren, P.W.H.; Lokhorst, H.M.; Mutis, T. The therapeutic human CD38 antibody daratumumab improves the anti-myeloma effect of newly emerging multi-drug therapies. Blood Cancer J. 2011, 1, e41. [Google Scholar] [CrossRef]
- Overdijk, M.B.; Verploegen, S.; Bögels, M.; Van Egmond, M.; Van Bueren, J.J.L.; Mutis, T.; Groen, R.W.J.; Breij, E.; Martens, A.C.M.; Bleeker, W.K.; et al. Antibody-mediated phagocytosis contributes to the anti-tumor activity of the therapeutic antibody daratumumab in lymphoma and multiple myeloma. mAbs 2015, 7, 311–320. [Google Scholar] [CrossRef]
- Overdijk, M.B.; Jansen, J.H.; Nederend, M.; Lammerts van Bueren, J.J.; Groen, R.W.; Parren, P.W.; Leusen, J.H.; Boross, P. The therapeutic CD38 monoclonal antibody daratumumab induces programmed cell death via fcgamma receptor-mediated cross-linking. J. Immunol. 2016, 197, 807–813. [Google Scholar] [CrossRef]
- Casneuf, T.; Xu, X.S.; Adams, H.C.; Axel, A.E.; Chiu, C.; Khan, I.; Ahmadi, T.; Yan, X.; Lonial, S.; Plesner, T.; et al. Effects of daratumumab on natural killer cells and impact on clinical outcomes in relapsed or refractory multiple myeloma. Blood Adv. 2017, 1, 2105–2114. [Google Scholar] [CrossRef] [PubMed]
- Southam, C.M.; Brunschwig, A.; Levin, A.G.; Dizon, Q.S. Effect of leukocytes on transplantability of human cancer. Cancer 1966, 19, 1743–1753. [Google Scholar] [CrossRef]
- Weiden, P.L.; Flournoy, N.; Thomas, E.D.; Prentice, R.; Fefer, A.; Buckner, C.D.; Storb, R. Antileukemic Effect of Graft-versus-Host Disease in Human Recipients of Allogeneic-Marrow Grafts. N. Engl. J. Med. 1979, 300, 1068–1073. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Packard, B.S.; Aebersold, P.M.; Solomon, D.; Topalian, S.L.; Toy, S.T.; Simon, P.; Lotze, M.T.; Yang, J.C.-H.; Seipp, C.A.; et al. Use of Tumor-Infiltrating Lymphocytes and Interleukin-2 in the Immunotherapy of Patients with Metastatic Melanoma. N. Engl. J. Med. 1988, 319, 1676–1680. [Google Scholar] [CrossRef]
- Mikucki, M.E.; Fisher, D.T.; Matsuzaki, J.; Skitzki, J.J.; Gaulin, N.B.; Muhitch, J.B.; Ku, A.W.; Frelinger, J.G.; Odunsi, K.; Gajewski, T.F.; et al. Non-redundant requirement for CXCR3 signalling during tumoricidal T-cell trafficking across tumour vascular checkpoints. Nat. Commun. 2015, 6, 7458. [Google Scholar] [CrossRef]
- Garrido, F.; Aptsiauri, N.; Doorduijn, E.M.; Lora, A.M.G.; Van Hall, T. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr. Opin. Immunol. 2016, 39, 44–51. [Google Scholar] [CrossRef]
- Jena, B.; Dotti, G.; Cooper, L.J.N. Redirecting T-cell specificity by introducing a tumor-specific chimeric antigen receptor. Blood 2010, 116, 1035–1044. [Google Scholar] [CrossRef]
- Herrera, L.; Santos, S.; Vesga, M.A.; Anguita, J.; Martin-Ruiz, I.; Carrascosa, T.; Juan, M.; Eguizabal, C. Adult peripheral blood and umbilical cord blood NK cells are good sources for effective CAR therapy against CD19 positive leukemic cells. Sci. Rep. 2019, 9, 18729. [Google Scholar] [CrossRef]
- Masuyama, J.-I.; Murakami, T.; Iwamoto, S.; Fujita, S. Ex vivo expansion of natural killer cells from human peripheral blood mononuclear cells co-stimulated with anti-CD3 and anti-CD52 monoclonal antibodies. Cytotherapy 2016, 18, 80–90. [Google Scholar] [CrossRef]
- Nianias, A.; Themeli, M. Induced pluripotent stem cell (iPSC)-derived lymphocytes for adoptive cell immunotherapy: Recent advances and challenges. Curr. Hematol. Malig. Rep. 2019, 14, 261–268. [Google Scholar] [CrossRef]
- Herrera, L.; Salcedo, J.M.; Santos, S.; Vesga, M. Ángel; Borrego, F.; Eguizabal, C. OP9 Feeder Cells Are Superior to M2-10B4 Cells for the Generation of Mature and Functional Natural Killer Cells from Umbilical Cord Hematopoietic Progenitors. Front. Immunol. 2017, 8, 755. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, S.; Kusakawa, S.; Kuroda, T.; Miura, T.; Tano, K.; Takada, N.; Matsuyama, S.; Matsuyama, A.; Nasu, M.; Umezawa, A.; et al. Tumorigenicity-associated characteristics of human iPS cell lines. PLoS ONE 2018, 13, e0205022. [Google Scholar] [CrossRef] [PubMed]
- Koehl, U.; Esser, R.; Zimmermann, S.; Tonn, T.; Kotchetkov, R.; Bartling, T.; Sörensen, J.; Grüttner, H.-P.; Bader, P.; Seifried, E.; et al. Ex vivo Expansion of Highly Purified NK Cells for Immunotherapy after Haploidentical Stem Cell Transplantation in Children. Klinische Pädiatrie 2005, 217, 345–350. [Google Scholar] [CrossRef]
- Shi, J.; Tricot, G.; Szmania, S.; Rosen, N.; Garg, T.K.; Malaviarachchi, P.A.; Moreno, A.; Dupont, B.; Hsu, K.C.; Baxter-Lowe, L.A.; et al. Infusion of haplo-identical killer immunoglobulin-like receptor ligand mismatched NK cells for relapsed myeloma in the setting of autologous stem cell transplantation. Br. J. Haematol. 2008, 143, 641–653. [Google Scholar] [CrossRef]
- Kweon, S.; Phan, M.-T.T.; Chun, S.; Yu, H.; Kim, J.; Kim, S.; Lee, J.; Ali, A.K.; Lee, S.-H.; Kim, S.-K.; et al. Expansion of Human NK Cells Using K562 Cells Expressing OX40 Ligand and Short Exposure to IL-21. Front. Immunol. 2019, 10, 879. [Google Scholar] [CrossRef]
- Koehl, U.; Brehm, C.; Huenecke, S.; Zimmermann, S.-Y.; Kloess, S.; Bremm, M.; Ullrich, E.; Soerensen, J.; Quaiser, A.; Erben, S.; et al. Clinical Grade Purification and Expansion of NK Cell Products for an Optimized Manufacturing Protocol. Front. Oncol. 2013, 3, 118. [Google Scholar] [CrossRef]
- Iliopoulou, E.G.; Kountourakis, P.; Karamouzis, M.V.; Doufexis, D.; Ardavanis, A.; Baxevanis, C.N.; Rigatos, G.; Papamichail, M.; Perez, S.A. A phase I trial of adoptive transfer of allogeneic natural killer cells in patients with advanced non-small cell lung cancer. Cancer Immunol. Immunother. 2010, 59, 1781–1789. [Google Scholar] [CrossRef]
- Carlsten, M.; Childs, R.W. Genetic Manipulation of NK Cells for Cancer Immunotherapy: Techniques and Clinical Implications. Front. Immunol. 2015, 6, 266. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef]
- Kyle, R.A.; Rajkumar, S.V. Multiple myeloma. Blood 2008, 111, 2962–2972. [Google Scholar] [CrossRef]
- Rajkumar, S.V. Multiple myeloma: 2020 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2020, 95, 548–567. [Google Scholar] [CrossRef]
- Li, S.; Gong, T.; Kou, C.; Fu, A.; Bolanos, R.; Liu, J. Clinical Outcomes Associated With Chronic Kidney Disease in Elderly Medicare Patients with Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2021. [Google Scholar] [CrossRef]
- Shragai, T.; Gatt, M.E.; Shaulov, A.; Katodritou, E.; Triantafyllou, T.; Lavi, N.; Pouli, A.; Sioni, A.; Vaxman, I.; Zektser, M.; et al. Characteristics and outcome of multiple myeloma patients presenting with anaemia only: A retrospective multi-centre study. Leuk. Res. 2021, 101, 106498. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V.; Kumar, S. Multiple Myeloma: Diagnosis and Treatment. Mayo Clin. Proc. 2016, 91, 101–119. [Google Scholar] [CrossRef] [PubMed]
- Bataille, R.; Manolagas, S.C.; Berenson, J.R. Pathogenesis and management of bone lesions in multiple myeloma. Hematol. Clin. N. Am. 1997, 11, 349–361. [Google Scholar] [CrossRef]
- Kumar, S.K.; Dispenzieri, A.; Lacy, M.Q.; Gertz, M.A.; Buadi, F.K.; Pandey, S.C.; Kapoor, P.; Dingli, D.; Hayman, S.R.; Leung, N.; et al. Continued improvement in survival in multiple myeloma: Changes in early mortality and outcomes in older patients. Leuk. 2014, 28, 1122–1128. [Google Scholar] [CrossRef]
- Weisel, K.; Majer, I.; DeCosta, L.; Oriol, A.; Goldschmidt, H.; Ludwig, H.; Campioni, M.; Szabo, Z.; Dimopoulos, M. Carfilzomib and dexamethasone versus eight cycles of bortezomib and dexamethasone in patients with relapsed or refractory multiple myeloma: An indirect comparison using data from the phase 3 ENDEAVOR and CASTOR trials. Leuk. Lymphoma 2019, 61, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, A.; Chanan-Khan, A.; Weisel, K.; Nooka, A.K.; Masszi, T.; Beksac, M.; Spicka, I.; Hungria, V.; Munder, M.; Mateos, M.V.; et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N. Engl. J. Med. 2016, 375, 754–766. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Oriol, A.; Nahi, H.; San-Miguel, J.; Bahlis, N.J.; Usmani, S.Z.; Rabin, N.; Orlowski, R.Z.; Komarnicki, M.; Suzuki, K.; et al. Daratumumab, Lenalidomide, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 375, 1319–1331. [Google Scholar] [CrossRef] [PubMed]
- Kararoudi, M.N.; Nagai, Y.; Elmas, E.; Pereira, M.D.S.F.; Ali, S.A.; Imus, P.H.; Wethington, D.; Borrello, I.M.; Lee, D.A.; Ghiaur, G. CD38 deletion of human primary NK cells eliminates daratumumab-induced fratricide and boosts their effector activity. Blood 2020, 136, 2416–2427. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Motais, B.; Charvátová, S.; Walek, Z.; Hrdinka, M.; Smolarczyk, R.; Cichoń, T.; Czapla, J.; Giebel, S.; Šimíček, M.; Jelínek, T.; et al. Selection, Expansion, and Unique Pretreatment of Allogeneic Human Natural Killer Cells with Anti-CD38 Monoclonal Antibody for Efficient Multiple Myeloma Treatment. Cells 2021, 10, 967. https://doi.org/10.3390/cells10050967
Motais B, Charvátová S, Walek Z, Hrdinka M, Smolarczyk R, Cichoń T, Czapla J, Giebel S, Šimíček M, Jelínek T, et al. Selection, Expansion, and Unique Pretreatment of Allogeneic Human Natural Killer Cells with Anti-CD38 Monoclonal Antibody for Efficient Multiple Myeloma Treatment. Cells. 2021; 10(5):967. https://doi.org/10.3390/cells10050967
Chicago/Turabian StyleMotais, Benjamin, Sandra Charvátová, Zuzana Walek, Matouš Hrdinka, Ryszard Smolarczyk, Tomasz Cichoń, Justyna Czapla, Sebastian Giebel, Michal Šimíček, Tomáš Jelínek, and et al. 2021. "Selection, Expansion, and Unique Pretreatment of Allogeneic Human Natural Killer Cells with Anti-CD38 Monoclonal Antibody for Efficient Multiple Myeloma Treatment" Cells 10, no. 5: 967. https://doi.org/10.3390/cells10050967
APA StyleMotais, B., Charvátová, S., Walek, Z., Hrdinka, M., Smolarczyk, R., Cichoń, T., Czapla, J., Giebel, S., Šimíček, M., Jelínek, T., Ševčíková, T., Sobotka, J., Kořístek, Z., Hájek, R., & Bagó, J. R. (2021). Selection, Expansion, and Unique Pretreatment of Allogeneic Human Natural Killer Cells with Anti-CD38 Monoclonal Antibody for Efficient Multiple Myeloma Treatment. Cells, 10(5), 967. https://doi.org/10.3390/cells10050967