
Risk Stratification and Treatment in Smoldering Multiple Myeloma


Abstract
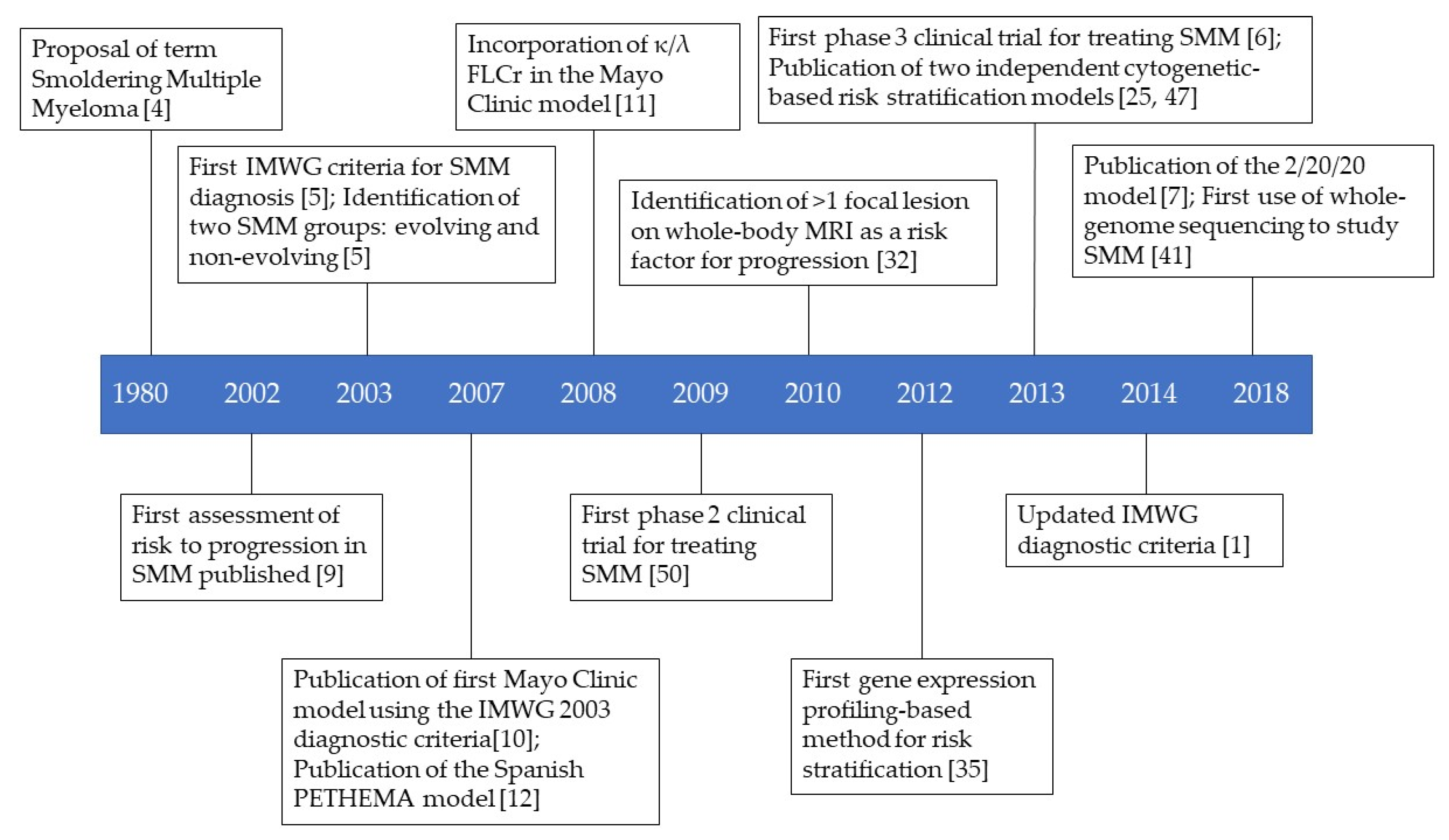
:1. Introduction
2. Risk Assessment Models
2.1. Clinical Markers
2.1.1. Baseline Clinical Measurements
2.1.2. Evolving Clinical Measurements
2.1.3. Imaging Approaches
2.2. Genetic-Based Models
2.2.1. DNA/RNA Sequencing Approaches and Gene Expression Profiling (GEP)
2.2.2. Cytogenetic Approaches
3. Treatment of Smoldering Multiple Myeloma
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rajkumar, S.V.; Dimopoulos, M.A.; Palumbo, A.; Blade, J.; Merlini, G.; Mateos, M.V.; Kumar, S.; Hillengass, J.; Kastritis, E.; Richardson, P.; et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014, 15, e538–e548. [Google Scholar] [CrossRef]
- Kyle, R.A.; Durie, B.G.; Rajkumar, S.V.; Landgren, O.; Blade, J.; Merlini, G.; Kroger, N.; Einsele, H.; Vesole, D.H.; Dimopoulos, M.; et al. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering (asymptomatic) multiple myeloma: IMWG consensus perspectives risk factors for progression and guidelines for monitoring and management. Leukemia 2010, 24, 1121–1127. [Google Scholar] [CrossRef] [Green Version]
- Rosinol, L.; Blade, J.; Esteve, J.; Aymerich, M.; Rozman, M.; Montoto, S.; Gine, E.; Nadal, E.; Filella, X.; Queralt, R.; et al. Smoldering multiple myeloma: Natural history and recognition of an evolving type. Br. J. Haematol. 2003, 123, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Kyle, R.A.; Greipp, P.R. Smoldering Multiple Myeloma. N. Engl. J. Med. 1980, 302, 1347–1349. [Google Scholar] [CrossRef]
- International Myeloma Working Group. Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: A report of the International Myeloma Working Group. Br. J. Haematol. 2003, 121, 749–757. [Google Scholar] [CrossRef] [Green Version]
- Mateos, M.V.; Hernandez, M.T.; Giraldo, P.; de la Rubia, J.; de Arriba, F.; Lopez Corral, L.; Rosinol, L.; Paiva, B.; Palomera, L.; Bargay, J.; et al. Lenalidomide plus dexamethasone for high-risk smoldering multiple myeloma. N. Engl. J. Med. 2013, 369, 438–447. [Google Scholar] [CrossRef] [Green Version]
- Lakshman, A.; Rajkumar, S.V.; Buadi, F.K.; Binder, M.; Gertz, M.A.; Lacy, M.Q.; Dispenzieri, A.; Dingli, D.; Fonder, A.L.; Hayman, S.R.; et al. Risk stratification of smoldering multiple myeloma incorporating revised IMWG diagnostic criteria. Blood Cancer J. 2018, 8, 59. [Google Scholar] [CrossRef]
- Mateos, M.V.; Kumar, S.; Dimopoulos, M.A.; Gonzalez-Calle, V.; Kastritis, E.; Hajek, R.; De Larrea, C.F.; Morgan, G.J.; Merlini, G.; Goldschmidt, H.; et al. International Myeloma Working Group risk stratification model for smoldering multiple myeloma (SMM). Blood Cancer J. 2020, 10, 102. [Google Scholar] [CrossRef]
- Cesana, C.; Klersy, C.; Barbarano, L.; Nosari, A.M.; Crugnola, M.; Pungolino, E.; Gargantini, L.; Granata, S.; Valentini, M.; Morra, E. Prognostic factors for malignant transformation in monoclonal gammopathy of undetermined significance and smoldering multiple myeloma. J. Clin. Oncol. 2002, 20, 1625–1634. [Google Scholar] [CrossRef]
- Kyle, R.A.; Remstein, E.D.; Therneau, T.M.; Dispenzieri, A.; Kurtin, P.J.; Hodnefield, J.M.; Larson, D.R.; Plevak, M.F.; Jelinek, D.F.; Fonseca, R.; et al. Clinical course and prognosis of smoldering (asymptomatic) multiple myeloma. N. Engl. J. Med. 2007, 356, 2582–2590. [Google Scholar] [CrossRef]
- Dispenzieri, A.; Kyle, R.A.; Katzmann, J.A.; Therneau, T.M.; Larson, D.; Benson, J.; Clark, R.J.; Melton, L.J., 3rd; Gertz, M.A.; Kumar, S.K.; et al. Immunoglobulin free light chain ratio is an independent risk factor for progression of smoldering (asymptomatic) multiple myeloma. Blood 2008, 111, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Perez-Persona, E.; Vidriales, M.B.; Mateo, G.; Garcia-Sanz, R.; Mateos, M.V.; de Coca, A.G.; Galende, J.; Martin-Nunez, G.; Alonso, J.M.; de Las Heras, N.; et al. New criteria to identify risk of progression in monoclonal gammopathy of uncertain significance and smoldering multiple myeloma based on multiparameter flow cytometry analysis of bone marrow plasma cells. Blood 2007, 110, 2586–2592. [Google Scholar] [CrossRef]
- Cherry, B.M.; Korde, N.; Kwok, M.; Manasanch, E.E.; Bhutani, M.; Mulquin, M.; Zuchlinski, D.; Yancey, M.A.; Maric, I.; Calvo, K.R.; et al. Modeling progression risk for smoldering multiple myeloma: Results from a prospective clinical study. Leuk. Lymphoma 2013, 54, 2215–2218. [Google Scholar] [CrossRef] [PubMed]
- Larsen, J.T.; Kumar, S.K.; Dispenzieri, A.; Kyle, R.A.; Katzmann, J.A.; Rajkumar, S.V. Serum free light chain ratio as a biomarker for high-risk smoldering multiple myeloma. Leukemia 2013, 27, 941–946. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, G.; Kyle, R.A.; Larson, D.R.; Witzig, T.E.; Kumar, S.; Dispenzieri, A.; Morice, W.G.; Rajkumar, S.V. High levels of peripheral blood circulating plasma cells as a specific risk factor for progression of smoldering multiple myeloma. Leukemia 2013, 27, 680–685. [Google Scholar] [CrossRef]
- Gonsalves, W.I.; Rajkumar, S.V.; Dispenzieri, A.; Dingli, D.; Timm, M.M.; Morice, W.G.; Lacy, M.Q.; Buadi, F.K.; Go, R.S.; Leung, N.; et al. Quantification of circulating clonal plasma cells via multiparametric flow cytometry identifies patients with smoldering multiple myeloma at high risk of progression. Leukemia 2017, 31, 130–135. [Google Scholar] [CrossRef] [Green Version]
- Kastritis, E.; Terpos, E.; Moulopoulos, L.; Spyropoulou-Vlachou, M.; Kanellias, N.; Eleftherakis-Papaiakovou, E.; Gkotzamanidou, M.; Migkou, M.; Gavriatopoulou, M.; Roussou, M.; et al. Extensive bone marrow infiltration and abnormal free light chain ratio identifies patients with asymptomatic myeloma at high risk for progression to symptomatic disease. Leukemia 2013, 27, 947–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waxman, A.J.; Mick, R.; Garfall, A.L.; Cohen, A.; Vogl, D.T.; Stadtmauer, E.A.; Weiss, B.M. Classifying ultra-high risk smoldering myeloma. Leukemia 2015, 29, 751–753. [Google Scholar] [CrossRef]
- Wu, V.; Moshier, E.; Leng, S.; Barlogie, B.; Cho, H.J.; Jagannath, S.; Madduri, D.; Mazumdar, M.; Parekh, S.; Chari, A. Risk stratification of smoldering multiple myeloma: Predictive value of free light chains and group-based trajectory modeling. Blood Adv. 2018, 2, 1470–1479. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez de la Calle, V.; Garcia-Sanz, R.; Sobejano, E.; Ocio, E.M.; Puig, N.; Gutierrez, N.C.; Melón, A.; Gonzalez, J.; Garcia de Coca, A.; Hernandez, J.M.; et al. Bence Jones Proteinuria in Smoldering Multiple Myeloma As Predictor Marker of Progression to Symptomatic Multiple Myeloma. Blood 2014, 124, 3369. [Google Scholar] [CrossRef]
- Sorrig, R.; Klausen, T.W.; Salomo, M.; Vangsted, A.J.; Ostergaard, B.; Gregersen, H.; Frolund, U.C.; Andersen, N.F.; Helleberg, C.; Andersen, K.T.; et al. Smoldering multiple myeloma risk factors for progression: A Danish population-based cohort study. Eur. J. Haematol. 2016, 97, 303–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paiva, B. CTCs as powerful tools in all stages of MM. In Proceedings of the 18th International Myeloma Workshop Conference, Vienna, Austria, 8–11 September 2021. [Google Scholar]
- Aljama, M.A.; Sidiqi, M.H.; Lakshman, A.; Dispenzieri, A.; Jevremovic, D.; Gertz, M.A.; Lacy, M.Q.; Buadi, F.K.; Dingli, D.; Muchtar, E.; et al. Plasma cell proliferative index is an independent predictor of progression in smoldering multiple myeloma. Blood Adv. 2018, 2, 3149–3154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajek, R.; Sandecka, V.; Spicka, I.; Raab, M.; Goldschmidt, H.; Beck, S.; Minarik, J.; Pavlicek, P.; Radocha, J.; Heindorfer, A.; et al. Identification of patients with smouldering multiple myeloma at ultra-high risk of progression using serum parameters: The Czech Myeloma Group model. Br. J. Haematol. 2020, 190, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Neben, K.; Jauch, A.; Hielscher, T.; Hillengass, J.; Lehners, N.; Seckinger, A.; Granzow, M.; Raab, M.S.; Ho, A.D.; Goldschmidt, H.; et al. Progression in smoldering myeloma is independently determined by the chromosomal abnormalities del(17p), t(4;14), gain 1q, hyperdiploidy, and tumor load. J. Clin. Oncol. 2013, 31, 4325–4332. [Google Scholar] [CrossRef]
- Vasco-Mogorron, M.A.; Campillo, J.A.; Periago, A.; Cabanas, V.; Berenguer, M.; Garcia-Garay, M.C.; Gimeno, L.; Soto-Ramirez, M.F.; Martinez-Hernandez, M.D.; Muro, M.; et al. Proliferation to Apoptosis Tumor Cell Ratio as a Biomarker to Improve Clinical Management of Pre-Malignant and Symptomatic Plasma Cell Neoplasms. Int. J. Mol. Sci. 2021, 22, 3895. [Google Scholar] [CrossRef]
- Visram, A.; Soof, C.; Rajkumar, S.V.; Kumar, S.K.; Bujarski, S.; Spektor, T.M.; Kyle, R.A.; Berenson, J.R.; Dispenzieri, A. Serum BCMA levels predict outcomes in MGUS and smoldering myeloma patients. Blood Cancer J. 2021, 11, 120. [Google Scholar] [CrossRef]
- Fernandez de Larrea, C.; Isola, I.; Pereira, A.; Cibeira, M.T.; Magnano, L.; Tovar, N.; Rodriguez-Lobato, L.G.; Calvo, X.; Arostegui, J.I.; Diaz, T.; et al. Evolving M-protein pattern in patients with smoldering multiple myeloma: Impact on early progression. Leukemia 2018, 32, 1427–1434. [Google Scholar] [CrossRef]
- Ravi, P.; Kumar, S.; Larsen, J.T.; Gonsalves, W.; Buadi, F.; Lacy, M.Q.; Go, R.; Dispenzieri, A.; Kapoor, P.; Lust, J.A.; et al. Evolving changes in disease biomarkers and risk of early progression in smoldering multiple myeloma. Blood Cancer J. 2016, 6, e454. [Google Scholar] [CrossRef] [Green Version]
- Atrash, S.; Robinson, M.; Slaughter, D.; Aneralla, A.; Brown, T.; Robinson, J.; Ndiaye, A.; Sprouse, C.; Zhang, Q.; Symanowski, J.T.; et al. Evolving changes in M-protein and hemoglobin as predictors for progression of smoldering multiple myeloma. Blood Cancer J. 2018, 8, 107. [Google Scholar] [CrossRef]
- Gran, C.; Luong, V.; Bruchfeld, J.B.; Liwing, J.; Afram, G.; Lund, J.; Usmani, S.; Alici, E.; Nahi, H. Dynamic follow-up of smoldering multiple myeloma identifies a subset of patients at high risk of progression. Am. J. Hematol. 2021, 96, E63–E65. [Google Scholar] [CrossRef]
- Hillengass, J.; Fechtner, K.; Weber, M.A.; Bauerle, T.; Ayyaz, S.; Heiss, C.; Hielscher, T.; Moehler, T.M.; Egerer, G.; Neben, K.; et al. Prognostic significance of focal lesions in whole-body magnetic resonance imaging in patients with asymptomatic multiple myeloma. J. Clin. Oncol. 2010, 28, 1606–1610. [Google Scholar] [CrossRef]
- Kastritis, E.; Moulopoulos, L.A.; Terpos, E.; Koutoulidis, V.; Dimopoulos, M.A. The prognostic importance of the presence of more than one focal lesion in spine MRI of patients with asymptomatic (smoldering) multiple myeloma. Leukemia 2014, 28, 2402–2403. [Google Scholar] [CrossRef] [PubMed]
- Zamagni, E.; Nanni, C.; Gay, F.; Pezzi, A.; Patriarca, F.; Bello, M.; Rambaldi, I.; Tacchetti, P.; Hillengass, J.; Gamberi, B.; et al. 18F-FDG PET/CT focal, but not osteolytic, lesions predict the progression of smoldering myeloma to active disease. Leukemia 2016, 30, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Wennmann, M.; Kintzele, L.; Piraud, M.; Menze, B.H.; Hielscher, T.; Hofmanninger, J.; Wagner, B.; Kauczor, H.U.; Merz, M.; Hillengass, J.; et al. Volumetry based biomarker speed of growth: Quantifying the change of total tumor volume in whole-body magnetic resonance imaging over time improves risk stratification of smoldering multiple myeloma patients. Oncotarget 2018, 9, 25254–25264. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Corral, L.; Sarasquete, M.E.; Bea, S.; Garcia-Sanz, R.; Mateos, M.V.; Corchete, L.A.; Sayagues, J.M.; Garcia, E.M.; Blade, J.; Oriol, A.; et al. SNP-based mapping arrays reveal high genomic complexity in monoclonal gammopathies, from MGUS to myeloma status. Leukemia 2012, 26, 2521–2529. [Google Scholar] [CrossRef] [Green Version]
- Williams, G.T.; Farzaneh, F. Are snoRNAs and snoRNA host genes new players in cancer? Nat. Rev. Cancer 2012, 12, 84–88. [Google Scholar] [CrossRef]
- Dhodapkar, M.V.; Sexton, R.; Waheed, S.; Usmani, S.; Papanikolaou, X.; Nair, B.; Petty, N.; Shaughnessy, J.D., Jr.; Hoering, A.; Crowley, J.; et al. Clinical, genomic, and imaging predictors of myeloma progression from asymptomatic monoclonal gammopathies (SWOG S0120). Blood 2014, 123, 78–85. [Google Scholar] [CrossRef] [Green Version]
- Shaughnessy, J.D., Jr.; Zhan, F.; Burington, B.E.; Huang, Y.; Colla, S.; Hanamura, I.; Stewart, J.P.; Kordsmeier, B.; Randolph, C.; Williams, D.R.; et al. A validated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes mapping to chromosome 1. Blood 2007, 109, 2276–2284. [Google Scholar] [CrossRef] [Green Version]
- Khan, R.; Dhodapkar, M.; Rosenthal, A.; Heuck, C.; Papanikolaou, X.; Qu, P.; van Rhee, F.; Zangari, M.; Jethava, Y.; Epstein, J.; et al. Four genes predict high risk of progression from smoldering to symptomatic multiple myeloma (SWOG S0120). Haematologica 2015, 100, 1214–1221. [Google Scholar] [CrossRef] [Green Version]
- Bolli, N.; Maura, F.; Minvielle, S.; Gloznik, D.; Szalat, R.; Fullam, A.; Martincorena, I.; Dawson, K.J.; Samur, M.K.; Zamora, J.; et al. Genomic patterns of progression in smoldering multiple myeloma. Nat. Commun. 2018, 9, 3363. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wen, W.; Bao, J.; Kuhs, K.L.; Cai, Q.; Long, J.; Shu, X.O.; Zheng, W.; Guo, X. Integrative genomic analyses of APOBEC-mutational signature, expression and germline deletion of APOBEC3 genes, and immunogenicity in multiple cancer types. BMC Med. Genom. 2019, 12, 131. [Google Scholar] [CrossRef] [Green Version]
- Bustoros, M.; Sklavenitis-Pistofidis, R.; Park, J.; Redd, R.; Zhitomirsky, B.; Dunford, A.J.; Salem, K.; Tai, Y.T.; Anand, S.; Mouhieddine, T.H.; et al. Genomic Profiling of Smoldering Multiple Myeloma Identifies Patients at a High Risk of Disease Progression. J. Clin. Oncol. 2020, 38, 2380–2389. [Google Scholar] [CrossRef]
- Zavidij, O.; Haradhvala, N.J.; Mouhieddine, T.H.; Sklavenitis-Pistofidis, R.; Cai, S.; Reidy, M.; Rahmat, M.; Flaifel, A.; Ferland, B.; Su, N.K.; et al. Single-cell RNA sequencing reveals compromised immune microenvironment in precursor stages of multiple myeloma. Nat. Cancer 2020, 1, 493–506. [Google Scholar] [CrossRef]
- Kawano, Y.; Zavidij, O.; Park, J.; Moschetta, M.; Kokubun, K.; Mouhieddine, T.H.; Manier, S.; Mishima, Y.; Murakami, N.; Bustoros, M.; et al. Blocking IFNAR1 inhibits multiple myeloma-driven Treg expansion and immunosuppression. J. Clin. Investig. 2018, 128, 2487–2499. [Google Scholar] [CrossRef]
- Oben, B.; Froyen, G.; Maclachlan, K.H.; Leongamornlert, D.; Abascal, F.; Zheng-Lin, B.; Yellapantula, V.; Derkach, A.; Geerdens, E.; Diamond, B.T.; et al. Whole-genome sequencing reveals progressive versus stable myeloma precursor conditions as two distinct entities. Nat. Commun. 2021, 12, 1861. [Google Scholar] [CrossRef]
- Rajkumar, S.V.; Gupta, V.; Fonseca, R.; Dispenzieri, A.; Gonsalves, W.I.; Larson, D.; Ketterling, R.P.; Lust, J.A.; Kyle, R.A.; Kumar, S.K. Impact of primary molecular cytogenetic abnormalities and risk of progression in smoldering multiple myeloma. Leukemia 2013, 27, 1738–1744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rangel-Pozzo, A.; Yu, P.L.I.; La, L.S.; Asbaghi, Y.; Sisdelli, L.; Tammur, P.; Tamm, A.; Punab, M.; Klewes, L.; Louis, S.; et al. Telomere Architecture Correlates with Aggressiveness in Multiple Myeloma. Cancers 2021, 13, 1969. [Google Scholar] [CrossRef]
- Vermolen, B.J.; Garini, Y.; Mai, S.; Mougey, V.; Fest, T.; Chuang, T.C.; Chuang, A.Y.; Wark, L.; Young, I.T. Characterizing the three-dimensional organization of telomeres. Cytom. A 2005, 67, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Lust, J.A.; Lacy, M.Q.; Zeldenrust, S.R.; Dispenzieri, A.; Gertz, M.A.; Witzig, T.E.; Kumar, S.; Hayman, S.R.; Russell, S.J.; Buadi, F.K.; et al. Induction of a chronic disease state in patients with smoldering or indolent multiple myeloma by targeting interleukin 1{beta}-induced interleukin 6 production and the myeloma proliferative component. Mayo Clin. Proc. 2009, 84, 114–122. [Google Scholar] [CrossRef]
- Hilbert, D.M.; Kopf, M.; Mock, B.A.; Kohler, G.; Rudikoff, S. Interleukin 6 is essential for in vivo development of B lineage neoplasms. J. Exp. Med. 1995, 182, 243–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, Y.; Donovan, K.A.; Kline, M.P.; Gornet, M.K.; Moon-Tasson, L.L.; Lacy, M.Q.; Dispenzieri, A.; Gertz, M.A.; Greipp, P.R.; Lust, J.A. Identification of two groups of smoldering multiple myeloma patients who are either high or low producers of interleukin-1. J. Interferon Cytokine Res. 2006, 26, 83–95. [Google Scholar] [CrossRef]
- Mateos, M.V.; Hernandez, M.T.; Giraldo, P.; de la Rubia, J.; de Arriba, F.; Corral, L.L.; Rosinol, L.; Paiva, B.; Palomera, L.; Bargay, J.; et al. Lenalidomide plus dexamethasone versus observation in patients with high-risk smouldering multiple myeloma (QuiRedex): Long-term follow-up of a randomised, controlled, phase 3 trial. Lancet Oncol. 2016, 17, 1127–1136. [Google Scholar] [CrossRef]
- Mateos, M.V.; Martinez-Lopez, J.; Rodriguez Otero, P.; Gonzalez-Calle, V.; Gonzalez, M.S.; Oriol, A.; Gutierrez, N.C.; Paiva, B.; Tamayo, R.R.; Dachs, L.R.; et al. Curative Strategy (GEM-CESAR) for High-Risk Smoldering Myeloma (SMM): Carfilzomib, Lenalidomide and Dexamethasone (KRd) As Induction Followed By HDT-ASCT, Consolidation with Krd and Maintenance with Rd. Blood 2019, 134, 781. [Google Scholar] [CrossRef]
- Witzig, T.E.; Laumann, K.M.; Lacy, M.Q.; Hayman, S.R.; Dispenzieri, A.; Kumar, S.; Reeder, C.B.; Roy, V.; Lust, J.A.; Gertz, M.A.; et al. A phase III randomized trial of thalidomide plus zoledronic acid versus zoledronic acid alone in patients with asymptomatic multiple myeloma. Leukemia 2013, 27, 220–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korde, N.; Roschewski, M.; Zingone, A.; Kwok, M.; Manasanch, E.E.; Bhutani, M.; Tageja, N.; Kazandjian, D.; Mailankody, S.; Wu, P.; et al. Treatment With Carfilzomib-Lenalidomide-Dexamethasone With Lenalidomide Extension in Patients With Smoldering or Newly Diagnosed Multiple Myeloma. JAMA Oncol. 2015, 1, 746–754. [Google Scholar] [CrossRef] [Green Version]
- Ghobrial, I.M.; Badros, A.Z.; Vredenburgh, J.J.; Matous, J.; Caola, A.M.; Savell, A.; Henrick, P.; Paba-Prada, C.E.; Schlossman, R.L.; Laubach, J.P.; et al. Phase II Trial of Combination of Elotuzumab, Lenalidomide, and Dexamethasone in High-Risk Smoldering Multiple Myeloma. Blood 2016, 128, 976. [Google Scholar] [CrossRef]
- Fancher, K.M.; Bunk, E.J. Elotuzumab: The First Monoclonal Antibody for the Treatment of Multiple Myeloma. J. Adv. Pract. Oncol. 2016, 7, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Mailankody, S.; Kazandjian, D.; Korde, N.; Roschewski, M.; Manasanch, E.; Bhutani, M.; Tageja, N.; Kwok, M.; Zhang, Y.; Zingone, A.; et al. Baseline mutational patterns and sustained MRD negativity in patients with high-risk smoldering myeloma. Blood Adv. 2017, 1, 1911–1918. [Google Scholar] [CrossRef] [Green Version]
- Nooka, A.K.; Wang, M.L.; Yee, A.J.; Kaufman, J.L.; Bae, J.; Peterkin, D.; Richardson, P.G.; Raje, N.S. Assessment of Safety and Immunogenicity of PVX-410 Vaccine With or Without Lenalidomide in Patients With Smoldering Multiple Myeloma: A Nonrandomized Clinical Trial. JAMA Oncol. 2018, 4, e183267. [Google Scholar] [CrossRef]
- Landgren, C.O.; Chari, A.; Cohen, Y.C.; Spencer, A.; Voorhees, P.; Estell, J.A.; Sandhu, I.; Jenner, M.W.; Williams, C.; Cavo, M.; et al. Daratumumab monotherapy for patients with intermediate-risk or high-risk smoldering multiple myeloma: A randomized, open-label, multicenter, phase 2 study (CENTAURUS). Leukemia 2020, 34, 1840–1852. [Google Scholar] [CrossRef] [PubMed]
- Lonial, S.; Jacobus, S.; Fonseca, R.; Weiss, M.; Kumar, S.; Orlowski, R.Z.; Kaufman, J.L.; Yacoub, A.M.; Buadi, F.K.; O’Brien, T.; et al. Randomized Trial of Lenalidomide Versus Observation in Smoldering Multiple Myeloma. J. Clin. Oncol. 2020, 38, 1126–1137. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Model | Risk Groups | Level of Risk | Reference |
|---|---|---|---|
| Kyle et al. (2007) | BMPC ≥ 10% M-protein ≥ 3 g/dL | High | [10] |
| BMPC ≥ 10% M-protein < 3 g/dL | Intermediate | ||
| BMPC < 10% M-protein ≥ 3 g/dL | Low | ||
| Dispenzieri et al. | BMPC ≥ 10% M-protein ≥ 3 g/dL κ/λ FLCr < 0.125 or > 8 | High | [11] |
| BMPC ≥ 10% or M-protein ≥ 3 g/dL and one other of above | Intermediate | ||
| BMPC ≥ 10% or M-protein ≥ 3 g/dL | Low | ||
| Pérez-Persona et al. | presence of immunoparesis aPC/BMPC ≥ 95% | High | [12] |
| presence of immunoparesis or aPC/BMPC ≥ 95% | Intermediate | ||
| absence of immunoparesis aPC/BMPC < 95% | Low | ||
| Larsen et al. | i:u FLCr ≥ 100 | High | [14] |
| i:u FLCr < 100 | Low | ||
| Bianchi et al. | cPCs 1 M-protein ≥ 3 g/dL | High | [15] |
| cPCs 1 or M-protein ≥ 3 g/dL | Intermediate | ||
| absence of cPCs 2 M-protein < 3 g/dL | Low | ||
| Kastritis et al. (2013) | BM infiltration ≥ 60% i:u FLCr ≥ 100 | High | [17] |
| BM infiltration ≥ 60% or i:u FLCr ≥ 100 | High-intermediate | ||
| BM infiltration < 60% i:u FLCr < 100 | Low | ||
| Waxman et al. | 2 or all of: BMPC ≥ 40% i:u FLCr ≥ 50 albumin concentration ≤ 3.5 | High | [18] |
| BMPC ≥ 40% and/or i:u FLCr ≥ 50 and/or albumin concentration ≤ 3.5 | Intermediate | ||
| BMPC < 40% i:u FLCr <50 albumin concentration > 3.5 | Low | ||
| Gonzalez de la Calle et al. | BJ proteinuria > 500 mg/24 h | High | [20] |
| BJ proteinuria 251–500 mg/24 h | High-intermediate | ||
| BJ proteinuria 1–250 mg/24 h | Low-Intermediate | ||
| BJ proteinuria = 0 mg/24 h | Low | ||
| Sørrig et al. | presence of immunoparesis M-protein ≥ 3 g/dL | High | [21] |
| presence of immunoparesis or M-protein ≥ 3 g/dL | Intermediate | ||
| absence of immunoparesis M-protein < 3 g/dL | Low | ||
| Lakshman et al. | 2 or all of: M-protein > 2 BMPC > 20% i:u FLCr > 20 | High | [7] |
| M-protein > 2 and/or BMPC > 20% and/or i:u FLCr > 20 | Intermediate | ||
| M-protein ≤ 2 BMPC ≤ 20% i:u FLCr ≤ 20 | Low | ||
| Aljama et al. | PCPI > 0.5% | High | [23] |
| PCPI ≤ 0.5% | Low | ||
| Hàjek et al. | presence of immunoparesis M-protein ≥ 2.3 g/dL i:u FLCr > 30 | High | [24] |
| 2 of: presence of immunoparesis M-protein ≥ 2.3 g/dL i:u FLCr > 30 | Intermediate | ||
| presence of immunoparesis and/or M-protein ≥ 2.3 g/dL and/or i:u FLCr > 30 | Low-intermediate | ||
| absence of immunoparesis M-protein < 2.3 g/dL i:u FLCr ≤ 30 | Low | ||
| Vasco-Mogorrón et al. | proliferation to apoptosis ratio ≥1.27 | High | [26] |
| proliferation to apoptosis ratio <1.27 | Low | ||
| Visram et al. | sBCMA ≥ 127 ng/mL | High | [27] |
| sBCMA < 127 ng/mL | Low |
| Model | Characteristics of the Evolving Type | Reference |
|---|---|---|
| Rosiñol et al. | Progressive increase in M-protein Higher IgA frequency | [3] |
| Fernandez de Larrea et al. | 10% increase of M-protein within one year with baseline M-protein concentration of ≥30 g/L Or <30 g/L baseline M-protein plus a progressive increase of M-protein over three years | [28] |
| Ravi et al. and Atrash et al. | ≥10% increase in M-protein within six months and/or ≥25% increase within the first year with a minimum absolute increase of 5 g/L Decrease of ≥0.5 g/dl hemoglobin within one year | [28,30] |
| Wu et al. | >64% increase in M-protein >169% increase in edFLC >1.57 g/dl decrease in hemoglobin all within one year | [19] |
| Gran et al. | ≥5 g/L increase in M-protein ≥4.5 increase in eFLCr both from SMM diagnosis up to 6 months prior to MM diagnosis | [31] |
| Model | Criteria for High-Risk Group | Reference |
|---|---|---|
| Hillengass et al. | >1 focal lesion on whole-body MRI | [32] |
| Kastritis et al. (2013) | >1 focal lesion on whole-body MRI Abnormal FLC ratio of ≥100 (or ≤1/100) BM infiltration of ≥60% | [17] |
| Zamagni et al. | PET/CT positivity | [34] |
| Wennmann et al. | Speed of tumor growth at cutoff of 114 mm3/month | [35] |
| Model | Criteria for High-Risk | Reference |
|---|---|---|
| López-Corral et al. | Mutation in four C/D box snoRNA (SNORD) genes (SNORD25, SNORD27, SNORD30 and SNORD31) | [36] |
| Dhodapkar et al. | 70-gene signature (GEP70) > 0.26 M-protein ≥ 3g/dL iFLC > 25 mg/dL | [38] |
| Khan et al. | GEP4 with a cut-off at 9.28 | [40] |
| Bustoros et al. | Enrichment for APOBEC associated mutations | [43] |
| Model | Characteristic | Model | Reference |
|---|---|---|---|
| Bolli et al. | Retained the subclonal architecture during progression to MM | static progression model | [41] |
| Changes in the subclonal architecture during progression to MM | spontaneous evolution model | ||
| Oben et al. | Higher number of genetic myeloma-defining events including “chromothripsis”, template insertions, mutations in driver genes, aneuploidy, and canonical APOBEC mutational activity | Evolving | [46] |
| Lower mutational burden | Stable |
| Risk Factor | Score 1 |
|---|---|
| i:u FLCr | - |
| 0–10 | 0 |
| 10–25 | 2 |
| 25–40 | 3 |
| >40 | 5 |
| M-protein concentration (g/dL) | - |
| 0–1.5 | 0 |
| 1.5–3 | 3 |
| >3 | 4 |
| BMPC% | - |
| 0–15 | 0 |
| 15–20 | 2 |
| 20–30 | 3 |
| 30–40 | 5 |
| >40 | 6 |
| FISH abnormality | 2 |
| Model | Risk Groups | Level of Risk | Reference |
|---|---|---|---|
| Rajkumar et al. (2013) | t(4;14) or del(17p) | High | [47] |
| trisomies 1 | Intermediate | ||
| one of: t(11;14) MAF translocations IgH translocations 2 monosomy13/del(13q) 1 trisomies and IgH translocations | Standard | ||
| no abnormalities detected 3 | Low | ||
| Neben et al. | one of: del(17p13) t(4;14) +1q21 | High | [25] |
| without: del(17p13) t(4;14) +1q21 | Standard | ||
| Mateos et al. (2020) | 3 or all of: M-protein > 2 BMPC > 20% i:u FLCr > 20 relevant cytogenetic abnormality 4 | High | [8] |
| 2 of: M-protein > 2 BMPC > 20% i:u FLCr > 20 relevant cytogenetic abnormality 4 | Intermediate | ||
| one of: M-protein > 2 BMPC > 20% i:u FLCr > 20 relevant cytogenetic abnormality 4 | Low-intermediate | ||
| M-protein ≤ 2 BMPC ≤ 20% i:u FLCr ≤ 20 absence of relevant cytogenetic abnormality 4 | Low | ||
| Rangel-Pozzo et al. | Low values of 3D telomeric parameters for telomeric profiles 5 | High | [48] |
| High values of 3D telomeric parameters for telomeric profiles 5 | Low |
| Group | Treatment Tested | Reference |
|---|---|---|
| Lust et al. | Interleukin 1 (IL-1) with inhibitors | [50] |
| Mateos et al. (2013) | Lenalidomide and dexamethasone followed by lenalidomide maintenance or observation | [6] |
| Mateos et al. (2016) | Lenalidomide and dexamethasone followed by lenalidomide and dexamethasone maintenance or observation | [53] |
| Mateos et al. (2019) | GEM-CESAR: combination treatment with carfilzomib, lenalidomide and dexamethasone (KRd), followed by high-dose therapy-autologous stem cell transplantation (HDT-ASCT) and KRd consolidation. Treatment continued with lenalidomide and dexamethasone maintenance | [54] |
| Witzig et al. | Thalidomide plus zoledronic acid versus zoledronic acid alone | [55] |
| Korde et al. and Mailankody et al. | Carfilzomib, lenalidomide and dexamethasone followed by a lenalidomide extension | [56,59] |
| Ghobrial et al. | Elotuzumab versus lenalidomide and dexamethasone | [57] |
| Nooka et al. | PVX-410 multiseptated vaccine with or without lenalidomide | [60] |
| Landgren et al. | Daratumumab with extended intense, extended intermediate, or short dosing schedules | [61] |
| Lonial et al. | Lenalidomide single agent versus observation | [62] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lussier, T.; Schoebe, N.; Mai, S. Risk Stratification and Treatment in Smoldering Multiple Myeloma. Cells 2022, 11, 130. https://doi.org/10.3390/cells11010130
Lussier T, Schoebe N, Mai S. Risk Stratification and Treatment in Smoldering Multiple Myeloma. Cells. 2022; 11(1):130. https://doi.org/10.3390/cells11010130
Chicago/Turabian StyleLussier, Tyler, Natalie Schoebe, and Sabine Mai. 2022. "Risk Stratification and Treatment in Smoldering Multiple Myeloma" Cells 11, no. 1: 130. https://doi.org/10.3390/cells11010130
APA StyleLussier, T., Schoebe, N., & Mai, S. (2022). Risk Stratification and Treatment in Smoldering Multiple Myeloma. Cells, 11(1), 130. https://doi.org/10.3390/cells11010130