
Alzheimer and Purinergic Signaling: Just a Matter of Inflammation?


 ,
, 
Abstract
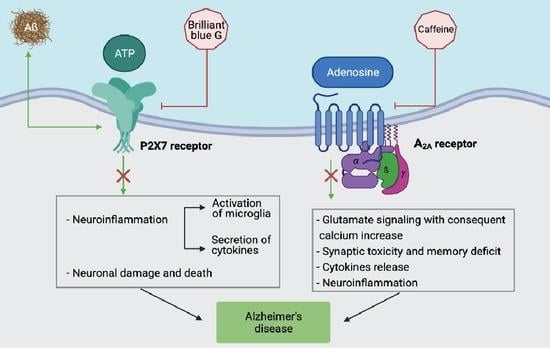
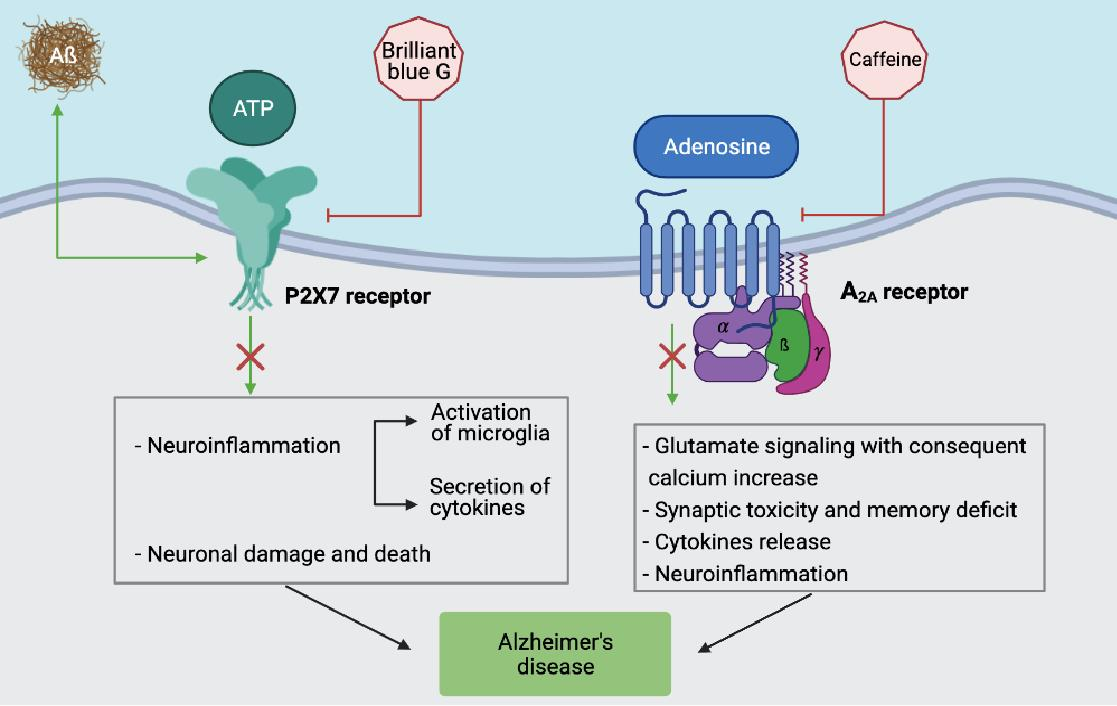
1. A Brief Update on Alzheimer’s Disease (AD)
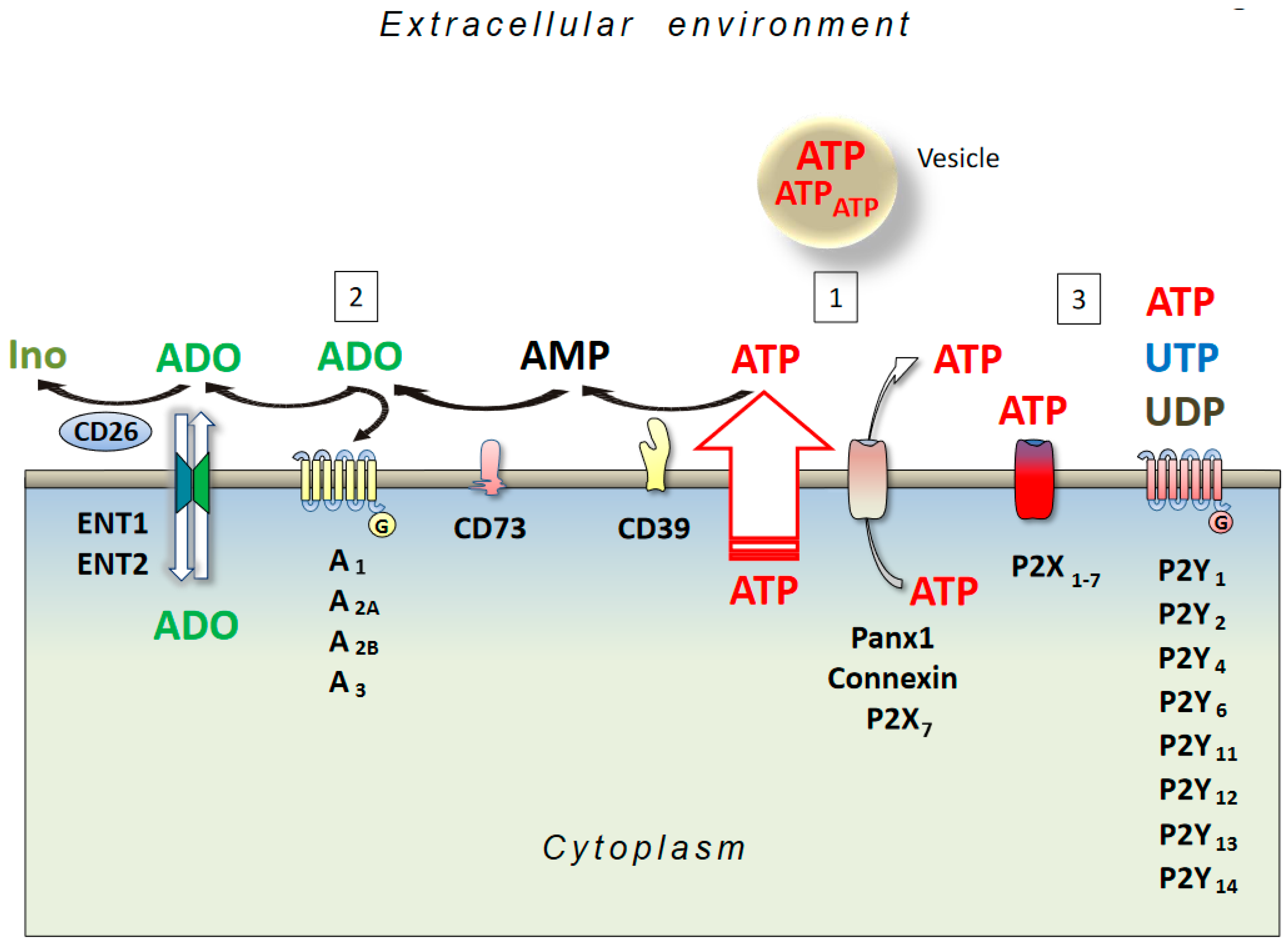
2. Adenosine and AD
3. ATP and AD
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders: DSM-5, 5th ed.; American Psychiatric Publishing: Arlinghton, VA, USA, 2013. [Google Scholar]
- Sachdev, P.S.; Mohan, A.; Taylor, L.; Jeste, D.V. DSM-5 and mental disorders in older individuals: An overview. Harv. Rev. Psychiatry 2015, 23, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Hanseeuw, B.J.; Betensky, R.A.; Jacobs, H.I.L.; Schultz, A.P.; Sepulcre, J.; Becker, J.A.; Cosio, D.M.O.; Farrell, M.; Quiroz, Y.T.; Mormino, E.C.; et al. Association of Amyloid and Tau With Cognition in Preclinical Alzheimer Disease: A Longitudinal Study. JAMA Neurol. 2019, 76, 915–924. [Google Scholar] [CrossRef]
- Elahi, F.M.; Miller, B.L. A clinicopathological approach to the diagnosis of dementia. Nat. Rev. Neurol. 2017, 13, 457–476. [Google Scholar] [CrossRef] [PubMed]
- Sepulveda, F.J.; Parodi, J.; Peoples, R.W.; Opazo, C.; Aguayo, L.G. Synaptotoxicity of Alzheimer Beta Amyloid Can Be Explained by Its Membrane Perforating Property. PLoS ONE 2010, 5, e11820. [Google Scholar] [CrossRef] [PubMed]
- Kandel, N.; Matos, J.O.; Tatulian, S.A. Structure of amyloid β25–35 in lipid environment and cholesterol-dependent membrane pore formation. Sci. Rep. 2019, 9, 2689. [Google Scholar] [CrossRef]
- Nortley, R.; Korte, N.; Izquierdo, P.; Hirunpattarasilp, C.; Mishra, A.; Jaunmuktane, Z.; Kyrargyri, V.; Pfeiffer, T.; Khennouf, L.; Madry, C.; et al. Amyloid β oligomers constrict human capillaries in Alzheimer’s disease via signaling to pericytes. Science 2019, 365, eaav9518. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Jack, C.R.; Wiste, H.J.; Schwarz, C.G.; Lowe, V.J.; Senjem, M.L.; Vemuri, P.; Weigand, S.D.; Therneau, T.M.; Knopman, D.S.; Gunter, J.L.; et al. Longitudinal tau PET in ageing and Alzheimer’s disease. Brain 2018, 141, 1517–1528. [Google Scholar] [CrossRef]
- Perez-Nievas, B.G.; Stein, T.D.; Tai, H.-C.; Dols-Icardo, O.; Scotton, T.C.; Barroeta-Espar, I.; Fernandez-Carballo, L.; De Munain, E.L.; Perez, J.; Marquie, M.; et al. Dissecting phenotypic traits linked to human resilience to Alzheimer’s pathology. Brain 2013, 136, 2510–2526. [Google Scholar] [CrossRef]
- Mirra, S.S.; Heyman, A.; McKeel, D.; Sumi, S.M.; Crain, B.J.; Brownlee, L.M.; Vogel, F.S.; Hughes, J.P.; Van Belle, G.; Berg, L.; et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD): Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 1991, 41, 479–486. [Google Scholar] [CrossRef]
- Braak, H.; Alafuzoff, I.; Arzberger, T.; Kretzschmar, H.; Del Tredici, K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006, 112, 389–404. [Google Scholar] [CrossRef] [PubMed]
- Montine, T.J.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; Mirra, S.S.; et al. National Institute on Aging–Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: A practical approach. Acta Neuropathol. 2011, 123, 1–11. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Betensky, R.A.; Frosch, M.P.; Hyman, B.T. Plaque-Associated Local Toxicity Increases over the Clinical Course of Alzheimer Disease. Am. J. Pathol. 2016, 186, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef]
- Völgyi, K.; Badics, K.; Sialana, F.J.; Gulyássy, P.; Udvari, E.B.; Kis, V.; Drahos, L.; Lubec, G.; Kékesi, K.A.; Juhász, G. Early Presymptomatic Changes in the Proteome of Mitochondria-Associated Membrane in the APP/PS1 Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 7839–7857. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Nahon-Crystal, E.; Shteinfer-Kuzmine, A.; Gupta, R. VDAC1, mitochondrial dysfunction, and Alzheimer’s disease. Pharmacol. Res. 2018, 131, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Chételat, G.; Arbizu, J.; Barthel, H.; Garibotto, V.; Law, I.; Morbelli, S.; van de Giessen, E.; Agosta, F.; Barkhof, F.; Brooks, D.J.; et al. Amyloid-PET and 18F-FDG-PET in the diagnostic investigation of Alzheimer’s disease and other dementias. Lancet Neurol. 2020, 19, 951–962. [Google Scholar] [CrossRef]
- Dubois, B.; Hampel, H.; Feldman, H.H.; Scheltens, P.; Aisen, P.; Andrieu, S.; Bakardjian, H.; Benali, H.; Bertram, L.; Blennow, K.; et al. Preclinical Alzheimer’s disease: Definition, natural history, and diagnostic criteria. Alzheimer’s Dement. 2016, 12, 292–323. [Google Scholar] [CrossRef]
- Molinuevo, J.L.; Ayton, S.; Batrla, R.; Bednar, M.M.; Bittner, T.; Cummings, J.; Fagan, A.M.; Hampel, H.; Mielke, M.M.; Mikulskis, A.; et al. Current state of Alzheimer’s fluid biomarkers. Acta Neuropathol. 2018, 136, 821–853. [Google Scholar] [CrossRef]
- Lewczuk, P.; Ermann, N.; Andreasson, U.; Schultheis, C.; Podhorna, J.; Spitzer, P.; Maler, J.M.; Kornhuber, J.; Blennow, K.; Zetterberg, H. Plasma neurofilament light as a potential biomarker of neurodegeneration in Alzheimer’s disease. Alzheimer’s Res. Ther. 2018, 10, 71. [Google Scholar] [CrossRef] [PubMed]
- Vermunt, L.; Sikkes, S.A.; Hout, A.V.D.; Handels, R.; Bos, I.; Van Der Flier, W.M.; Kern, S.; Ousset, P.-J.; Maruff, P.; Skoog, I.; et al. Duration of preclinical, prodromal, and dementia stages of Alzheimer’s disease in relation to age, sex, and APOE genotype. Alzheimer’s Dement. 2019, 15, 888–898. [Google Scholar] [CrossRef] [PubMed]
- Williamson, J.; Goldman, J.; Marder, K.S. Genetic Aspects of Alzheimer Disease. Neurologist 2009, 15, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Livingston, J.M.; McDonald, M.W.; Gagnon, T.; Jeffers, M.S.; Gomez-Smith, M.; Antonescu, S.; Cron, G.O.; Boisvert, C.; Lacoste, B.; Corbett, D. Influence of metabolic syndrome on cerebral perfusion and cognition. Neurobiol. Dis. 2020, 137, 104756. [Google Scholar] [CrossRef] [PubMed]
- Lourida, I.; Hannon, E.; Littlejohns, T.J.; Langa, K.M.; Hyppönen, E.; Kuzma, E.; Llewellyn, D.J. Association of Lifestyle and Genetic Risk With Incidence of Dementia. JAMA 2019, 322, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s disease drug development pipeline: 2020. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2020, 6, e12050. [Google Scholar] [CrossRef]
- Bittar, A.; Bhatt, N.; Kayed, R. Advances and considerations in AD tau-targeted immunotherapy. Neurobiol. Dis. 2020, 134, 104707. [Google Scholar] [CrossRef]
- Hara, Y.; McKeehan, N.; Fillit, H.M. Translating the biology of aging into novel therapeutics for Alzheimer disease. Neurology 2019, 92, 84–93. [Google Scholar] [CrossRef]
- Cummings, J.L.; Cohen, S.; Van Dyck, C.H.; Brody, M.; Curtis, C.; Cho, W.; Ward, M.; Friesenhahn, M.; Rabe, C.; Brunstein, F.; et al. ABBY. Neurology 2018, 90, e1889–e1897. [Google Scholar] [CrossRef]
- Sperling, R.A.; Jack, C.R.; Aisen, P.S. Testing the Right Target and Right Drug at the Right Stage. Sci. Transl. Med. 2011, 3, 111cm33. [Google Scholar] [CrossRef]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef] [PubMed]
- Poloni, T.E.; Medici, V.; Carlos, A.F.; Davin, A.; Ceretti, A.; Mangieri, M.; Cassini, P.; Vaccaro, R.; Zaccaria, D.; Abbondanza, S.; et al. Abbiategrasso Brain Bank Protocol for Collecting, Processing and Characterizing Aging Brains. J. Vis. Exp. 2020, 160, 60296. [Google Scholar] [CrossRef] [PubMed]
- Newby, A.C. Adenosine and the concept of ‘retaliatory metabolites’. Trends Biochem. Sci. 1984, 9, 42–44. [Google Scholar] [CrossRef]
- Drury, A.N.; Szent-Györgyi, A. The physiological activity of adenine compounds with especial reference to their action upon the mammalian heart1. J. Physiol. 1929, 68, 213–237. [Google Scholar] [CrossRef] [PubMed]
- Ballesteros-Yáñez, I.; Castillo, C.A.; Merighi, S.; Gessi, S. The Role of Adenosine Receptors in Psychostimulant Addiction. Front. Pharmacol. 2018, 8, 985. [Google Scholar] [CrossRef] [PubMed]
- Borea, P.A.; Gessi, S.; Merighi, S.; Varani, K. Adenosine as a Multi-Signalling Guardian Angel in Human Diseases: When, Where and How Does it Exert its Protective Effects? Trends Pharmacol. Sci. 2016, 37, 419–434. [Google Scholar] [CrossRef]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pharmacology of Adenosine Receptors: The State of the Art. Physiol. Rev. 2018, 98, 1591–1625. [Google Scholar] [CrossRef]
- Gonçalves, F.Q.; Lopes, J.P.; Silva, H.B.; Lemos, C.; Silva, A.C.; Gonçalves, N.; Tomé, Â.R.; Ferreira, S.; Canas, P.M.; Rial, D.; et al. Synaptic and memory dysfunction in a β-amyloid model of early Alzheimer’s disease depends on increased formation of ATP-derived extracellular adenosine. Neurobiol. Dis. 2019, 132, 104570. [Google Scholar] [CrossRef]
- Angulo, E.; Casadó, V.; Mallol, J.; Canela, E.I.; Viñals, F.; Ferrer, I.; Lluis, C.; Franco, R. A1 Adenosine Receptors Accumulate in Neurodegenerative Structures in Alzheimer’s Disease and Mediate Both Amyloid Precursor Protein Processing and Tau Phosphorylation and Translocation. Brain Pathol. 2006, 13, 440–451. [Google Scholar] [CrossRef]
- Cieślak, M.; Wojtczak, A. Role of purinergic receptors in the Alzheimer’s disease. Purinergic Signal. 2018, 14, 331–344. [Google Scholar] [CrossRef]
- Mendiola-Precoma, J.; Padilla, K.; Rodríguez-Cruz, A.; Berumen, L.C.; Miledi, R.; García-Alcocer, G. Theobromine-Induced Changes in A1 Purinergic Receptor Gene Expression and Distribution in a Rat Brain Alzheimer’s Disease Model. J. Alzheimer’s Dis. 2017, 55, 1273–1283. [Google Scholar] [CrossRef]
- Semwal, B.C.; Garabadu, D. 5-N-ethyl Carboxamidoadenosine Stimulates Adenosine-2b Receptor-Mediated Mitogen-Activated Protein Kinase Pathway to Improve Brain Mitochondrial Function in Amyloid Beta-Induced Cognitive Deficit Mice. NeuroMol. Med. 2020, 22, 542–556. [Google Scholar] [CrossRef]
- Semwal, B.C.; Garabadu, D. Amyloid beta (1–42) downregulates adenosine-2b receptors in addition to mitochondrial impairment and cholinergic dysfunction in memory-sensitive mouse brain regions. J. Recept. Signal Transduct. 2020, 40, 531–540. [Google Scholar] [CrossRef]
- Coppi, E.; Cherchi, F.; Fusco, I.; Failli, P.; Vona, A.; Dettori, I.; Gaviano, L.; Lucarini, E.; Jacobson, K.A.; Tosh, D.K.; et al. Adenosine A3 receptor activation inhibits pronociceptive N-type Ca2+ currents and cell excitability in dorsal root ganglion neurons. Pain 2019, 160, 1103–1118. [Google Scholar] [CrossRef]
- Lucarini, E.; Coppi, E.; Micheli, L.; Parisio, C.; Vona, A.; Cherchi, F.; Pugliese, A.M.; Pedata, F.; Failli, P.; Palomino, S.; et al. Acute visceral pain relief mediated by A3AR agonists in rats: Involvement of N-type voltage-gated calcium channels. Pain 2020, 161. [Google Scholar] [CrossRef]
- Salvemini, D.; Jacobson, K.A. Highly selective A3 adenosine receptor agonists relieve chronic neuropathic pain. Expert Opin. Ther. Patents 2017, 27, 967. [Google Scholar] [CrossRef]
- Ferreira-Silva, J.; Aires, I.D.; Boia, R.; Ambrósio, A.F.; Santiago, A.R. Activation of Adenosine A3 Receptor Inhibits Microglia Reactivity Elicited by Elevated Pressure. Int. J. Mol. Sci. 2020, 21, 7218. [Google Scholar] [CrossRef]
- Li, P.; Li, X.; Deng, P.; Wang, D.; Bai, X.; Li, Y.; Luo, C.; Belguise, K.; Wang, X.; Wei, X.; et al. Activation of adenosine A3 receptor reduces early brain injury by alleviating neuroinflammation after subarachnoid hemorrhage in elderly rats. Aging 2020, 13, 694–713. [Google Scholar] [CrossRef]
- Farr, S.A.; Cuzzocrea, S.; Esposito, E.; Campolo, M.; Niehoff, M.L.; Doyle, T.M.; Salvemini, D. Adenosine A3 receptor as a novel therapeutic target to reduce secondary events and improve neurocognitive functions following traumatic brain injury. J. Neuroinflamm. 2020, 17, 339. [Google Scholar] [CrossRef]
- Merighi, S.; Poloni, T.E.; Pelloni, L.; Pasquini, S.; Varani, K.; Vincenzi, F.; Borea, P.A.; Gessi, S. An Open Question: Is the A2A Adenosine Receptor a Novel Target for Alzheimer’s Disease Treatment? Front. Pharmacol. 2021, 12. [Google Scholar] [CrossRef]
- Rebola, N.; Canas, P.; Oliveira, C.; Cunha, R. Different synaptic and subsynaptic localization of adenosine A2A receptors in the hippocampus and striatum of the rat. Neuroscience 2005, 132, 893–903. [Google Scholar] [CrossRef]
- Orr, A.G.; Orr, A.L.; Li, X.-J.; Gross, R.E.; Traynelis, S.F. Adenosine A2A receptor mediates microglial process retraction. Nat. Neurosci. 2009, 12, 872–878. [Google Scholar] [CrossRef]
- Rebola, N.; Simões, A.P.; Canas, P.M.; Tomé, A.R.; Andrade, G.; Barry, C.E.; Agostinho, P.M.; Lynch, M.A.; Cunha, R.A. Adenosine A2A receptors control neuroinflammation and consequent hippocampal neuronal dysfunction. J. Neurochem. 2011, 117, 100–111. [Google Scholar] [CrossRef]
- Cunha, R.A.; Johansson, B.; Van Der Ploeg, I.; Sebastião, A.M.; Ribeiro, J.A.; Fredholm, B.B. Evidence for functionally important adenosine A2a receptors in the rat hippocampus. Brain Res. 1994, 649, 208–216. [Google Scholar] [CrossRef]
- Temido-Ferreira, M.; Ferreira, D.G.; Batalha, V.L.; Marques-Morgado, I.; Coelho, J.E.; Pereira, P.; Gomes, R.; Pinto, A.; Carvalho, S.; Canas, P.M.; et al. Age-related shift in LTD is dependent on neuronal adenosine A2A receptors interplay with mGluR5 and NMDA receptors. Mol. Psychiatry 2020, 25, 1876–1900. [Google Scholar] [CrossRef]
- Horgusluoglu-Moloch, E.; Nho, K.; Risacher, S.L.; Kim, S.; Foroud, T.; Shaw, L.M.; Trojanowski, J.Q.; Aisen, P.S.; Petersen, R.C.; Jack, C.R.; et al. Targeted neurogenesis pathway-based gene analysis identifies ADORA2A associated with hippocampal volume in mild cognitive impairment and Alzheimer’s disease. Neurobiol. Aging 2017, 60, 92–103. [Google Scholar] [CrossRef]
- Canas, P.M.; Porciúncula, L.O.; Cunha, G.M.A.; Silva, C.G.; Machado, N.J.; Oliveira, J.M.A.; Oliveira, C.R.; Cunha, R.A. Adenosine A2A Receptor Blockade Prevents Synaptotoxicity and Memory Dysfunction Caused by β-Amyloid Peptides via p38 Mitogen-Activated Protein Kinase Pathway. J. Neurosci. 2009, 29, 14741–14751. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimer’s Disease Is a Synaptic Failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef]
- Scheff, S.W.; Price, D.A.; Schmitt, F.A.; DeKosky, S.T.; Mufson, E.J. Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology 2007, 68, 1501–1508. [Google Scholar] [CrossRef]
- Scheff, S.W.; Price, D.A.; Ansari, M.A.; Roberts, K.N.; Schmitt, F.A.; Ikonomovic, M.D.; Mufson, E.J. Synaptic Change in the Posterior Cingulate Gyrus in the Progression of Alzheimer’s Disease. J. Alzheimer’s Dis. 2014, 43, 1073–1090. [Google Scholar] [CrossRef]
- Lopes, L.V.; Cunha, R.A.; Ribeiro, J.A. Increase in the Number, G Protein Coupling, and Efficiency of Facilitatory Adenosine A2A Receptors in the Limbic Cortex, but not Striatum, of Aged Rats. J. Neurochem. 2002, 73, 1733–1738. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Rial, D.; Canas, P.M.; Yoo, J.-H.; Li, W.; Zhou, X.; Wang, Y.; Van Westen, G.J.P.; Payen, M.-P.; Augusto, E.; et al. Optogenetic activation of intracellular adenosine A2A receptor signaling in the hippocampus is sufficient to trigger CREB phosphorylation and impair memory. Mol. Psychiatry 2015, 20, 1339–1349. [Google Scholar] [CrossRef]
- Pagnussat, N.; Almeida, A.S.; Marques, D.M.; Nunes, F.S.; Chenet, G.C.; Botton, P.H.S.; Mioranzza, S.; Loss, C.M.; Cunha, R.A.; Porciuncula, L.O. Adenosine A2Areceptors are necessary and sufficient to trigger memory impairment in adult mice. Br. J. Pharmacol. 2015, 172, 3831–3845. [Google Scholar] [CrossRef] [PubMed]
- Albasanz, J.L.; Perez, S.M.; Barrachina, M.; Ferrer, I.; Martín, M. RESEARCH ARTICLE: Up-regulation of Adenosine Receptors in the Frontal Cortex in Alzheimer’s Disease. Brain Pathol. 2008, 18, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Orr, A.G.; Hsiao, E.C.; Wang, M.M.; Ho, K.; Kim, D.H.; Wang, X.; Guo, W.; Kang, J.; Yu, G.-Q.; Adame, A.; et al. Astrocytic adenosine receptor A2A and Gs-coupled signaling regulate memory. Nat. Neurosci. 2015, 18, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Arendash, G.; Schleif, W.; Rezai-Zadeh, K.; Jackson, E.; Zacharia, L.; Cracchiolo, J.; Shippy, D.; Tan, J. Caffeine protects Alzheimer’s mice against cognitive impairment and reduces brain β-amyloid production. Neuroscience 2006, 142, 941–952. [Google Scholar] [CrossRef]
- Espinosa, J.; Rocha, A.; Nunes, F.; Costa, M.S.; Schein, V.; Kazlauckas, V.; Kalinine, E.; Souza, D.O.; Cunha, R.A.; Porciúncula, L.O. Caffeine Consumption Prevents Memory Impairment, Neuronal Damage, and Adenosine A2A Receptors Upregulation in the Hippocampus of a Rat Model of Sporadic Dementia. J. Alzheimer’s Dis. 2013, 34, 509–518. [Google Scholar] [CrossRef]
- Merighi, S.; Battistello, E.; Casetta, I.; Gragnaniello, D.; Poloni, T.E.; Medici, V.; Cirrincione, A.; Varani, K.; Vincenzi, F.; Borea, P.A.; et al. Upregulation of Cortical A2A Adenosine Receptors is Reflected in Platelets of Patients with Alzheimer’s Disease. J. Alzheimer’s Dis. 2021, 80, 1105–1117. [Google Scholar] [CrossRef]
- da Silva, S.V.; Haberl, M.G.; Zhang, P.; Bethge, P.; Lemos, C.; Gonçalves, N.; Gorlewicz, A.; Malezieux, M.; Gonçalves, F.Q.; Grosjean, N.; et al. Early synaptic deficits in the APP/PS1 mouse model of Alzheimer’s disease involve neuronal adenosine A2A receptors. Nat. Commun. 2016, 7, 11915. [Google Scholar] [CrossRef]
- Cunha, R.A. How does adenosine control neuronal dysfunction and neurodegeneration? J. Neurochem. 2016, 139, 1019–1055. [Google Scholar] [CrossRef]
- Shen, L.L.; Li, W.W.; Xu, Y.L.; Gao, S.H.; Xu, M.Y.; Bu, X.L.; Liu, Y.H.; Wang, J.; Zhu, J.; Zeng, F.; et al. Neurotrophin receptor p75 mediates amyloid β-induced tau pathology. Neurobiol. Dis. 2019, 132, 104567. [Google Scholar] [CrossRef]
- Dall’Igna, O.P.; Fett, P.; Gomes, M.W.; Souza, D.O.; Cunha, R.A.; Lara, D.R. Caffeine and adenosine A2a receptor antagonists prevent β-amyloid (25–35)-induced cognitive deficits in mice. Exp. Neurol. 2007, 203, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Orr, A.G.; Lo, I.; Schumacher, H.; Ho, K.; Gill, M.; Guo, W.; Kim, D.H.; Knox, A.; Saito, T.; Saido, T.C.; et al. Istradefylline reduces memory deficits in aging mice with amyloid pathology. Neurobiol. Dis. 2018, 110, 29–36. [Google Scholar] [CrossRef]
- Laurent, C.; Burnouf, S.; Ferry, B.; Batalha, V.L.; Coelho, J.E.; Baqi, Y.; Malik, E.; Mariciniak, E.; Parrot, S.; Van Der Jeugd, A.; et al. A2A adenosine receptor deletion is protective in a mouse model of Tauopathy. Mol. Psychiatry 2016, 21, 97–107. [Google Scholar] [CrossRef]
- Carvalho, K.; Faivre, E.; Pietrowski, M.J.; Marques, X.; Gomez-Murcia, V.; Deleau, A.; Huin, V.; Hansen, J.N.; Kozlov, S.; Danis, C.; et al. Exacerbation of C1q dysregulation, synaptic loss and memory deficits in tau pathology linked to neuronal adenosine A2A receptor. Brain 2019, 142, 3636–3654. [Google Scholar] [CrossRef] [PubMed]
- Santiago, A.R.; Baptista, F.I.; Santos, P.F.; Cristovão, G.; Ambrosio, A.F.; Cunha, R.A.; Gomes, C.A. Role of Microglia Adenosine A2AReceptors in Retinal and Brain Neurodegenerative Diseases. Mediat. Inflamm. 2014, 2014, 465694. [Google Scholar] [CrossRef]
- Illes, P.; Rubini, P.; Ulrich, H.; Zhao, Y.; Tang, Y. Regulation of Microglial Functions by Purinergic Mechanisms in the Healthy and Diseased CNS. Cells 2020, 9, 1108. [Google Scholar] [CrossRef]
- Franco, R.; Reyes-Resina, I.; Aguinaga, D.; Lillo, A.; Jiménez, J.; Raïch, I.; Borroto-Escuela, D.O.; Ferreiro-Vera, C.; Canela, E.I.; De Medina, V.S.; et al. Potentiation of cannabinoid signaling in microglia by adenosine A 2A receptor antagonists. Glia 2019, 67, 2410–2423. [Google Scholar] [CrossRef]
- Colella, M.; Zinni, M.; Pansiot, J.; Cassanello, M.; Mairesse, J.; Ramenghi, L.; Baud, O. Modulation of Microglial Activation by Adenosine A2a Receptor in Animal Models of Perinatal Brain Injury. Front. Neurol. 2018, 9, 605. [Google Scholar] [CrossRef]
- Franco, R.; Rivas-Santisteban, R.; Casanovas, M.; Lillo, A.; Saura, C.A.; Navarro, G. Adenosine A2A Receptor Antagonists Affects NMDA Glutamate Receptor Function. Potential to Address Neurodegeneration in Alzheimer’s Disease. Cells 2020, 9, 1075. [Google Scholar] [CrossRef]
- Rebola, N.; Lujan, R.; Cunha, R.A.; Mulle, C. Adenosine A2A Receptors Are Essential for Long-Term Potentiation of NMDA-EPSCs at Hippocampal Mossy Fiber Synapses. Neuron 2008, 57, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Mouro, F.M.; Rombo, D.M.; Dias, R.B.; Ribeiro, J.A.; Sebastião, A.M. Adenosine A2A receptors facilitate synaptic NMDA currents in CA1 pyramidal neurons. Br. J. Pharmacol. 2018, 175, 4386–4397. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.V.; Hansen, J.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Chambers, E.S.; Akbar, A.N. Can blocking inflammation enhance immunity during aging? J. Allergy Clin. Immunol. 2020, 145, 1323–1331. [Google Scholar] [CrossRef]
- Yates, S.L.; Burgess, L.H.; Kocsis-Angle, J.; Antal, J.M.; Dority, M.D.; Embury, P.B.; Piotrkowski, A.M.; Brunden, K.R. Amyloid β and Amylin Fibrils Induce Increases in Proinflammatory Cytokine and Chemokine Production by THP-1 Cells and Murine Microglia. J. Neurochem. 2000, 74, 1017–1025. [Google Scholar] [CrossRef]
- Shieh, C.-H.; Heinrich, A.; Serchov, T.; Van Calker, D.; Biber, K. P2X7-dependent, but differentially regulated release of IL-6, CCL2, and TNF-α in cultured mouse microglia. Glia 2014, 62, 592–607. [Google Scholar] [CrossRef]
- Calovi, S.; Mut-Arbona, P.; Sperlágh, B. Microglia and the Purinergic Signaling System. Neuroscience 2019, 405, 137–147. [Google Scholar] [CrossRef]
- Koizumi, S.; Shigemoto-Mogami, Y.; Nasu-Tada, K.; Shinozaki, Y.; Ohsawa, K.; Tsuda, M.; Joshi, B.V.; Jacobson, K.A.; Kohsaka, S.; Inoue, K. UDP acting at P2Y6 receptors is a mediator of microglial phagocytosis. Nature 2007, 446, 1091–1095. [Google Scholar] [CrossRef]
- Koizumi, S.; Ohsawa, K.; Inoue, K.; Kohsaka, S. Purinergic receptors in microglia: Functional modal shifts of microglia mediated by P2 and P1 receptors. Glia 2012, 61, 47–54. [Google Scholar] [CrossRef]
- Kim, S.Y.; Moon, J.H.; Lee, H.G.; Kim, S.U.; Beom Lee, Y.B. ATP released from beta-amyloid-stimulated microglia induces reactive oxygen species production in an autocrine fashion. Exp. Mol. Med. 2007, 39, 820–827. [Google Scholar] [CrossRef]
- Rampe, D.; Wang, L.; Ringheim, G.E. P2X7 receptor modulation of β-amyloid- and LPS-induced cytokine secretion from human macrophages and microglia. J. Neuroimmunol. 2004, 147, 56–61. [Google Scholar] [CrossRef]
- Sanz, J.M.; Chiozzi, P.; Ferrari, D.; Colaianna, M.; Idzko, M.; Falzoni, S.; Fellin, R.; Trabace, L.; Di Virgilio, F. Activation of microglia by amyloid β requires P2X7 receptor expression. J. Immunol. 2009, 182, 4378–4385. [Google Scholar] [CrossRef]
- Idzko, M.; Ferrari, D.; Eltzschig, H.K. Nucleotide signalling during inflammation. Nature 2014, 509, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Labasi, J.M.; Petrushova, N.; Donovan, C.; McCurdy, S.; Lira, P.; Payette, M.M.; Brissette, W.; Wicks, J.R.; Audoly, L.; Gabel, C.A. Absence of the P2X7Receptor Alters Leukocyte Function and Attenuates an Inflammatory Response. J. Immunol. 2002, 168, 6436–6445. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.K.; McLarnon, J.G. Block of purinergic P2X7 receptor is neuroprotective in an animal model of Alzheimer’s disease. NeuroReport 2008, 19, 1715–1719. [Google Scholar] [CrossRef]
- Hide, I.; Tanaka, M.; Inoue, A.; Nakajima, K.; Kohsaka, S.; Inoue, K.; Nakata, Y. Extracellular ATP Triggers Tumor Necrosis Factor-α Release from Rat Microglia. J. Neurochem. 2002, 75, 965–972. [Google Scholar] [CrossRef]
- Simi, A.; Tsakiri, N.; Wang, P.; Rothwell, N.J. Interleukin-1 and inflammatory neurodegeneration. Biochem. Soc. Trans. 2007, 35, 1122–1126. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.S.; Paris, D.; Mathura, V.; Quadros, A.N.; Crawford, F.C.; Mullan, M.J. Inflammatory cytokine levels correlate with amyloid load in transgenic mouse models of Alzheimer’s disease. J. Neuroinflamm. 2005, 2, 9. [Google Scholar] [CrossRef]
- Parajuli, B.; Sonobe, Y.; Horiuchi, H.; Takeuchi, H.; Mizuno, T.; Suzumura, A. Oligomeric amyloid β induces IL-1β processing via production of ROS: Implication in Alzheimer’s disease. Cell Death Dis. 2013, 4, e975. [Google Scholar] [CrossRef]
- Martin, E.; Amar, M.; Dalle, C.; Youssef, I.; Boucher, C.; Le Duigou, C.; Brückner, M.; Prigent, A.; Sazdovitch, V.; Halle, A.; et al. New role of P2X7 receptor in an Alzheimer’s disease mouse model. Mol. Psychiatry 2019, 24, 108–125. [Google Scholar] [CrossRef]
- Able, S.; Fish, R.; Bye, H.; Booth, L.; Logan, Y.; Nathaniel, C.; Hayter, P.; Katugampola, S. Receptor localization, native tissue binding and ex vivo occupancy for centrally penetrant P2X7 antagonists in the rat. Br. J. Pharmacol. 2010, 162, 405–414. [Google Scholar] [CrossRef]
- Eskelinen, M.H.; Ngandu, T.; Tuomilehto, J.; Soininen, H.; Kivipelto, M. Midlife Coffee and Tea Drinking and the Risk of Late-Life Dementia: A Population-Based CAIDE Study. J. Alzheimer’s Dis. 2009, 16, 85–91. [Google Scholar] [CrossRef]
- Eskelinen, M.H.; Kivipelto, M. Caffeine as a Protective Factor in Dementia and Alzheimer’s Disease. J. Alzheimer’s Dis. 2010, 20, S167–S174. [Google Scholar] [CrossRef]
- Gelber, R.P.; Petrovitch, H.; Masaki, K.H.; Ross, G.W.; White, L.R. Coffee Intake in Midlife and Risk of Dementia and its Neuropathologic Correlates. J. Alzheimer’s Dis. 2011, 23, 607–615. [Google Scholar] [CrossRef]
- Liu, Q.-P.; Wu, Y.-F.; Cheng, H.-Y.; Xia, T.; Ding, H.; Wang, H.; Wang, Z.-M.; Xu, Y. Habitual coffee consumption and risk of cognitive decline/dementia: A systematic review and meta-analysis of prospective cohort studies. Nutrition 2016, 32, 628–636. [Google Scholar] [CrossRef] [PubMed]
- Reyes, C.M.; Cornelis, M.C. Caffeine in the Diet: Country-Level Consumption and Guidelines. Nutrition 2018, 10, 1772. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.; Costa, J.; Santos, J.; Vaz-Carneiro, A.; Lunet, N. Caffeine Intake and Dementia: Systematic Review and Meta-Analysis. J. Alzheimer’s Dis. 2010, 20, S187–S204. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, K.; Tomata, Y.; Kaiho, Y.; Honkura, K.; Sugawara, Y.; Tsuji, I. Association between Coffee Consumption and Incident Risk of Disabling Dementia in Elderly Japanese: The Ohsaki Cohort 2006 Study. J. Alzheimer’s Dis. 2015, 50, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Li, S.; Sun, J.; Li, Y.; Zhang, D. Association of Coffee, Decaffeinated Coffee and Caffeine Intake from Coffee with Cognitive Performance in Older Adults: National Health and Nutrition Examination Survey (NHANES) 2011–2014. Nutrients 2020, 12, 840. [Google Scholar] [CrossRef]
- Iranpour, S.; Saadati, H.M.; Koohi, F.; Sabour, S. Association between caffeine intake and cognitive function in adults; effect modification by sex: Data from National Health and Nutrition Examination Survey (NHANES) 2013–2014. Clin. Nutr. 2020, 39, 2158–2168. [Google Scholar] [CrossRef]
- Yu, L.; Coelho, J.E.; Zhang, X.; Fu, Y.; Tillman, A.; Karaoz, U.; Fredholm, B.B.; Weng, Z.; Chen, J.-F. Uncovering multiple molecular targets for caffeine using a drug target validation strategy combining A2A receptor knockout mice with microarray profiling. Physiol. Genom. 2009, 37, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Lopes, J.P.; Pliássova, A.; Cunha, R.A. The physiological effects of caffeine on synaptic transmission and plasticity in the mouse hippocampus selectively depend on adenosine A1 and A2A receptors. Biochem. Pharmacol. 2019, 166, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, K.A.; Gao, Z.; Matricon, P.; Eddy, M.T.; Carlsson, J. Adenosine A2A receptor antagonists: From caffeine to selective non-xanthines. Br. J. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Maia, L.; De Mendonca, A. Does caffeine intake protect from Alzheimer’s disease? Eur. J. Neurol. 2002, 9, 377–382. [Google Scholar] [CrossRef]
- Lindsay, J.; Laurin, D.; Verreault, R.; Hébert, R.; Helliwell, B.; Hill, G.B.; McDowell, I. Risk Factors for Alzheimer’s Disease: A Prospective Analysis from the Canadian Study of Health and Aging. Am. J. Epidemiol. 2002, 156, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Van Gelder, B.M.; Buijsse, B.; Tijhuis, M.; Kalmijn, S.; Giampaoli, S.; Nissinen, A.; Kromhout, D. Coffee consumption is inversely associated with cognitive decline in elderly European men: The FINE Study. Eur. J. Clin. Nutr. 2006, 61, 226–232. [Google Scholar] [CrossRef]
- Ritchie, K.; Carrière, I.; De Mendonca, A.; Portet, F.; Dartigues, J.-F.; Rouaud, O.; Barberger-Gateau, P.; Ancelin, M.-L. The neuroprotective effects of caffeine: A prospective population study (the Three City Study). Neurology 2007, 69, 536–545. [Google Scholar] [CrossRef]
- Cao, C.; Loewenstein, D.A.; Lin, X.; Zhang, C.; Wang, L.; Duara, R.; Wu, Y.; Giannini, A.; Bai, G.; Cai, J.; et al. High Blood Caffeine Levels in MCI Linked to Lack of Progression to Dementia. J. Alzheimer’s Dis. 2012, 30, 559–572. [Google Scholar] [CrossRef]
- Potter, H.; Woodcock, J.H.; Boyd, T.D.; Coughlan, C.M.; O’Shaughnessy, J.R.; Borges, M.T.; Thaker, A.A.; Raj, B.A.; Adamszuk, K.; Scott, D.; et al. Safety and efficacy of sargramostim (GM-CSF) in the treatment of Alzheimer’s disease. Alzheimer’s Dement. 2021, 7, e12158. [Google Scholar] [CrossRef]
- Laske, C.; Stellos, K.; Stransky, E.; Leyhe, T.; Gawaz, M. Decreased Plasma Levels of Granulocyte-Colony Stimulating Factor (G-CSF) in Patients with Early Alzheimer’s Disease. J. Alzheimer’s Dis. 2009, 17, 115–123. [Google Scholar] [CrossRef]
- Zotova, E.; Bharambe, V.; Cheaveau, M.; Morgan, W.; Holmes, C.; Harris, S.; Neal, J.W.; Love, S.; Nicoll, J.A.R.; Boche, D. Inflammatory components in human Alzheimer’s disease and after active amyloid-β42 immunization. Brain 2013, 136, 2677–2696. [Google Scholar] [CrossRef]
- Mejias, N.H.; Martinez, C.C.; Stephens, M.E.; de Rivero Vaccari, J.P. Contribution of the inflammasome to inflammaging. J. Inflamm. (Lond). 2018, 15, 23. [Google Scholar] [CrossRef] [PubMed]
- Borota, D.; Murray, E.; Keceli, G.; Chang, A.; Watabe, J.M.; Ly, M.; Toscano, J.P.; Yassa, M.A. Post-study caffeine administration enhances memory consolidation in humans. Nat. Neurosci. 2014, 17, 201–203. [Google Scholar] [CrossRef] [PubMed]
- Favila, S.E.; Kuhl, B.A. Stimulating memory consolidation. Nat. Neurosci. 2014, 17, 151–152. [Google Scholar] [CrossRef]
- Arendash, G.W.; Mori, T.; Cao, C.; Mamcarz, M.; Runfeldt, M.; Dickson, A.; Rezai-Zadeh, K.; Tane, J.; Citron, B.A.; Lin, X.; et al. Caffeine reverses cognitive impairment and decreases brain amyloid-β levels in aged Alzheimer’s disease mice. J. Alzheimer’s Dis. 2009, 17, 661–680. [Google Scholar] [CrossRef] [PubMed]
- Canas, P.M.; Porciúncula, L.O.; Simões, A.P.; Augusto, E.; Silva, H.B.; Machado, N.J.; Gonçalves, N.; Alfaro, T.M.; Gonçalves, F.Q.; Araujo, I.; et al. Neuronal Adenosine A2A Receptors Are Critical Mediators of Neurodegeneration Triggered by Convulsions. eNeuro 2018, 5. [Google Scholar] [CrossRef]
- Cao, C.; Cirrito, J.R.; Lin, X.; Wang, L.; Verges, D.K.; Dickson, A.; Mamcarz, M.; Zhang, C.; Mori, T.; Arendash, G.W.; et al. Caffeine Suppresses Amyloid-β Levels in Plasma and Brain of Alzheimer’s Disease Transgenic Mice. J. Alzheimer’s Dis. 2009, 17, 681–697. [Google Scholar] [CrossRef] [PubMed]
- Laurent, C.; Eddarkaoui, S.; Derisbourg, M.; Leboucher, A.; Demeyer, D.; Carrier, S.; Schneider, M.; Hamdane, M.; Müller, C.E.; Buée, L.; et al. Beneficial effects of caffeine in a transgenic model of Alzheimer’s disease-like tau pathology. Neurobiol. Aging 2014, 35, 2079–2090. [Google Scholar] [CrossRef]
- Kolahdouzan, M.; Hamadeh, M.J. The neuroprotective effects of caffeine in neurodegenerative diseases. CNS Neurosci. Ther. 2017, 23, 272–290. [Google Scholar] [CrossRef]
- Costa, M.; Botton, P.; Mioranzza, S.; Souza, D.; Porciúncula, L. Caffeine prevents age-associated recognition memory decline and changes brain-derived neurotrophic factor and tirosine kinase receptor (TrkB) content in mice. Neuroscience 2008, 153, 1071–1078. [Google Scholar] [CrossRef]
- Kaster, M.P.; Machado, N.J.; Silva, H.B.; Nunes, A.; Ardais, A.P.; Santana, M.; Baqi, Y.; Müller, C.E.; Rodrigues, A.L.S.; Porciúncula, L.O.; et al. Caffeine acts through neuronal adenosine A2A receptors to prevent mood and memory dysfunction triggered by chronic stress. Proc. Natl. Acad. Sci. USA 2015, 112, 7833–7838. [Google Scholar] [CrossRef] [PubMed]
- Janitschke, D.; Nelke, C.; Lauer, A.A.; Regner, L.; Winkler, J.; Thiel, A.; Grimm, H.S.; Hartmann, T.; Grimm, M.O.W. Effect of Caffeine and Other Methylxanthines on Aβ-Homeostasis in SH-SY5Y Cells. Biomolecules 2019, 9, 689. [Google Scholar] [CrossRef] [PubMed]
- Sinyor, B.; Mineo, J.; Ochner, C. Alzheimer’s Disease, Inflammation, and the Role of Antioxidants. J. Alzheimer’s Dis. Rep. 2020, 4, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Travassos, M.; Santana, I.; Baldeiras, I.; Tsolaki, M.; Gkatzima, O.; Sermin, G.; Yener, G.G.; Simonsen, A.; Hasselbalch, S.G.; Kapaki, E.; et al. Does Caffeine Consumption Modify Cerebrospinal Fluid Amyloid-β Levels in Patients with Alzheimer’s Disease? J. Alzheimer’s Dis. 2015, 47, 1069–1078. [Google Scholar] [CrossRef] [PubMed]
- Moreira, A.; Diógenes, M.J.; De Mendonça, A.; Lunet, N.; Barros, H. Chocolate Consumption is Associated with a Lower Risk of Cognitive Decline. J. Alzheimer’s Dis. 2016, 53, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pathological overproduction: The bad side of adenosine. Br. J. Pharmacol. 2017, 174, 1945–1960. [Google Scholar] [CrossRef]
- Chen, J.-F.; Cunha, R.A. The belated US FDA approval of the adenosine A2A receptor antagonist istradefylline for treatment of Parkinson’s disease. Purinergic Signal. 2020, 16, 167–174. [Google Scholar] [CrossRef]
- Von Kügelgen, I.; Hoffmann, K. Pharmacology and structure of P2Y receptors. Neuropharmacology 2016, 104, 50–61. [Google Scholar] [CrossRef]
- Boeynaems, J.-M.; Communi, D.; Gonzalez, N.S.; Robaye, B. Overview of the P2 Receptors. Semin. Thromb. Hemost. 2005, 31, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Illes, P.; Ribeiro, J. Neuronal P2 Receptors of the Central Nervous System. Curr. Top. Med. Chem. 2004, 4, 831–838. [Google Scholar] [CrossRef]
- Neary, J.T.; Kang, Y.; Shi, Y.-F.; Tran, M.D.; Wanner, I.B. P2 Receptor Signalling, Proliferation of Astrocytes, and Expression of Molecules Involved in Cell-Cell Interactions. In Purinergic Signalling in Neuron–Glia Interactions; Novartis Foundation Symposia; John Wiley & Sons: Hoboken, NJ, USA, 2006; Volume 276, pp. 131–147. [Google Scholar] [CrossRef]
- Tsuda, M.; Shigemoto-Mogami, Y.; Koizumi, S.; Mizokoshi, A.; Kohsaka, S.; Salter, M.W.; Inoue, K. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nat. Cell Biol. 2003, 424, 778–783. [Google Scholar] [CrossRef] [PubMed]
- Andrejew, R.; Oliveira-Giacomelli, Á.; Ribeiro, D.E.; Glaser, T.; Arnaud-Sampaio, V.F.; Lameu, C.; Ulrich, H. The P2X7 Receptor: Central Hub of Brain Diseases. Front. Mol. Neurosci. 2020, 13, 124. [Google Scholar] [CrossRef] [PubMed]
- Sperlágh, B.; Vizi, E.S.; Wirkner, K.; Illes, P. P2X7 receptors in the nervous system. Prog. Neurobiol. 2006, 78, 327–346. [Google Scholar] [CrossRef]
- Sluyter, R. Significance of P2X7 Receptor Variants to Human Health and Disease. Recent Patents DNA Gene Seq. 2011, 5, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Sengoku, R. Aging and Alzheimer’s disease pathology. Neuropathology 2020, 40, 22–29. [Google Scholar] [CrossRef]
- Woods, L.T.; Ajit, D.; Camden, J.M.; Erb, L.; Weisman, G.A. Purinergic receptors as potential therapeutic targets in Alzheimer’s disease. Neuropharmacology 2016, 104, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. Calcium signalling and Alzheimer’s disease. Neurochem. Res. 2011, 36, 1149–1156. [Google Scholar] [CrossRef]
- Pham, C.; Hérault, K.; Oheim, M.; Maldera, S.; Vialou, V.; Cauli, B.; Li, D. Astrocytes respond to a neurotoxic Aβ fragment with state-dependent Ca2+ alteration and multiphasic transmitter release. Acta Neuropathol. Commun. 2021, 9, 44. [Google Scholar] [CrossRef]
- Jin, H.; Han, J.; Resing, D.; Liu, H.; Yue, X.; Miller, R.L.; Schoch, K.M.; Miller, T.M.; Perlmutter, J.S.; Egan, T.M.; et al. Synthesis and in vitro characterization of a P2X7 radioligand [123I]TZ6019 and its response to neuroinflammation in a mouse model of Alzheimer disease. Eur. J. Pharmacol. 2018, 820, 8–17. [Google Scholar] [CrossRef]
- McLarnon, J.G.; Ryu, M.J.K.; Walker, D.G.; Choi, B.H.B. Upregulated Expression of Purinergic P2X7Receptor in Alzheimer Disease and Amyloid-β Peptide-Treated Microglia and in Peptide-Injected Rat Hippocampus. J. Neuropathol. Exp. Neurol. 2006, 65, 1090–1097. [Google Scholar] [CrossRef]
- Gallart-Palau, X.; Serra, A.; Lee, B.S.T.; Guo, X.; Sze, S.K. Brain ureido degenerative protein modifications are associated with neuroinflammation and proteinopathy in Alzheimer’s disease with cerebrovascular disease. J. Neuroinflamm. 2017, 14, 175. [Google Scholar] [CrossRef] [PubMed]
- Savio, L.E.B.; Mello, P.D.A.; Da Silva, C.G.; Coutinho-Silva, R. The P2X7 receptor in inflammatory diseases: Angel or demon? Front. Pharmacol. 2018, 9, 52. [Google Scholar] [CrossRef]
- Sanz, J.M.; Falzoni, S.; Rizzo, R.; Cipollone, F.; Zuliani, G.; Di Virgilio, F. Possible protective role of the 489C>T P2X7R polymorphism in Alzheimer’s disease. Exp. Gerontol. 2014, 60, 117–119. [Google Scholar] [CrossRef] [PubMed]
- Perregaux, D.G.; McNiff, P.; Laliberte, R.; Conklyn, M.; Gabel, C.A. ATP Acts as an Agonist to Promote Stimulus-Induced Secretion of IL-1β and IL-18 in Human Blood. J. Immunol. 2000, 165, 4615–4623. [Google Scholar] [CrossRef] [PubMed]
- Solle, M.; Labasi, J.; Perregaux, D.G.; Stam, E.; Petrushova, N.; Koller, B.H.; Griffiths, R.J.; Gabel, C.A. Altered Cytokine Production in Mice Lacking P2X7Receptors. J. Biol. Chem. 2001, 276, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Browne, S.E. When too much ATP is a bad thing: A pivotal role for P2X7 receptors in motor neuron degeneration. J. Neurochem. 2013, 126, 301–304. [Google Scholar] [CrossRef] [PubMed]
- Kong, Q.; Wang, M.; Liao, Z.; Camden, J.M.; Yu, S.; Simonyi, A.; Sun, G.Y.; Gonzalez, F.A.; Erb, L.; Seye, C.I.; et al. P2X7 nucleotide receptors mediate caspase-8/9/3-dependent apoptosis in rat primary cortical neurons. Purinergic Signal. 2005, 1, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Delarasse, C.; Auger, R.; Gonnord, P.; Fontaine, B.; Kanellopoulos, J.M. The Purinergic Receptor P2X7 Triggers α-Secretase-dependent Processing of the Amyloid Precursor Protein. J. Biol. Chem. 2011, 286, 2596–2606. [Google Scholar] [CrossRef]
- León-Otegui, M.; Gómez-Villafuertes, R.; Díaz-Hernández, J.I.; Díaz-Hernández, M.; Miras-Portugal, M.T.; Gualix, J. Opposite effects of P2X7 and P2Y2nucleotide receptors on α-secretase-dependent APP processing in Neuro-2a cells. FEBS Lett. 2011, 585, 2255–2262. [Google Scholar] [CrossRef]
- Diaz-Hernandez, J.I.; Gomez-Villafuertes, R.; León-Otegui, M.; Hontecillas-Prieto, L.; Del Puerto, A.; Trejo, J.L.; Lucas, J.J.; Garrido, J.J.; Gualix, J.; Miras-Portugal, M.T.; et al. In vivo P2X7 inhibition reduces amyloid plaques in Alzheimer’s disease through GSK3β and secretases. Neurobiol. Aging 2012, 33, 1816–1828. [Google Scholar] [CrossRef]
- Miras-Portugal, M.T.; Diaz-Hernandez, J.I.; Gomez-Villafuertes, R.; Diaz-Hernandez, M.; Artalejo, A.R.; Gualix, J. Role of P2X7 and P2Y2 receptors on α-secretase-dependent APP processing: Control of amyloid plaques formation “in vivo” by P2X7 receptor. Comput. Struct. Biotechnol. J. 2015, 13, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Sáez-Orellana, F.; Fuentes-Fuentes, M.C.; Godoy, P.A.; Silva-Grecchi, T.; Panes, J.D.; Guzmán, L.; Yévenes, G.E.; Gavilán, J.; Egan, T.M.; Aguayo, L.G.; et al. P2X receptor overexpression induced by soluble oligomers of amyloid beta peptide potentiates synaptic failure and neuronal dyshomeostasis in cellular models of Alzheimer’s disease. Neuropharmacology 2018, 128, 366–378. [Google Scholar] [CrossRef]
- Varma, R.; Chai, Y.; Troncoso, J.; Gu, J.; Xing, H.; Stojilkovic, S.S.; Mattson, M.P.; Haughey, N.J. Amyloid-β Induces a Caspase-mediated Cleavage of P2X4 to Promote Purinotoxicity. Neuromol. Med. 2009, 11, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Erb, L.; Woods, L.T.; Khalafalla, M.G.; Weisman, G.A. Purinergic signaling in Alzheimer’s disease. Brain Res. Bull. 2019, 151, 25–37. [Google Scholar] [CrossRef]
- Kong, Q.; Peterson, T.S.; Baker, O.; Stanley, E.; Camden, J.; Seye, C.I.; Erb, L.; Simonyi, A.; Wood, W.G.; Sun, G.Y.; et al. Interleukin-1beta enhances nucleotide-induced and alpha-secretase-dependent amyloid precursor protein processing in rat primary cortical neurons via up-regulation of the P2Y(2) receptor. J. Neurochem. 2009, 109, 1300–1310. [Google Scholar] [CrossRef]
- Reichenbach, N.; Delekate, A.; Breithausen, B.; Keppler, K.; Poll, S.; Schulte, T.; Peter, J.; Plescher, M.; Hansen, J.N.; Blank, N.; et al. P2Y1 receptor blockade normalizes network dysfunction and cognition in an Alzheimer’s disease model. J. Exp. Med. 2018, 215, 1649–1663. [Google Scholar] [CrossRef] [PubMed]
- Delekate, A.; Füchtemeier, M.; Schumacher, T.; Ulbrich, C.; Foddis, M.; Petzold, G.C. Metabotropic P2Y1 receptor signalling mediates astrocytic hyperactivity in vivo in an Alzheimer’s disease mouse model. Nat. Commun. 2014, 5, 5422. [Google Scholar] [CrossRef]
- Kim, H.J.; Ajit, D.; Peterson, T.S.; Wang, Y.; Camden, J.M.; Wood, W.G.; Sun, G.Y.; Erb, L.; Petris, M.; Weisman, G.A. Nucleotides released from Aβ₁₋₄₂ -treated microglial cells increase cell migration and Aβ₁₋₄₂ uptake through P2Y₂ receptor activation. J. Neurochem. 2012, 121, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-Q.; Chen, C.; Dou, Y.; Wu, H.-J.; Liu, Y.-J.; Lou, H.-F.; Zhang, J.-M.; Li, X.-M.; Wang, H.; Duan, S. P2Y4 Receptor-Mediated Pinocytosis Contributes to Amyloid Beta-Induced Self-Uptake by Microglia. Mol. Cell. Biol. 2013, 33, 4282–4293. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Thawkar, B.S.; Kaur, G. Inhibitors of NF-κB and P2X7/NLRP3/Caspase 1 pathway in microglia: Novel therapeutic opportunities in neuroinflammation induced early-stage Alzheimer’s disease. J. Neuroimmunol. 2019, 326, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Temido-Ferreira, M.; Coelho, J.E.; Pousinha, P.A.; Lopes, L.V. Novel Players in the Aging Synapse: Impact on Cognition. J. Caffeine Adenosine Res. 2019, 9, 104–127. [Google Scholar] [CrossRef] [PubMed]
- Haynes, S.E.; Hollopeter, G.; Yang, G.; Kurpius, D.; Dailey, M.E.; Gan, W.-B.; Julius, D. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat. Neurosci. 2006, 9, 1512–1519. [Google Scholar] [CrossRef]
- Arulkumaran, N.; Unwin, R.J.; Tam, F.W.K. A potential therapeutic role for P2X7 receptor (P2X7R) antagonists in the treatment of inflammatory diseases. Expert Opin. Investig. Drugs 2011, 20, 897–915. [Google Scholar] [CrossRef] [PubMed]
- Chrovian, C.C.; Rech, J.C.; Bhattacharya, A.; Letavic, M.A. P2X7 Antagonists as Potential Therapeutic Agents for the Treatment of CNS Disorders. Prog. Med. Chem. 2014, 53, 65–100. [Google Scholar] [CrossRef] [PubMed]
- Rech, J.C.; Bhattacharya, A.; Letavic, M.A.; Savall, B.M. The evolution of P2X7 antagonists with a focus on CNS indications. Bioorg. Med. Chem. Lett. 2016, 26, 3838–3845. [Google Scholar] [CrossRef] [PubMed]
- Labrousse, V.; Costes, L.; Aubert, A.; Darnaudéry, M.; Ferreira, G.; Amédée, T.; Layé, S. Impaired interleukin-1β and c-Fos expression in the hippocampus is associated with a spatial memory deficit in P2X7 receptor-deficient mice. PLoS ONE 2009, 4, e6006. [Google Scholar] [CrossRef] [PubMed]
- Gelin, C.F.; Bhattacharya, A.; Letavic, M.A. P2X7 receptor antagonists for the treatment of systemic inflammatory disorders. Prog. Med. Chem. 2020, 59, 63–99. [Google Scholar] [CrossRef]
- Takenouchi, T.; Sekiyama, K.; Sekigawa, A.; Fujita, M.; Waragai, M.; Sugama, S.; Iwamaru, Y.; Kitani, H.; Hashimoto, M. P2X7 Receptor Signaling Pathway as a Therapeutic Target for Neurodegenerative Diseases. Arch. Immunol. Ther. Exp. 2010, 58, 91–96. [Google Scholar] [CrossRef]
- Domingos, L.; Hott, S.; Terzian, A.; Resstel, L. P2X7 purinergic receptors participate in the expression and extinction processes of contextual fear conditioning memory in mice. Neuropharmacology 2018, 128, 474–481. [Google Scholar] [CrossRef]
- Borzelleca, J.; Depukat, K.; Hallagan, J. Lifetime toxicity/carcinogenicity studies of FD & C blue No. 1 (Brilliant blue FCF) in rats and mice. Food Chem. Toxicol. 1990, 28, 221–234. [Google Scholar] [CrossRef]
- Gao, M.; Wang, M.; Green, M.A.; Hutchins, G.D.; Zheng, Q.-H. Synthesis of [11C]GSK1482160 as a new PET agent for targeting P2X7 receptor. Bioorg. Med. Chem. Lett. 2015, 25, 1965–1970. [Google Scholar] [CrossRef] [PubMed]
- Territo, P.R.; Meyer, J.A.; Peters, J.S.; Riley, A.A.; McCarthy, B.P.; Gao, M.; Wang, M.; Green, M.A.; Zheng, Q.-H.; Hutchins, G.D. Characterization of 11 C-GSK1482160 for Targeting the P2X7 Receptor as a Biomarker for Neuroinflammation. J. Nucl. Med. 2017, 58, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Baudelet, D.; Lipka, E.; Millet, R.; Ghinet, A. Involvement of the P2X7 purinergic receptor in inflammation: An update of antagonists series since 2009 and their promising therapeutic potential. Curr. Med. Chem. 2015, 22, 713–729. [Google Scholar] [CrossRef]
- Murphy, N.; Cowley, T.R.; Richardson, J.C.; Virley, D.; Upton, N.; Walter, D.; Lynch, M.A. The Neuroprotective Effect of a Specific P2X7 Receptor Antagonist Derives from its Ability to Inhibit Assembly of the NLRP3 Inflammasome in Glial Cells. Brain Pathol. 2011, 22, 295–306. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Ceusters, M. Targeting neuroinflammation with brain penetrant P2X7 antagonists as novel therapeutics for neuropsychiatric disorders. Neuropsychopharmacology 2020, 45, 234–235. [Google Scholar] [CrossRef] [PubMed]
- Parvathenani, L.K.; Tertyshnikova, S.; Greco, C.R.; Roberts, S.B.; Robertson, B.; Posmantur, R. P2X7 Mediates Superoxide Production in Primary Microglia and Is Up-regulated in a Transgenic Mouse Model of Alzheimer’s Disease. J. Biol. Chem. 2003, 278, 13309–13317. [Google Scholar] [CrossRef] [PubMed]
- Letavic, M.A.; Savall, B.M.; Allison, B.D.; Aluisio, L.; Andres, J.I.; De Angelis, M.; Ao, H.; Beauchamp, D.A.; Bonaventure, P.; Bryant, S.; et al. 4-Methyl-6,7-dihydro-4H-triazolo[4,5-c]pyridine-Based P2X7 Receptor Antagonists: Optimization of Pharmacokinetic Properties Leading to the Identification of a Clinical Candidate. J. Med. Chem. 2017, 60, 4559–4572. [Google Scholar] [CrossRef]
- Danino, O.; Grossman, S.; Fischer, B. ATP-γ-S-(α,β-CH2) protects against oxidative stress and amyloid beta toxicity in neuronal culture. Biochem. Biophys. Res. Commun. 2015, 460, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Espada, S.; Ortega, F.; Molina-Jijón, E.; Rojo, A.I.; Pérez-Sen, R.; Pedraza-Chaverri, J.; Miras-Portugal, M.T.; Cuadrado, A. The purinergic P2Y13 receptor activates the Nrf2/HO-1 axis and protects against oxidative stress-induced neuronal death. Free Radic. Biol. Med. 2010, 49, 416–426. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Purinergic Receptor | Cell Type | Response | Role in AD | Refrerences |
|---|---|---|---|---|
| A1 | Neuron | TAU translocation | positive | [36] |
| Non-Am. APP Processing | Positive | [36] | ||
| A2A | Neuron | Aβ-Induced Neurotoxiciy | Negative | [54,59,60,66,68,69] |
| Neuron | Increased Aβ accumulation | Negative | [84,85] | |
| Neuron | Tau pathology | Negative | [71,72] | |
| Neuron | Memory damage | Negative | [84] | |
| Astrocytes | Memory function | Negative | [62] | |
| Microglia | Neuroinflammation | Negative | [75,76] | |
| A2B | Neuron | Mitochondrial function | Positive | [39,40] |
| A3 | Microglia | Decrease of inflammation | Positive | [44] |
| P2X4 | Neuron | Neurotoxicity | Negative | [86,87] |
| P2X7 | Microglia | Inflammation | Negative | [88,89,90,91,92,93,94] |
| Neuron | Amyloidogenic | Negative | [95,96,97] | |
| P2Y1 | Astrocyte | Hyper-Activation | Negative | [98] |
| Neuron | Synaptic damage | Negative | [98] | |
| P2Y2 | Neuron | Neurite Development | Positive | [95] |
| Neuron | Non-Am. APP Processing | Positive | [99] | |
| P2Y4 | Microglia | Uptake Aβ1-42 | Positive | [100] |
| P2Y6 | Microglia | Phagocytosis | Positive | [86] |
| P2Y12 | Microglia | Migration | Positive | [101] |
| P2Y13 | Neuron | Decrease ROS-induced death | Positive | [102] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merighi, S.; Poloni, T.E.; Terrazzan, A.; Moretti, E.; Gessi, S.; Ferrari, D. Alzheimer and Purinergic Signaling: Just a Matter of Inflammation? Cells 2021, 10, 1267. https://doi.org/10.3390/cells10051267
Merighi S, Poloni TE, Terrazzan A, Moretti E, Gessi S, Ferrari D. Alzheimer and Purinergic Signaling: Just a Matter of Inflammation? Cells. 2021; 10(5):1267. https://doi.org/10.3390/cells10051267
Chicago/Turabian StyleMerighi, Stefania, Tino Emanuele Poloni, Anna Terrazzan, Eva Moretti, Stefania Gessi, and Davide Ferrari. 2021. "Alzheimer and Purinergic Signaling: Just a Matter of Inflammation?" Cells 10, no. 5: 1267. https://doi.org/10.3390/cells10051267
APA StyleMerighi, S., Poloni, T. E., Terrazzan, A., Moretti, E., Gessi, S., & Ferrari, D. (2021). Alzheimer and Purinergic Signaling: Just a Matter of Inflammation? Cells, 10(5), 1267. https://doi.org/10.3390/cells10051267