
Chromosome Instability, Aging and Brain Diseases



{kind=link}
Abstract
1. Introduction
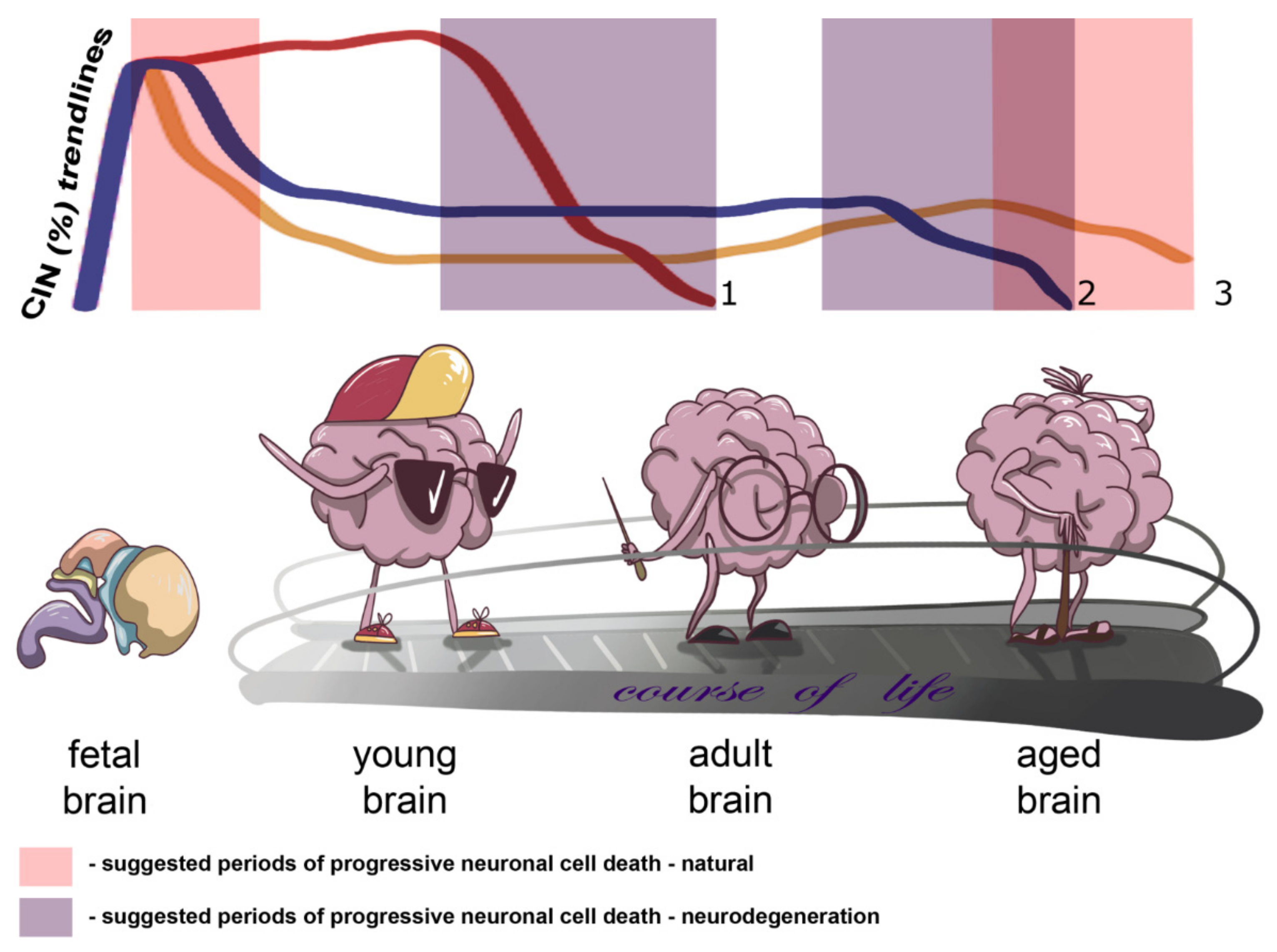
2. CIN in the Human Brain: An Ontogenetic View
3. CIN in the Diseased Brain: An Aging Perspective
4. CIN in the Aged Brain: The Shape of Things to Come
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jacobs, P.A.; Court Brown, W.M.; Doll, R. Distribution of human chromosome counts in relation to age. Nature 1961, 191, 1178–1180. [Google Scholar] [CrossRef] [PubMed]
- Russell, L.M.; Strike, P.; Browne, C.E.; Jacobs, P.A. X chromosome loss and ageing. Cytogenet. Genome Res. 2007, 116, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Yurov, Y.B.; Vorsanova, S.G.; Iourov, I.Y. Ontogenetic variation of the human genome. Curr. Genom. 2010, 11, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Guo, X. Decoding and rejuvenating human ageing genomes: Lessons from mosaic chromosomal alterations. Ageing Res. Rev. 2021, 68, 101342. [Google Scholar] [CrossRef]
- Simonetti, G.; Bruno, S.; Padella, A.; Tenti, E.; Martinelli, G. Aneuploidy: Cancer strength or vulnerability? Int. J. Cancer 2019, 144, 8–25. [Google Scholar] [CrossRef]
- Heng, J.; Heng, H.H. Genome chaos: Creating new genomic information essential for cancer macroevolution. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef]
- Iourov, I.Y.; Vorsanova, S.G.; Yurov, Y.B.; Kutsev, S.I. Ontogenetic and pathogenetic views on somatic chromosomal mosaicism. Genes 2019, 10, 379. [Google Scholar] [CrossRef]
- Kennedy, S.R.; Loeb, L.A.; Herr, A.J. Somatic mutations in aging, cancer and neurodegeneration. Mech. Ageing Dev. 2012, 133, 118–126. [Google Scholar] [CrossRef]
- Chow, H.M.; Herrup, K. Genomic integrity and the ageing brain. Nat. Rev. Neurosci. 2015, 16, 672–684. [Google Scholar] [CrossRef]
- Vijg, J.; Dong, X.; Milholland, B.; Zhang, L. Genome instability: A conserved mechanism of ageing? Essays Biochem. 2017, 61, 305–315. [Google Scholar] [CrossRef]
- Helbling-Leclerc, A.; Garcin, C.; Rosselli, F. Beyond DNA repair and chromosome instability—Fanconi anaemia as a cellular senescence-associated syndrome. Cell Death Differ. 2021, 28, 1159–1173. [Google Scholar] [CrossRef]
- Iourov, I.Y.; Vorsanova, S.G.; Liehr, T.; Kolotii, A.D.; Yurov, Y.B. Increased chromosome instability dramatically disrupts neural genome integrity and mediates cerebellar degeneration in the ataxia-telangiectasia brain. Hum. Mol. Genet. 2009, 18, 2656–2669. [Google Scholar] [CrossRef]
- Hou, Y.; Song, H.; Croteau, D.L.; Akbari, M.; Bohr, V.A. Genome instability in Alzheimer disease. Mech. Ageing Dev. 2017, 161, 83–94. [Google Scholar] [CrossRef]
- Iourov, I.Y.; Vorsanova, S.G.; Liehr, T.; Yurov, Y.B. Aneuploidy in the normal, Alzheimer’s disease and ataxia-telangiectasia brain: Differential expression and pathological meaning. Neurobiol. Dis. 2009, 34, 212–220. [Google Scholar] [CrossRef]
- Shepherd, C.E.; Yang, Y.; Halliday, G.M. Region- and cell-specific aneuploidy in brain aging and neurodegeneration. Neuroscience 2018, 374, 326–334. [Google Scholar] [CrossRef]
- Potter, H.; Chial, H.J.; Caneus, J.; Elos, M.; Elder, N.; Borysov, S.; Granic, A. Chromosome instability and mosaic aneuploidy in neurodegenerative and neurodevelopmental disorders. Front. Genet. 2019, 10, 1092. [Google Scholar] [CrossRef]
- Zhang, L.; Vijg, J. Somatic mutagenesis in mammals and its implications for human disease and aging. Annu. Rev. Genet. 2018, 52, 397–419. [Google Scholar] [CrossRef]
- Jourdon, A.; Fasching, L.; Scuderi, S.; Abyzov, A.; Vaccarino, F.M. The role of somatic mosaicism in brain disease. Curr. Opin. Genet. Dev. 2020, 65, 84–90. [Google Scholar] [CrossRef]
- Rohrback, S.; Siddoway, B.; Liu, C.S.; Chun, J. Genomic mosaicism in the developing and adult brain. Dev. Neurobiol. 2018, 78, 1026–1048. [Google Scholar] [CrossRef]
- Graham, E.J.; Vermeulen, M.; Vardarajan, B.; Bennett, D.; De Jager, P.; Pearse, R.V., II; Young-Pearse, T.L.; Mostafavi, S. Somatic mosaicism of sex chromosomes in the blood and brain. Brain Res. 2019, 1721, 146345. [Google Scholar] [CrossRef]
- Yurov, Y.B.; Iourov, I.Y.; Monakhov, V.V.; Soloviev, I.V.; Vostrikov, V.M.; Vorsanova, S.G. The variation of aneuploidy frequency in the developing and adult human brain revealed by an interphase FISH study. J. Histochem. Cytochem. 2005, 53, 385–390. [Google Scholar] [CrossRef]
- Yurov, Y.B.; Iourov, I.Y.; Vorsanova, S.G.; Liehr, T.; Kolotii, A.D.; Kutsev, S.I.; Pellestor, F.; Beresheva, A.K.; Demidova, I.A.; Kravets, V.S.; et al. Aneuploidy and confined chromosomal mosaicism in the developing human brain. PLoS ONE 2007, 2, e558. [Google Scholar] [CrossRef]
- Rohrback, S.; April, C.; Kaper, F.; Rivera, R.R.; Liu, C.S.; Siddoway, B.; Chun, J. Submegabase copy number variations arise during cerebral cortical neurogenesis as revealed by single-cell whole-genome sequencing. Proc. Natl. Acad. Sci. USA 2018, 115, 10804–10809. [Google Scholar] [CrossRef]
- Sekar, S.; Tomasini, L.; Proukakis, C.; Bae, T.; Manlove, L.; Jang, Y.; Scuderi, S.; Zhou, B.; Kalyva, M.; Amiri, A.; et al. Complex mosaic structural variations in human fetal brains. Genome Res. 2020, 30, 1695–1704. [Google Scholar] [CrossRef]
- Yurov, Y.B.; Vorsanova, S.G.; Iourov, I.Y. GIN′n′CIN hypothesis of brain aging: Deciphering the role of somatic genetic instabilities and neural aneuploidy during ontogeny. Mol. Cytogenet. 2009, 2, 23. [Google Scholar] [CrossRef]
- Wilhelm, T.; Said, M.; Naim, V. DNA replication stress and chromosomal instability: Dangerous liaisons. Genes 2020, 11, 642. [Google Scholar] [CrossRef]
- Sah, E.; Krishnamurthy, S.; Ahmidouch, M.Y.; Gillispie, G.J.; Milligan, C.; Orr, M.E. The cellular senescence stress response in post-mitotic brain cells: Cell survival at the expense of tissue degeneration. Life 2021, 11, 229. [Google Scholar] [CrossRef]
- Iourov, I.Y.; Vorsanova, S.G.; Yurov, Y.B. Chromosomal mosaicism goes global. Mol. Cytogenet. 2008, 1, 26. [Google Scholar] [CrossRef]
- Vijg, J. Somatic mutations, genome mosaicism, cancer and aging. Curr. Opin. Genet. Dev. 2014, 26, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, J.; Osei-Owusu, I.A.; Avigdor, B.E.; Tupler, R.; Pevsner, J. Mosaicism in human health and disease. Annu. Rev. Genet. 2020, 54, 487–510. [Google Scholar] [CrossRef]
- Fielder, E.; von Zglinicki, T.; Jurk, D. The DNA damage response in neurons: Die by apoptosis or survive in a senescence-like state? J. Alzheimers Dis. 2017, 60, S107–S131. [Google Scholar] [CrossRef] [PubMed]
- Ueberham, U.; Arendt, T. Genomic indexing by somatic gene recombination of mRNA/ncRNA—Does it play a role in genomic mosaicism, memory formation, and Alzheimer’s disease? Front. Genet. 2020, 11, 370. [Google Scholar] [CrossRef] [PubMed]
- Iourov, I.Y.; Vorsanova, S.G.; Kurinnaia, O.S.; Zelenova, M.A.; Vasin, K.S.; Yurov, Y.B. Causes and consequences of genome instability in psychiatric and neurodegenerative diseases. Mol. Biol. 2021, 55, 37–46. [Google Scholar] [CrossRef]
- Arendt, T.; Brückner, M.K.; Lösche, A. Regional mosaic genomic heterogeneity in the elderly and in Alzheimer’s disease as a correlate of neuronal vulnerability. Acta Neuropathol. 2015, 130, 501–510. [Google Scholar] [CrossRef]
- Bajic, V.P.; Essack, M.; Zivkovic, L.; Stewart, A.; Zafirovic, S.; Bajic, V.B.; Gojobori, T.; Isenovic, E.; Spremo-Potparevic, B. The X Files: “The mystery of X chromosome instability in Alzheimer’s disease”. Front. Genet. 2020, 10, 1368. [Google Scholar] [CrossRef]
- Guerreiro, R.; Gibbons, E.; Tábuas-Pereira, M.; Kun-Rodrigues, C.; Santo, G.C.; Bras, J. Genetic architecture of common non-Alzheimer’s disease dementias. Neurobiol. Dis. 2020, 142, 104946. [Google Scholar] [CrossRef]
- Neuner, S.M.; Tcw, J.; Goate, A.M. Genetic architecture of Alzheimer’s disease. Neurobiol. Dis. 2020, 143, 104976. [Google Scholar] [CrossRef]
- Malik, B.; Currais, A.; Andres, A.; Towlson, C.; Pitsi, D.; Nunes, A.; Niblock, M.; Cooper, J.; Hortobágyi, T.; Soriano, S. Loss of neuronal cell cycle control as a mechanism of neurodegeneration in the presenilin-1 Alzheimer’s disease brain. Cell Cycle 2008, 7, 637–646. [Google Scholar] [CrossRef]
- Currais, A.; Hortobágyi, T.; Soriano, S. The neuronal cell cycle as a mechanism of pathogenesis in Alzheimer’s disease. Aging 2009, 1, 363–371. [Google Scholar] [CrossRef]
- Granic, A.; Padmanabhan, J.; Norden, M.; Potter, H. Alzheimer Abeta peptide induces chromosome mis-segregation and aneuploidy, including trisomy 21: Requirement for tau and APP. Mol. Biol. Cell 2010, 21, 511–520. [Google Scholar] [CrossRef]
- Iourov, I.Y.; Vorsanova, S.G.; Yurov, Y.B. Genomic landscape of the Alzheimer’s disease brain: Chromosome instability—Aneuploidy, but not tetraploidy—Mediates neurodegeneration. Neurodegener. Dis. 2011, 8, 35–37. [Google Scholar] [CrossRef]
- Nudelman, K.N.H.; McDonald, B.C.; Lahiri, D.K.; Saykin, A.J. Biological hallmarks of cancer in Alzheimer’s disease. Mol. Neurobiol. 2019, 56, 7173–7187. [Google Scholar] [CrossRef]
- Boeras, D.I.; Granic, A.; Padmanabhan, J.; Crespo, N.C.; Rojiani, A.M.; Potter, H. Alzheimer’s presenilin 1 causes chromosome missegregation and aneuploidy. Neurobiol. Aging 2008, 29, 319–328. [Google Scholar] [CrossRef]
- Bajic, V.; Spremo-Potparevic, B.; Zivkovic, L.; Isenovic, E.R.; Arendt, T. Cohesion and the aneuploid phenotype in Alzheimer’s disease: A tale of genome instability. Neurosci. Biobehav. Rev. 2015, 55, 365–374. [Google Scholar] [CrossRef]
- Snyder, H.M.; Bain, L.J.; Brickman, A.M.; Carrillo, M.C.; Esbensen, A.J.; Espinosa, J.M.; Fernandez, F.; Fortea, J.; Hartley, S.L.; Head, E.; et al. Further understanding the connection between Alzheimer’s disease and Down syndrome. Alzheimers Dement. 2020, 16, 1065–1077. [Google Scholar] [CrossRef]
- Yurov, Y.B.; Vorsanova, S.G.; Liehr, T.; Kolotii, A.D.; Iourov, I.Y. X chromosome aneuploidy in the Alzheimer’s disease brain. Mol. Cytogenet. 2014, 7, 20. [Google Scholar] [CrossRef]
- Lee, M.H.; Siddoway, B.; Kaeser, G.E.; Segota, I.; Rivera, R.; Romanow, W.J.; Liu, C.S.; Park, C.; Kennedy, G.; Long, T.; et al. Somatic APP gene recombination in Alzheimer’s disease and normal neurons. Nature 2018, 563, 639–645. [Google Scholar] [CrossRef]
- Lin, X.; Kapoor, A.; Gu, Y.; Chow, M.J.; Peng, J.; Zhao, K.; Tang, D. Contributions of DNA damage to Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 1666. [Google Scholar] [CrossRef]
- Van Leeuwen, L.A.; Hoozemans, J.J. Physiological and pathophysiological functions of cell cycle proteins in post-mitotic neurons: Implications for Alzheimer’s disease. Acta Neuropathol. 2015, 129, 511–525. [Google Scholar] [CrossRef]
- Yurov, Y.B.; Vorsanova, S.G.; Iourov, I.Y. The DNA replication stress hypothesis of Alzheimer’s disease. Sci. World J. 2011, 11, 2602–2612. [Google Scholar] [CrossRef]
- Guo, L.; Zhong, M.B.; Zhang, L.; Zhang, B.; Cai, D. Sex differences in Alzheimer’s disease: Insights from the multiomics landscape. Biol. Psychiatry 2021. [Google Scholar] [CrossRef]
- Yang, Y.; Shepherd, C.; Halliday, G. Aneuploidy in Lewy body diseases. Neurobiol. Aging 2015, 36, 1253–1260. [Google Scholar] [CrossRef]
- Caneus, J.; Granic, A.; Rademakers, R.; Dickson, D.W.; Coughlan, C.M.; Chial, H.J.; Potter, H. Mitotic defects lead to neuronal aneuploidy and apoptosis in frontotemporal lobar degeneration caused by MAPT mutations. Mol. Biol. Cell 2018, 29, 575–586. [Google Scholar] [CrossRef]
- Rossi, G.; Conconi, D.; Panzeri, E.; Redaelli, S.; Piccoli, E.; Paoletta, L.; Dalprà, L.; Tagliavini, F. Mutations in MAPT gene cause chromosome instability and introduce copy number variations widely in the genome. J. Alzheimers Dis. 2013, 33, 969–982. [Google Scholar] [CrossRef]
- Gentile, G.; La Cognata, V.; Cavallaro, S. The contribution of CNVs to the most common aging-related neurodegenerative diseases. Aging Clin. Exp. Res. 2020. [Google Scholar] [CrossRef]
- Sferra, A.; Nicita, F.; Bertini, E. Microtubule dysfunction: A common feature of neurodegenerative diseases. Int. J. Mol. Sci. 2020, 21, 7354. [Google Scholar] [CrossRef]
- Yurov, Y.B.; Vorsanova, S.G.; Iourov, I.Y. Chromosome instability in the neurodegenerating brain. Front. Genet. 2019, 10, 892. [Google Scholar] [CrossRef]
- Baker, D.J.; Petersen, R.C. Cellular senescence in brain aging and neurodegenerative diseases: Evidence and perspectives. J. Clin. Investig. 2018, 128, 1208–1216. [Google Scholar] [CrossRef]
- Martínez-Cué, C.; Rueda, N. Cellular senescence in neurodegenerative diseases. Front. Cell. Neurosci. 2020, 14, 16. [Google Scholar] [CrossRef]
- Saez-Atienzar, S.; Masliah, E. Cellular senescence and Alzheimer disease: The egg and the chicken scenario. Nat. Rev. Neurosci. 2020, 21, 433–444. [Google Scholar] [CrossRef]
- Van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Sikora, E.; Bielak-Zmijewska, A.; Dudkowska, M.; Krzystyniak, A.; Mosieniak, G.; Wesierska, M.; Wlodarczyk, J. Cellular senescence in brain aging. Front. Aging Neurosci. 2021, 13, 646924. [Google Scholar] [CrossRef] [PubMed]
- Flanary, B.E.; Sammons, N.W.; Nguyen, C.; Walker, D.; Streit, W.J. Evidence that aging and amyloid promote microglial cell senescence. Rejuvenation Res. 2007, 10, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Ogrodnik, M. Cellular aging beyond cellular senescence: Markers of senescence prior to cell cycle arrest in vitro and in vivo. Aging Cell 2021, 20, e13338. [Google Scholar] [CrossRef]
- Fricker, M.; Tolkovsky, A.M.; Borutaite, V.; Coleman, M.; Brown, G.C. Neuronal cell death. Physiol. Rev. 2018, 98, 813–880. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Barroso-Vilares, M.; Macedo, J.C.; Reis, M.; Warren, J.D.; Compton, D.; Logarinho, E. Small-molecule inhibition of aging-associated chromosomal instability delays cellular senescence. EMBO Rep. 2020, 21, e49248. [Google Scholar] [CrossRef]
- Vorsanova, S.G.; Yurov, Y.B.; Iourov, I.Y. Dynamic nature of somatic chromosomal mosaicism, genetic-environmental interactions and therapeutic opportunities in disease and aging. Mol. Cytogenet. 2020, 13, 16. [Google Scholar] [CrossRef]
- Faggioli, F.; Wang, T.; Vijg, J.; Montagna, C. Chromosome-specific accumulation of aneuploidy in the aging mouse brain. Hum. Mol. Genet. 2012, 21, 5246–5253. [Google Scholar] [CrossRef]
- Fischer, H.G.; Morawski, M.; Brückner, M.K.; Mittag, A.; Tarnok, A.; Arendt, T. Changes in neuronal DNA content variation in the human brain during aging. Aging Cell 2012, 11, 628–633. [Google Scholar] [CrossRef]
- Andriani, G.A.; Vijg, J.; Montagna, C. Mechanisms and consequences of aneuploidy and chromosome instability in the aging brain. Mech. Ageing Dev. 2017, 161, 19–36. [Google Scholar] [CrossRef]
- Macedo, J.C.; Vaz, S.; Logarinho, E. Mitotic dysfunction associated with aging hallmarks. Adv. Exp. Med. Biol. 2017, 1002, 153–188. [Google Scholar]
- Iourov, I.Y.; Vorsanova, S.G.; Yurov, Y.B. Somatic cell genomics of brain disorders: A new opportunity to clarify genetic-environmental interactions. Cytogenet. Genome Res. 2013, 139, 181–188. [Google Scholar] [CrossRef]
- Barroso-Vilares, M.; Logarinho, E. Chromosomal instability and pro-inflammatory response in aging. Mech. Ageing Dev. 2019, 182, 111118. [Google Scholar] [CrossRef]
- Hägg, S.; Jylhävä, J. Sex differences in biological aging with a focus on human studies. Elife 2021, 10, e63425. [Google Scholar] [CrossRef]
- Thadathil, N.; Hori, R.; Xiao, J.; Khan, M.M. DNA double-strand breaks: A potential therapeutic target for neurodegenerative diseases. Chromosome Res. 2019, 27, 345–364. [Google Scholar] [CrossRef]
- Iourov, I.Y.; Vorsanova, S.G.; Yurov, Y.B.; Zelenova, M.A.; Kurinnaia, O.S.; Vasin, K.S.; Kutsev, S.I. The cytogenomic “theory of everything”: Chromohelkosis may underlie chromosomal instability and mosaicism in disease and aging. Int. J. Mol. Sci. 2020, 21, 8328. [Google Scholar] [CrossRef]
- Pathak, R.U.; Soujanya, M.; Mishra, R.K. Deterioration of nuclear morphology and architecture: A hallmark of senescence and aging. Ageing Res. Rev. 2021, 67, 101264. [Google Scholar] [CrossRef]
- Petr, M.A.; Tulika, T.; Carmona-Marin, L.M.; Scheibye-Knudsen, M. Protecting the aging genome. Trends Cell Biol. 2020, 30, 117–132. [Google Scholar] [CrossRef]
- Iourov, I.Y.; Vorsanova, S.G.; Yurov, Y.B. Pathway-based classification of genetic diseases. Mol. Cytogenet. 2019, 12, 4. [Google Scholar] [CrossRef]
- Melo Pereira, S.; Ribeiro, R.; Logarinho, E. Approaches towards longevity: Reprogramming, senolysis, and improved mitotic competence as anti-aging therapies. Int. J. Mol. Sci. 2019, 20, 938. [Google Scholar] [CrossRef] [PubMed]
- Iourov, I.Y.; Vorsanova, S.G.; Yurov, Y.B. The variome concept: Focus on CNVariome. Mol. Cytogenet. 2019, 12, 52. [Google Scholar] [CrossRef] [PubMed]
- Heng, H.H. New data collection priority: Focusing on genome-based bioinformation. Res. Results Biomed. 2020, 6, 5–8. [Google Scholar] [CrossRef]
- Chronister, W.D.; Burbulis, I.E.; Wierman, M.B.; Wolpert, M.J.; Haakenson, M.F.; Smith, A.C.B.; Kleinman, J.E.; Hyde, T.M.; Weinberger, D.R.; Bekiranov, S.; et al. Neurons with complex karyotypes are rare in aged human neocortex. Cell Rep. 2019, 26, 825–835.e7. [Google Scholar] [CrossRef] [PubMed]
- Iourov, I.Y.; Vorsanova, S.G.; Yurov, Y.B. Single cell genomics of the brain: Focus on neuronal diversity and neuropsychiatric diseases. Curr. Genom. 2012, 13, 477–488. [Google Scholar] [CrossRef][Green Version]
- Medvedev, Z.A. An attempt at a rational classification of theories of ageing. Biol. Rev. Camb. Philos. Soc. 1990, 65, 375–398. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iourov, I.Y.; Yurov, Y.B.; Vorsanova, S.G.; Kutsev, S.I. Chromosome Instability, Aging and Brain Diseases. Cells 2021, 10, 1256. https://doi.org/10.3390/cells10051256
Iourov IY, Yurov YB, Vorsanova SG, Kutsev SI. Chromosome Instability, Aging and Brain Diseases. Cells. 2021; 10(5):1256. https://doi.org/10.3390/cells10051256
Chicago/Turabian StyleIourov, Ivan Y., Yuri B. Yurov, Svetlana G. Vorsanova, and Sergei I. Kutsev. 2021. "Chromosome Instability, Aging and Brain Diseases" Cells 10, no. 5: 1256. https://doi.org/10.3390/cells10051256
APA StyleIourov, I. Y., Yurov, Y. B., Vorsanova, S. G., & Kutsev, S. I. (2021). Chromosome Instability, Aging and Brain Diseases. Cells, 10(5), 1256. https://doi.org/10.3390/cells10051256