
Regulatory Effects of Cannabidiol on Mitochondrial Functions: A Review


Abstract
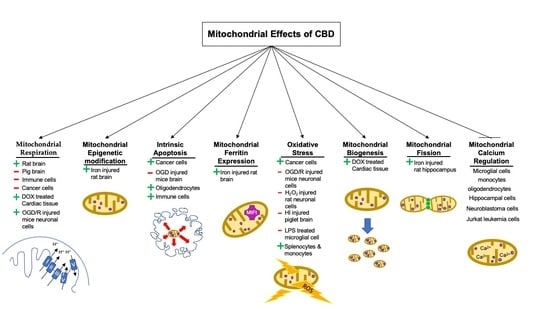
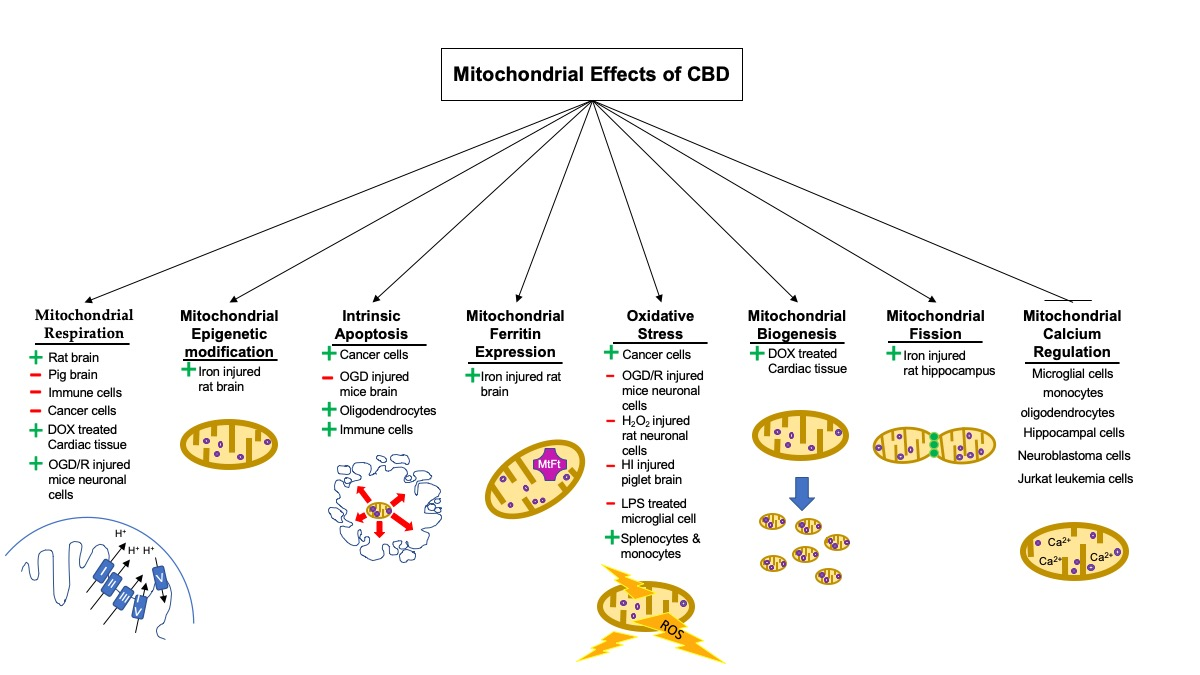
1. Introduction
2. Pharmacokinetics of CBD—A Brief Overview
3. CBD and Mitochondrial Respiration/Bioenergetics
4. CBD and Epigenetic Modifications of Mitochondrial DNA
5. CBD and Intrinsic Apoptosis
6. CBD and Oxidative Stress
7. CBD and Mitochondrial Regulation of Intracellular Calcium
8. CBD and Mitochondrial Fission and Fusion
9. CBD and Mitochondrial Biogenesis
10. CBD and Mitochondrial Ferritin Regulation
11. CBD and Monoamine Oxidase
12. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ujvary, I.; Hanus, L. Human Metabolites of Cannabidiol: A Review on Their Formation, Biological Activity, and Relevance in Therapy. Cannabis Cannabinoid Res. 2016, 1, 90–101. [Google Scholar] [CrossRef]
- Adams, R.; Hunt, M.; Clark, J.H. Structure of Cannabidiol, a Product Isolated from the Marihuana Extract of Minnesota Wild Hemp. I. J. Am. Chem. Soc. 1940, 62, 196–200. [Google Scholar] [CrossRef]
- Mechoulam, R.; Shvo, Y. Hashish—I: The structure of Cannabidiol. Tetrahedron 1963, 19, 2073–2078. [Google Scholar] [CrossRef]
- Perez-Reyes, M.; Timmons, M.C.; Davis, K.H.; Wall, E.M. A comparison of the pharmacological activity in man of intravenously administered delta9-tetrahydrocannabinol, cannabinol, and cannabidiol. Experientia 1973, 29, 1368–1369. [Google Scholar] [CrossRef]
- Zuardi, A.W.; Shirakawa, I.; Finkelfarb, E.; Karniol, I.G. Action of cannabidiol on the anxiety and other effects produced by delta 9-THC in normal subjects. Psychopharmacology 1982, 76, 245–250. [Google Scholar] [CrossRef]
- Cunha, J.M.; Carlini, E.A.; Pereira, A.E.; Ramos, O.L.; Pimentel, C.; Gagliardi, R.; Sanvito, W.L.; Lander, N.; Mechoulam, R. Chronic Administration of Cannabidiol to Healthy Volunteers and Epileptic Patients. Pharmacology 1980, 21, 175–185. [Google Scholar] [CrossRef]
- Consroe, P.; Martin, A.; Singh, V. Antiepileptic potential of cannabidiol analogs. J. Clin. Pharm. 1981, 21, 428S–436S. [Google Scholar] [CrossRef]
- Martin, A.R.; Consroe, P.; Kane, V.V. Structure-anticonvulsant activity relationships of cannabidiol analogs. Nida. Res. Monogr. 1987, 79, 48–58. [Google Scholar]
- Zuardi, A.W.; Antunes Rodrigues, J.; Cunha, J.M. Effects of cannabidiol in animal models predictive of antipsychotic activity. Psychopharmacology 1991, 104, 260–264. [Google Scholar] [CrossRef]
- Thomas, B.; Gilliam, A.F.; Burch, D.; Roche, M.; Seltzman, H. Comparative Receptor Binding Analyses of Cannabinoid Agonists and Antagonists. J. Pharmacol. Exp. Ther. 1998, 285, 285–292. [Google Scholar]
- Mechoulam, R.; Peters, M.; Murillo-Rodriguez, E.; Hanus, L.O. Cannabidiol–recent advances. Chem. Biodivers. 2007, 4, 1678–1692. [Google Scholar] [CrossRef]
- Thomas, A.; Baillie, G.L.; Phillips, A.M.; Razdan, R.K.; Ross, R.A.; Pertwee, R.G. Cannabidiol displays unexpectedly high potency as an antagonist of CB1 and CB2 receptor agonists in vitro. Br. J. Pharm. 2007, 150, 613–623. [Google Scholar] [CrossRef]
- Pertwee, R.G. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: Delta9-tetrahydrocannabinol, cannabidiol and delta9-tetrahydrocannabivarin. Br. J. Pharm. 2008, 153, 199–215. [Google Scholar] [CrossRef]
- Miller, S.; Daily, L.; Leishman, E.; Bradshaw, H.; Straiker, A. Δ9-Tetrahydrocannabinol and Cannabidiol Differentially Regulate Intraocular Pressure. Investig. Ophthalmol. Vis. Sci. 2018, 59, 5904–5911. [Google Scholar] [CrossRef]
- Gray, R.A.; Stott, C.G.; Jones, N.A.; Di Marzo, V.; Whalley, B.J. Anticonvulsive Properties of Cannabidiol in a Model of Generalized Seizure Are Transient Receptor Potential Vanilloid 1 Dependent. Cannabis Cannabinoid Res. 2020, 5, 145–149. [Google Scholar] [CrossRef]
- Russo, E.B.; Burnett, A.; Hall, B.; Parker, K.K. Agonistic properties of cannabidiol at 5-HT1a receptors. Neurochem. Res. 2005, 30, 1037–1043. [Google Scholar] [CrossRef]
- De Filippis, D.; Esposito, G.; Cirillo, C.; Cipriano, M.; De Winter, B.Y.; Scuderi, C.; Sarnelli, G.; Cuomo, R.; Steardo, L.; De Man, J.G.; et al. Cannabidiol reduces intestinal inflammation through the control of neuroimmune axis. PLoS ONE 2011, 6, e28159. [Google Scholar] [CrossRef]
- Ribeiro, A.; Ferraz-de-Paula, V.; Pinheiro, M.L.; Vitoretti, L.B.; Mariano-Souza, D.P.; Quinteiro-Filho, W.M.; Akamine, A.T.; Almeida, V.I.; Quevedo, J.; Dal-Pizzol, F.; et al. Cannabidiol, a non-psychotropic plant-derived cannabinoid, decreases inflammation in a murine model of acute lung injury: Role for the adenosine A(2A) receptor. Eur. J. Pharm. 2012, 678, 78–85. [Google Scholar] [CrossRef]
- Atalay, S.; Jarocka-Karpowicz, I.; Skrzydlewska, E. Antioxidative and Anti-Inflammatory Properties of Cannabidiol. Antioxidants 2019, 9, 21. [Google Scholar] [CrossRef]
- Ryan, D.; Drysdale, A.J.; Lafourcade, C.; Pertwee, R.G.; Platt, B. Cannabidiol targets mitochondria to regulate intracellular Ca2+ levels. J. Neurosci. 2009, 29, 2053–2063. [Google Scholar] [CrossRef]
- McKallip, R.J.; Jia, W.; Schlomer, J.; Warren, J.W.; Nagarkatti, P.S.; Nagarkatti, M. Cannabidiol-induced apoptosis in human leukemia cells: A novel role of cannabidiol in the regulation of p22phox and Nox4 expression. Mol. Pharm. 2006, 70, 897–908. [Google Scholar] [CrossRef]
- da Silva, V.K.; de Freitas, B.S.; Dornelles, V.C.; Kist, L.W.; Bogo, M.R.; Silva, M.C.; Streck, E.L.; Hallak, J.E.; Zuardi, A.W.; Crippa, J.A.S.; et al. Novel insights into mitochondrial molecular targets of iron-induced neurodegeneration: Reversal by cannabidiol. Brain Res. Bull. 2018, 139, 1–8. [Google Scholar] [CrossRef]
- Valvassori, S.S.; Bavaresco, D.V.; Scaini, G.; Varela, R.B.; Streck, E.L.; Chagas, M.H.; Hallak, J.E.; Zuardi, A.W.; Crippa, J.A.; Quevedo, J. Acute and chronic administration of cannabidiol increases mitochondrial complex and creatine kinase activity in the rat brain. Braz. J. Psychiatry 2013, 35, 380–386. [Google Scholar] [CrossRef]
- Hao, E.; Mukhopadhyay, P.; Cao, Z.; Erdelyi, K.; Holovac, E.; Liaudet, L.; Lee, W.S.; Hasko, G.; Mechoulam, R.; Pacher, P. Cannabidiol Protects against Doxorubicin-Induced Cardiomyopathy by Modulating Mitochondrial Function and Biogenesis. Mol. Med. 2015, 21, 38–45. [Google Scholar] [CrossRef]
- da Silva, V.K.; de Freitas, B.S.; da Silva Dornelles, A.; Nery, L.R.; Falavigna, L.; Ferreira, R.D.; Bogo, M.R.; Hallak, J.E.; Zuardi, A.W.; Crippa, J.A.; et al. Cannabidiol normalizes caspase 3, synaptophysin, and mitochondrial fission protein DNM1L expression levels in rats with brain iron overload: Implications for neuroprotection. Mol. Neurobiol. 2014, 49, 222–233. [Google Scholar] [CrossRef]
- Reddy, P.H. Role of mitochondria in neurodegenerative diseases: Mitochondria as a therapeutic target in Alzheimer’s disease. Cns Spectr. 2009, 14, 8–18. [Google Scholar] [CrossRef]
- Siasos, G.; Tsigkou, V.; Kosmopoulos, M.; Theodosiadis, D.; Simantiris, S.; Tagkou, N.M.; Tsimpiktsioglou, A.; Stampouloglou, P.K.; Oikonomou, E.; Mourouzis, K.; et al. Mitochondria and cardiovascular diseases-from pathophysiology to treatment. Ann. Transl. Med. 2018, 6, 256. [Google Scholar] [CrossRef]
- Pinti, M.V.; Fink, G.K.; Hathaway, Q.A.; Durr, A.J.; Kunovac, A.; Hollander, J.M. Mitochondrial dysfunction in type 2 diabetes mellitus: An organ-based analysis. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E268–E285. [Google Scholar] [CrossRef]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef]
- Millar, S.A.; Stone, N.L.; Yates, A.S.; O’Sullivan, S.E. A Systematic Review on the Pharmacokinetics of Cannabidiol in Humans. Front. Pharm. 2018, 9, 1365. [Google Scholar] [CrossRef]
- Ohlsson, A.; Lindgren, J.E.; Andersson, S.; Agurell, S.; Gillespie, H.; Hollister, L.E. Single-dose kinetics of deuterium-labelled cannabidiol in man after smoking and intravenous administration. Biomed. Env. Mass Spectrom. 1986, 13, 77–83. [Google Scholar] [CrossRef]
- Manini, A.F.; Yiannoulos, G.; Bergamaschi, M.M.; Hernandez, S.; Olmedo, R.; Barnes, A.J.; Winkel, G.; Sinha, R.; Jutras-Aswad, D.; Huestis, M.A.; et al. Safety and pharmacokinetics of oral cannabidiol when administered concomitantly with intravenous fentanyl in humans. J. Addict. Med. 2015, 9, 204–210. [Google Scholar] [CrossRef]
- Guy, G.W.; Robson, P.J. A Phase I, Open Label, Four-Way Crossover Study to Compare the Pharmacokinetic Profiles of a Single Dose of 20 mg of a Cannabis Based Medicine Extract (CBME) Administered on 3 Different Areas of the Buccal Mucosa and to Investigate the Pharmacokinetics of CBME per Oral in Healthy Male and Female Volunteers (GWPK0112). J. Cannabis Ther. 2004, 3, 79–120. [Google Scholar]
- Contin, M.; Mohamed, S.; Santucci, M.; Lodi, M.A.M.; Russo, E.; Mecarelli, O.; Cbd Lice Italy Study, G. Cannabidiol in Pharmacoresistant Epilepsy: Clinical Pharmacokinetic Data From an Expanded Access Program. Front. Pharm. 2021, 12, 637801. [Google Scholar] [CrossRef]
- Rimmerman, N.; Ben-Hail, D.; Porat, Z.; Juknat, A.; Kozela, E.; Daniels, M.P.; Connelly, P.S.; Leishman, E.; Bradshaw, H.B.; Shoshan-Barmatz, V.; et al. Direct modulation of the outer mitochondrial membrane channel, voltage-dependent anion channel 1 (VDAC1) by cannabidiol: A novel mechanism for cannabinoid-induced cell death. Cell Death Dis. 2013, 4, e949. [Google Scholar] [CrossRef]
- Fisar, Z.; Singh, N.; Hroudova, J. Cannabinoid-induced changes in respiration of brain mitochondria. Toxicol. Lett. 2014, 231, 62–71. [Google Scholar] [CrossRef]
- Singh, N.; Hroudova, J.; Fisar, Z. Cannabinoid-Induced Changes in the Activity of Electron Transport Chain Complexes of Brain Mitochondria. J. Mol. Neurosci. 2015, 56, 926–931. [Google Scholar] [CrossRef]
- Schultze, N.; Wanka, H.; Zwicker, P.; Lindequist, U.; Haertel, B. Mitochondrial functions of THP-1 monocytes following the exposure to selected natural compounds. Toxicology 2017, 377, 57–63. [Google Scholar] [CrossRef]
- Jeong, S.; Yun, H.K.; Jeong, Y.A.; Jo, M.J.; Kang, S.H.; Kim, J.L.; Kim, D.Y.; Park, S.H.; Kim, B.R.; Na, Y.J.; et al. Cannabidiol-induced apoptosis is mediated by activation of Noxa in human colorectal cancer cells. Cancer Lett. 2019, 447, 12–23. [Google Scholar] [CrossRef]
- Jeong, S.; Jo, M.J.; Yun, H.K.; Kim, D.Y.; Kim, B.R.; Kim, J.L.; Park, S.H.; Na, Y.J.; Jeong, Y.A.; Kim, B.G.; et al. Cannabidiol promotes apoptosis via regulation of XIAP/Smac in gastric cancer. Cell Death Dis. 2019, 10, 846. [Google Scholar] [CrossRef]
- Sun, S.; Hu, F.; Wu, J.; Zhang, S. Cannabidiol attenuates OGD/R-induced damage by enhancing mitochondrial bioenergetics and modulating glucose metabolism via pentose-phosphate pathway in hippocampal neurons. Redox. Biol. 2017, 11, 577–585. [Google Scholar] [CrossRef]
- Massi, P.; Vaccani, A.; Bianchessi, S.; Costa, B.; Macchi, P.; Parolaro, D. The non-psychoactive cannabidiol triggers caspase activation and oxidative stress in human glioma cells. Cell. Mol. Life Sci. 2006, 63, 2057–2066. [Google Scholar] [CrossRef]
- Olivas-Aguirre, M.; Torres-Lopez, L.; Valle-Reyes, J.S.; Hernandez-Cruz, A.; Pottosin, I.; Dobrovinskaya, O. Cannabidiol directly targets mitochondria and disturbs calcium homeostasis in acute lymphoblastic leukemia. Cell. Death Dis. 2019, 10, 779. [Google Scholar] [CrossRef]
- Shrivastava, A.; Kuzontkoski, P.M.; Groopman, J.E.; Prasad, A. Cannabidiol induces programmed cell death in breast cancer cells by coordinating the cross-talk between apoptosis and autophagy. Mol. Cancer 2011, 10, 1161–1172. [Google Scholar] [CrossRef]
- Castillo, A.; Tolon, M.R.; Fernandez-Ruiz, J.; Romero, J.; Martinez-Orgado, J. The neuroprotective effect of cannabidiol in an in vitro model of newborn hypoxic-ischemic brain damage in mice is mediated by CB(2) and adenosine receptors. Neurobiol. Dis. 2010, 37, 434–440. [Google Scholar] [CrossRef]
- Mato, S.; Victoria Sanchez-Gomez, M.; Matute, C. Cannabidiol induces intracellular calcium elevation and cytotoxicity in oligodendrocytes. Glia 2010, 58, 1739–1747. [Google Scholar] [CrossRef]
- Wu, H.Y.; Huang, C.H.; Lin, Y.H.; Wang, C.C.; Jan, T.R. Cannabidiol induced apoptosis in human monocytes through mitochondrial permeability transition pore-mediated ROS production. Free Radic. Biol. Med. 2018, 124, 311–318. [Google Scholar] [CrossRef]
- Lee, C.Y.; Wey, S.P.; Liao, M.H.; Hsu, W.L.; Wu, H.Y.; Jan, T.R. A comparative study on cannabidiol-induced apoptosis in murine thymocytes and EL-4 thymoma cells. Int. Immunopharmacol. 2008, 8, 732–740. [Google Scholar] [CrossRef]
- Kim, J.; Choi, J.Y.; Seo, J.; Choi, I.S. Neuroprotective Effect of Cannabidiol Against Hydrogen Peroxide in Hippocampal Neuron Culture. Cannabis Cannabinoid Res. 2021, 6, 40–47. [Google Scholar] [CrossRef]
- di Giacomo, V.; Chiavaroli, A.; Recinella, L.; Orlando, G.; Cataldi, A.; Rapino, M.; Di Valerio, V.; Ronci, M.; Leone, S.; Brunetti, L.; et al. Antioxidant and Neuroprotective Effects Induced by Cannabidiol and Cannabigerol in Rat CTX-TNA2 Astrocytes and Isolated Cortexes. Int. J. Mol. Sci. 2020, 21, 3575. [Google Scholar] [CrossRef]
- Dos-Santos-Pereira, M.; Guimaraes, F.S.; Del-Bel, E.; Raisman-Vozari, R.; Michel, P.P. Cannabidiol prevents LPS-induced microglial inflammation by inhibiting ROS/NF-kappaB-dependent signaling and glucose consumption. Glia 2020, 68, 561–573. [Google Scholar] [CrossRef]
- Wu, H.Y.; Chu, R.M.; Wang, C.C.; Lee, C.Y.; Lin, S.H.; Jan, T.R. Cannabidiol-induced apoptosis in primary lymphocytes is associated with oxidative stress-dependent activation of caspase-8. Toxicol. Appl. Pharm. 2008, 226, 260–270. [Google Scholar] [CrossRef]
- Bertram, R.; Pedersen, M.; Luciani, D.; Sherman, A. A simplified model for mitochondrial ATP production. J. Theor. Biol. 2006, 243, 575–586. [Google Scholar] [CrossRef]
- Sousa, J.S.; D’Imprima, E.; Vonck, J. Mitochondrial Respiratory Chain Complexes. In Membrane Protein Complexes: Structure and Function; Harris, J.R., Boekema, E.J., Eds.; Springer: Singapore, 2018; pp. 167–227. [Google Scholar]
- Bonora, M.; Patergnani, S.; Rimessi, A.; De Marchi, E.; Suski, J.M.; Bononi, A.; Giorgi, C.; Marchi, S.; Missiroli, S.; Poletti, F.; et al. ATP synthesis and storage. Purinergic Signal. 2012, 8, 343–357. [Google Scholar] [CrossRef]
- Sendoel, A.; Hengartner, M.O. Apoptotic cell death under hypoxia. Physiology 2014, 29, 168–176. [Google Scholar] [CrossRef]
- Eguchi, Y.; Shimizu, S.; Tsujimoto, Y. Intracellular ATP Levels Determine Cell Death Fate by Apoptosis or Necrosis. Cancer Res. 1997, 57, 1835–1840. [Google Scholar]
- Dupont, C.; Armant, D.R.; Brenner, C.A. Epigenetics: Definition, mechanisms and clinical perspective. Semin. Reprod. Med. 2009, 27, 351–357. [Google Scholar] [CrossRef]
- Stimpfel, M.; Jancar, N.; Virant-Klun, I. New Challenge: Mitochondrial Epigenetics? Stem. Cell Rev. Rep. 2018, 14, 13–26. [Google Scholar] [CrossRef]
- Manev, H.; Dzitoyeva, S. Progress in mitochondrial epigenetics. Biomol. Concepts 2013, 4, 381–389. [Google Scholar] [CrossRef]
- Coppede, F.; Stoccoro, A. Mitoepigenetics and Neurodegenerative Diseases. Front. Endocrinol. 2019, 10, 86. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Diaz-Mendoza, M.J.; Lorda-Diez, C.I.; Montero, J.A.; Garcia-Porrero, J.A.; Hurle, J.M. Interdigital cell death in the embryonic limb is associated with depletion of Reelin in the extracellular matrix. Cell Death Dis. 2013, 4, e800. [Google Scholar] [CrossRef]
- Brentnall, M.; Rodriguez-Menocal, L.; De Guevara, R.L.; Cepero, E.; Boise, L.H. Caspase-9, caspase-3 and caspase-7 have distinct roles during intrinsic apoptosis. BMC Cell Biol. 2013, 12. [Google Scholar] [CrossRef]
- Wu, C.C.; Bratton, S.B. Regulation of the intrinsic apoptosis pathway by reactive oxygen species. Antioxid. Redox Signal. 2013, 19, 546–558. [Google Scholar] [CrossRef]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef]
- Massi, P.; Solinas, M.; Cinquina, V.; Parolaro, D. Cannabidiol as potential anticancer drug. Br. J. Clin. Pharm. 2013, 75, 303–312. [Google Scholar] [CrossRef]
- Nagata, S.; Tanaka, M. Programmed cell deatg and the immune system. Nat. Rev. Immunol. 2017, 17, 333–340. [Google Scholar] [CrossRef]
- Lenaz, G. Role of mitochondria in oxidative stress and ageing. Biochim. Biophys. Acta 1998, 1366, 53–67. [Google Scholar] [CrossRef]
- Fernandez-Checa, J.C.; Garcia-Ruiz, C.; Colell, A.; Mari, M.; Miranda, M.; Ardite, E. Oxidative stress: Role of mitochondria and protection by glutathione. Biofactors 1997, 8, 7–11. [Google Scholar] [CrossRef]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Role of Cardiolipin in Mitochondrial Function and Dynamics in Health and Disease: Molecular and Pharmacological Aspects. Cells 2019, 8, 728. [Google Scholar] [CrossRef]
- Ansenberger-Fricano, K.; Ganini, D.; Mao, M.; Chatterjee, S.; Dallas, S.; Mason, R.P.; Stadler, K.; Santos, J.H.; Bonini, M.G. The peroxidase activity of mitochondrial superoxide dismutase. Free Radic. Biol. Med. 2013, 54, 116–124. [Google Scholar] [CrossRef]
- Margis, R.; Dunand, C.; Teixeira, F.K.; Margis-Pinheiro, M. Glutathione peroxidase family—An evolutionary overview. Febs. J. 2008, 275, 3959–3970. [Google Scholar] [CrossRef]
- Maccarrone, M.; Ullrich, V. Redox regulation in disease and ageing. Cell Death Differ 2004, 11, 949–951. [Google Scholar] [CrossRef][Green Version]
- Angelova, P.R.; Abramov, A.Y. Role of mitochondrial ROS in the brain: From physiology to neurodegeneration. Febs. Lett. 2018, 592, 692–702. [Google Scholar] [CrossRef]
- Harrison, D.; Griendling, K.K.; Landmesser, U.; Hornig, B.; Drexler, H. Role of oxidative stress in atherosclerosis. Am. J. Cardiol. 2003, 91, 7–11. [Google Scholar] [CrossRef]
- Harris, I.S.; DeNicola, G.M. The Complex Interplay between Antioxidants and ROS in Cancer. Trends. Cell. Biol. 2020, 30, 440–451. [Google Scholar] [CrossRef]
- Newsholme, P.; Cruzat, V.F.; Keane, K.N.; Carlessi, R.; de Bittencourt, P.I., Jr. Molecular mechanisms of ROS production and oxidative stress in diabetes. Biochem. J. 2016, 473, 4527–4550. [Google Scholar] [CrossRef]
- Booz, G.W. Cannabidiol as an emergent therapeutic strategy for lessening the impact of inflammation on oxidative stress. Free Radic. Biol. Med. 2011, 51, 1054–1061. [Google Scholar] [CrossRef]
- Pazos, M.R.; Mohammed, N.; Lafuente, H.; Santos, M.; Martinez-Pinilla, E.; Moreno, E.; Valdizan, E.; Romero, J.; Pazos, A.; Franco, R.; et al. Mechanisms of cannabidiol neuroprotection in hypoxic-ischemic newborn pigs: Role of 5HT(1A) and CB2 receptors. Neuropharmacology 2013, 71, 282–291. [Google Scholar] [CrossRef]
- Hamelink, C.; Hampson, A.; Wink, D.A.; Eiden, L.E.; Eskay, R.L. Comparison of cannabidiol, antioxidants, and diuretics in reversing binge ethanol-induced neurotoxicity. J. Pharm. Exp. 2005, 314, 780–788. [Google Scholar] [CrossRef]
- Pan, H.; Mukhopadhyay, P.; Rajesh, M.; Patel, V.; Mukhopadhyay, B.; Gao, B.; Hasko, G.; Pacher, P. Cannabidiol attenuates cisplatin-induced nephrotoxicity by decreasing oxidative/nitrosative stress, inflammation, and cell death. J. Pharm. Exp. 2009, 328, 708–714. [Google Scholar] [CrossRef]
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell. Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell. Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef]
- Westermann, B. Bioenergetic role of mitochondrial fusion and fission. Biochim. Biophys. Acta 2012, 1817, 1833–1838. [Google Scholar] [CrossRef]
- Westermann, B. Molecular machinery of mitochondrial fusion and fission. J. Biol. Chem. 2008, 283, 13501–13505. [Google Scholar] [CrossRef]
- Sheng, B.; Wang, X.; Su, B.; Lee, H.G.; Casadesus, G.; Perry, G.; Zhu, X. Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer’s disease. J. Neurochem. 2012, 120, 419–429. [Google Scholar] [CrossRef]
- Ren, J.; Pulakat, L.; Whaley-Connell, A.; Sowers, J.R. Mitochondrial biogenesis in the metabolic syndrome and cardiovascular disease. J. Mol. Med. 2010, 88, 993–1001. [Google Scholar] [CrossRef]
- Drysdale, J.; Arosio, P.; Invernizzi, R.; Cazzola, M.; Volz, A.; Corsi, B.; Biasiotto, G.; Levi, S. Mitochondrial ferritin: A new player in iron metabolism. Blood Cells Mol. Dis. 2002, 29, 376–383. [Google Scholar] [CrossRef]
- Levi, S.; Corsi, B.; Bosisio, M.; Invernizzi, R.; Volz, A.; Sanford, D.; Arosio, P.; Drysdale, J. A human mitochondrial ferritin encoded by an intronless gene. J. Biol. Chem. 2001, 276, 24437–24440. [Google Scholar] [CrossRef]
- Santambrogio, P.; Biasiotto, G.; Sanvito, F.; Olivieri, S.; Arosio, P.; Levi, S. Mitochondrial ferritin expression in adult mouse tissues. J. Histochem. Cytochem. 2007, 55, 1129–1137. [Google Scholar] [CrossRef]
- Gao, G.; Chang, Y.Z. Mitochondrial ferritin in the regulation of brain iron homeostasis and neurodegenerative diseases. Front. Pharm. 2014, 5, 19. [Google Scholar] [CrossRef]
- Schurr, A.; Livne, A. Differential inhibition of mitochondrial monoamine oxidase from brain by hashish components. Biochem. Pharmacol. 1976, 25, 1201–1203. [Google Scholar] [CrossRef]
- Schurr, A.; Porath, O.; Krup, M.; Livne, A. The effects of hashish components and their mode of action on monoamine oxidase from the brain. Biochem. Pharm. 1978, 27, 2513–2517. [Google Scholar] [CrossRef]
- Mazor, M.; Dvilansky, A.; Aharon, M.; Lazarovitz, Z.; Nathan, I. Effect of cannabinoids on the activity of monoamine oxidase in normal human platelets. Arch. Int. Physiol. Biochim. 1982, 90, 15–20. [Google Scholar] [CrossRef]
- Niesink, R.J.; van Laar, M.W. Does Cannabidiol Protect Against Adverse Psychological Effects of THC? Front. Psychiatry 2013, 4, 130. [Google Scholar] [CrossRef]

{kind=link}
| Study Title | CBD Treatment | Reference |
|---|---|---|
| Direct modulation of the outer mitochondrial membrane channel, voltage-dependent anion channel 1 (VDAC1) by cannabidiol: a novel mechanism for cannabinoid-induced cell death | 5 and 10 µM (30 min–6 h) | [35] |
| Cannabinoid-induced changes in respiration of brain mitochondria | 8.2 and 19.1 µM | [36] |
| Cannabinoid-induced changes in the activity of electron transport chain complexes of brain mitochondria | 50 µM (30 min) | [37] |
| Mitochondrial functions of THP-1 monocytes following exposure to selected natural compounds | 5, 7.5, 10, 10.68 15, 20, 21.64, 30, and 40 µM (0–24 h) | [38] |
| Cannabidiol-induced apoptosis is mediated by activation of Noxa in human colorectal cancer cells | 6 µM (0–2 weeks) | [39] |
| Cannabidiol promotes apoptosis via regulation of XIAP/Smac in gastric cancer | 4 and 10 µM (24 h) | [40] |
| Cannabidiol targets mitochondria to regulate intracellular Ca2+ levels | 1 µM (overnight, ~12 h) | [20] |
| Cannabidiol attenuates OGD/R-induced damage by enhancing mitochondrial bioenergetics and modulating glucose metabolism via pentose-phosphate pathway in hippocampal neurons | 5 µM (24 h) | [41] |
| The non-psychoactive cannabidiol triggers caspase activation and oxidative stress in human glioma cells | 25 µM (6–24 h) | [42] |
| Cannabidiol-induced apoptosis in human leukemia cells: a novel role of cannabidiol in the regulation of p22phox and Nox4 expression | 2.5 and 5 µM (24 h) | [21] |
| Cannabidiol directly targets mitochondria and disturbs calcium homeostasis in acute lymphoblastic leukemia | 1, 10, 30, 60, and 100 µM (0–72 h) | [43] |
| Cannabidiol induces programmed cell death in breast cancer cells by coordinating the cross-talk between apoptosis and autophagy | 7.5 and 10 µM (12–24 h) | [44] |
| The neuroprotective effect of cannabidiol in an in vitro model of newborn hypoxic–ischemic brain damage in mice is mediated by CB2 and adenosine receptors | 100 µM (30 min) | [45] |
| Cannabidiol induces intracellular calcium elevation and cytotoxicity in oligodendrocytes | 0.1, 1, and 10 µM (0–30 min) | [46] |
| Cannabidiol induced apoptosis in human monocytes through mitochondrial permeability transition pore-mediated ROS production | 16 µM (5 min–2 h) | [47] |
| A comparative study on cannabidiol-induced apoptosis in murine thymocytes and EL-4 thymoma cells | 4, 8, 12, and 16 µM (1–24 h) | [48] |
| Neuroprotective effects of cannabidiol against hydrogen peroxide in hippocampal neuron culture | 1, 5, 10, 15, and 30 µM (24 h) | [49] |
| Antioxidant and neuroprotective effects induced by cannabidiol and cannabigerol in rat CTX-TNA2 astrocytes and isolated cortexes | 1 µM (24–48 h) | [50] |
| Cannabidiol prevents LPS-induced microglial inflammation by inhibiting ROS/NF-κB-dependent signaling and glucose consumption | 1 and 10 µM (30 min–24 h) | [51] |
| Cannabidiol-induced apoptosis in primary lymphocytes is associated with oxidative stress-dependent activation of caspase-8 | 1, 2, 4, and 8 µM (15 min–12 h) | [52] |
| Tissue Type | CBD Treatment Range | Mitochondrial Respiration | Mechanism of Action | Reference |
|---|---|---|---|---|
| Prefrontal and cerebral tissue of the rat brain | 60 mg/kg/day (14 days) 60 mg/kg (acute) 15 mg/kg/day (14 days) 15 mg/kg (acute) | ↑complex I, II, III, and IV activity ↑complex I, II, III, and IV activity ↑complex II and IV activity No effect | ↑ intramitochondrial calcium | [23] |
| Isolated mitochondria from pig brain | IC50 = 8.2 ± 0.6 µM IC50 = 19.1 ± 1.1 µM (30 min) 50 µM (30 min) | ↓complex I activity ↓complex II activity ↓complex I, II/III, and IV activity | DNI | [36,37] |
| BV2 microglial cells | IC50 = 10 µM (1 h) | ↓mitochondrial membrane potential | ↓ VDAC1 conductance | [35] |
| THP monocytes | IC50 = 21.64 µM (24 h) IC10 = 10.68 µM (24 h) | ↓maximal respiration ↓ATP production No effect | ↓ ETC activity | [38] |
| OGD/R injured hippocampal cells | 5 µM (24 h) | ↑succinate dehydrogenase activity ↑ATP production/oxygen consumption rate ↑maximal respiration ↑spare respiratory capacity ↑basal respiration | ↑glucose consumption preservation of NADPH/NADP+ ratio activiation of glucose-6-phosphate dehydrogenase | [41] |
| DOX-treated cardiac tissue | 10 mg/kg/day IP injection (5 days) | ↑complex I and II activity | DNI | [24] |
| Patient-derived colorectal cancer cells | 6 µM (30 min–6 h) | ↓mitochondria membrane potential | ↑mitochondrial ROS | [39] |
| AGS gastric adenocarcinoma cells | 4 µM (24 h) | ↓basal respiration ↓ATP production ↓oxygen consumption | ↓expression of NADH dehydrogenase ubiquinone 1α subcomplex subunit 9 (complex I) ↓mitochondrial membrane potential | [40] |
| Tissue Type | CBD Treatment Range | Intrinsic Apoptosis | Mechanism of Action | Reference |
|---|---|---|---|---|
| U87 glioma cells | 25 µM (6–24 h) 10 µM (6–24 h) | ↑apoptosis No effect | ↑caspase 9 activation ↑caspase 3 activation ↑cytochrome c release ↑oxidative stress ↓GSH levels No effect | [42] |
| EL-4 murine lymphoma cells C57BL/6 mice injected with EL-4 | 1.25 µM (24 h) 2.5 and 5 µM (24 h) 12.5 mg/kg (1 day) 25 mg/kg (1 day) | No effect ↑apoptosis No effect ↑apoptosis | No effect Activation of CB2 No effect DNI | [21] |
| Jurkat human leukemia cells | 2.5 and 5 µM (24 h) | ↑apoptosis | Activation of CB2 ↑expression of Nox4 and p22phox ↓mitochondrial membrane potential ↑ROS ↑cytochrome c release ↑activation of caspase 9 ↑activation of caspase 3 ↓full-length BID ↑cleavage of PARP | [21] |
| Acute lymphoblastic leukemia of T lineage (Jurkat cells) | 30 µM (12 h) | ↑apoptosis | ↓mitochondrial membrane potential ↑caspase 9 activity ↑caspase 3 activity ↑C2+ influx into the mitochondria ↑cytochrome c release ↑mPTP opening | [43] |
| AGS gastric cancer cells | 4 µM (24 h) | ↑apoptosis | ↑ cleaved caspase 9 ↑ cleaved caspase 3 ↑ cleaved PARP ↓XIAP ↑SMAC | [40] |
| MKN45 gastric cancer cells | 10 µM (24 h) | ↑apoptosis | ↑ cleaved caspase 9 ↑ cleaved caspase 3 ↑ cleaved PARP ↓XIAP ↑SMAC | [40] |
| Patient-derived colorectal cancer cells | 6 µM (12 h–2 weeks) | ↑apoptosis | ↑ROS ↑NOXA | [39] |
| MDA-MB-231 breast cancer cells | 7.5 and 10 µM (12–24 h) | ↑apoptosis | ↑ROS ↑cleaved PARP ↑cleaved caspase 9 ↑ cleaved caspase 3 ↑ cleaved caspase 7 ↑t-BID ↑cytochrome c release | [44] |
| C57BL6 mice forebrain subjected to OGD injury | 100 µM (30 min) | ↓apoptosis | Interactions with the CB2 and A2a adenosine receptors ↓activation of caspase 9 | [45] |
| HT22 mouse hippocampal cell subjected to OGD/R injury | 5 µM (24 h) | ↓apoptosis | ↓caspase 3 activity ↓PARP activity ↓ROS ↑GSH content ↑SOD activity ↑GPX activity | [41] |
| Oligodendrocytes isolated from Dawley rat optic nerves | 0.1, 1, and 10 µM (20–30 min) | ↑apoptosis | ↑ROS ↑caspase 9 ↑Ca2+ outflux from the mitochondria to the cytosol | [46] |
| CD14+ human monocytes | 16 µM (5 min–2 h) | ↑apoptosis | ↑ ROS ↓MMP ↑mPTP opening | [47] |
| Thymocytes isolated from male BALB/c mice | 4, 8, 12, and 16 µM (4–24 h) | ↑apoptosis | ↑ROS | [48] |
| EL4 cells | 12 and 16 µM (1–24 h) | ↑apoptosis | ↑ROS | [48] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chan, J.Z.; Duncan, R.E. Regulatory Effects of Cannabidiol on Mitochondrial Functions: A Review. Cells 2021, 10, 1251. https://doi.org/10.3390/cells10051251
Chan JZ, Duncan RE. Regulatory Effects of Cannabidiol on Mitochondrial Functions: A Review. Cells. 2021; 10(5):1251. https://doi.org/10.3390/cells10051251
Chicago/Turabian StyleChan, John Zewen, and Robin Elaine Duncan. 2021. "Regulatory Effects of Cannabidiol on Mitochondrial Functions: A Review" Cells 10, no. 5: 1251. https://doi.org/10.3390/cells10051251
APA StyleChan, J. Z., & Duncan, R. E. (2021). Regulatory Effects of Cannabidiol on Mitochondrial Functions: A Review. Cells, 10(5), 1251. https://doi.org/10.3390/cells10051251