
Transcriptional CDK Inhibitors CYC065 and THZ1 Induce Apoptosis in Glioma Stem Cells Derived from Recurrent GBM

 ,
,  ,
, 

Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cell Culture
2.2. Stem Cell Population Characterisation
2.3. Western Blot Analysis
2.4. Cell Death Analysis Using Flow Cytometry
2.5. siRNA Transfection
2.6. Fluorometric Cell Viability and Cytotoxicity Detection
2.7. Fluorescence-Activated Cell Sorting (FACS)
2.8. Light Sheet Fluorescence Microscopy (LSFM)
2.9. Laser Scanning Confocal Microscopy
2.10. Fluorescence Imaging Using Nikon Eclipse TE2000 Microscope
2.11. TCGA Data Analysis
2.12. Statistics and Software
3. Results
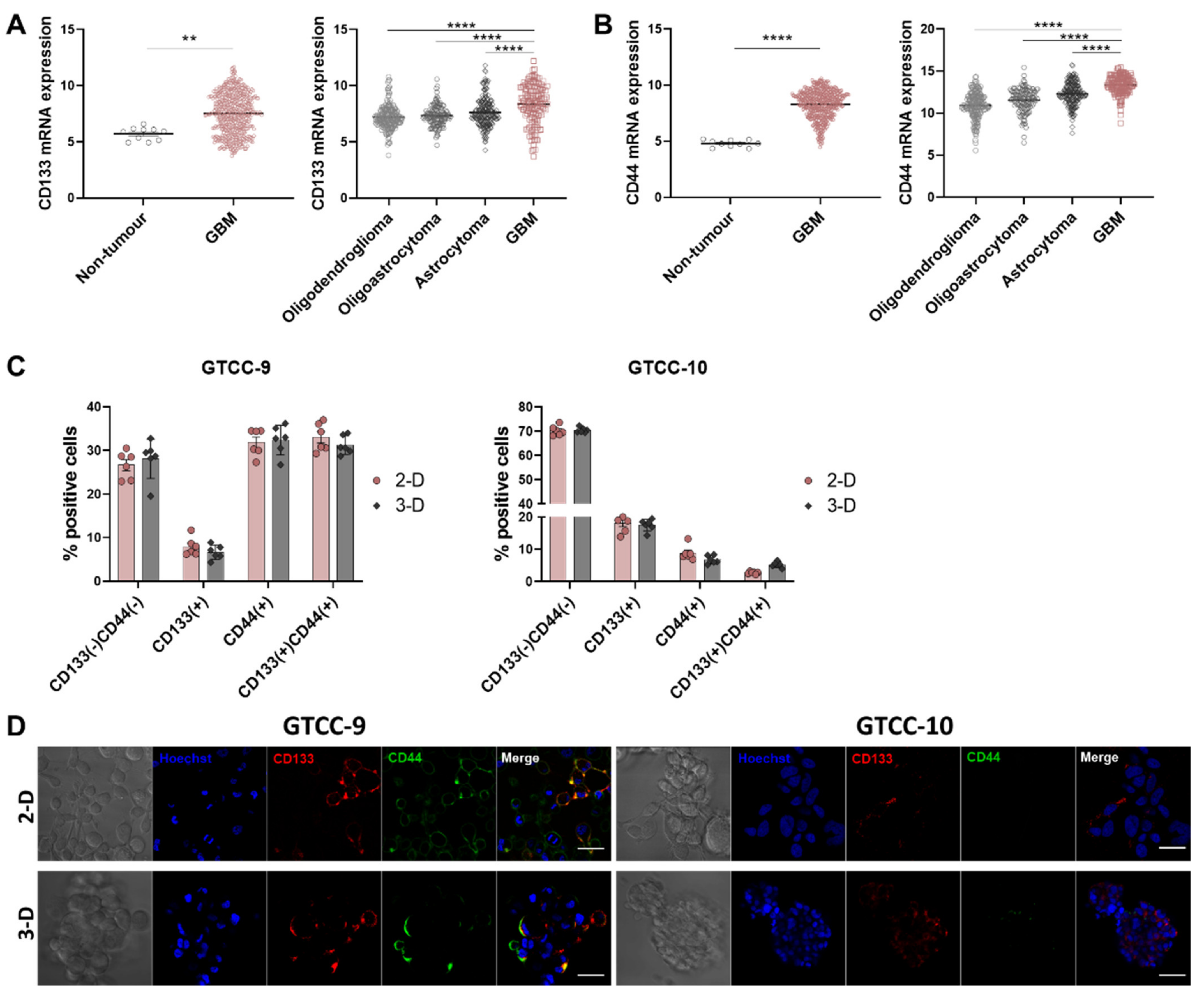
3.1. Expression of GSCs Markers, CD133 and CD44 Is Upregulated in GBM
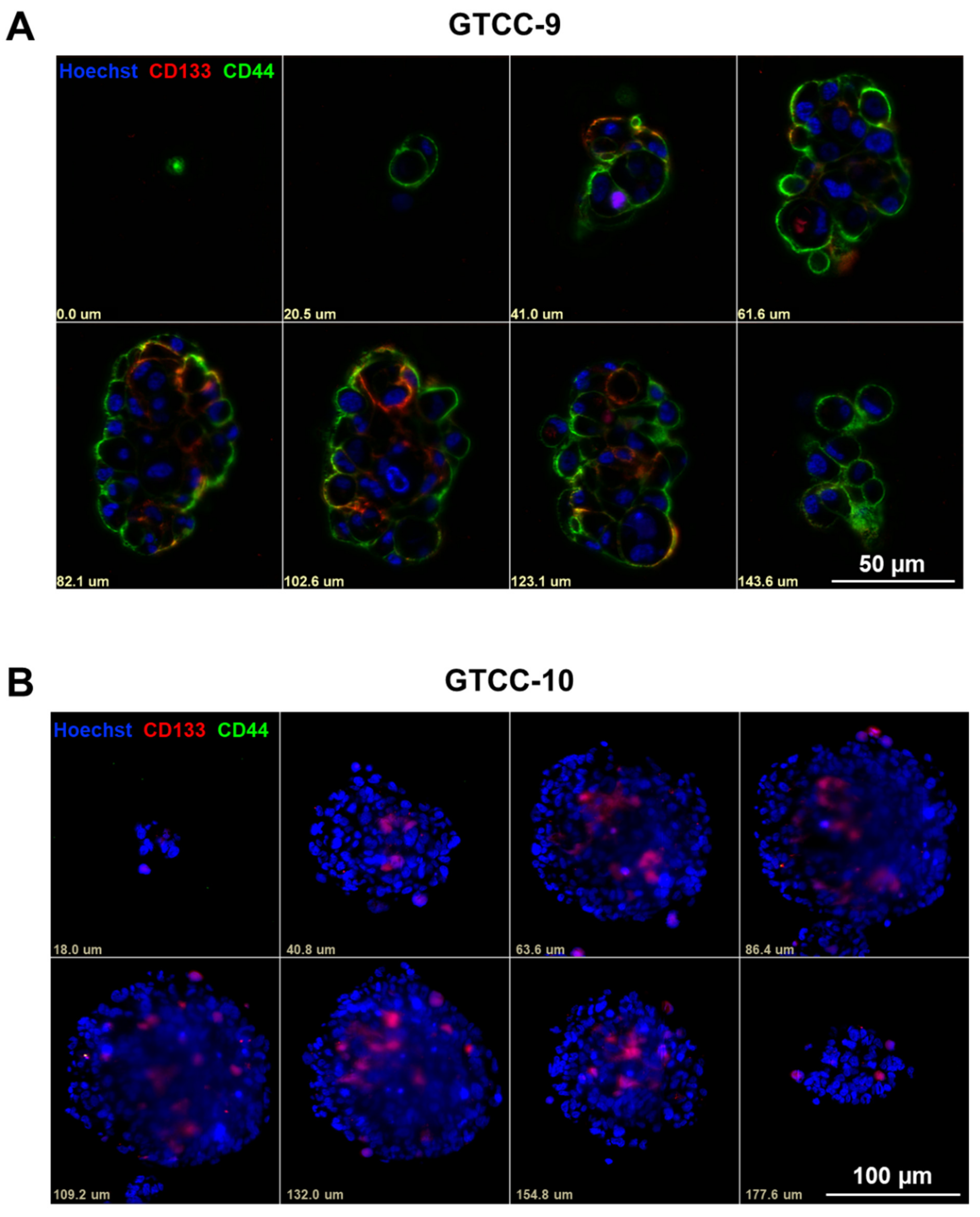
3.2. CD133 and CD44 GSC Biomarker-Negative Cells Convert into GSC Biomarker-Positive Cells and Contribute to the Enrichment in GSCs in Recurrent GBM
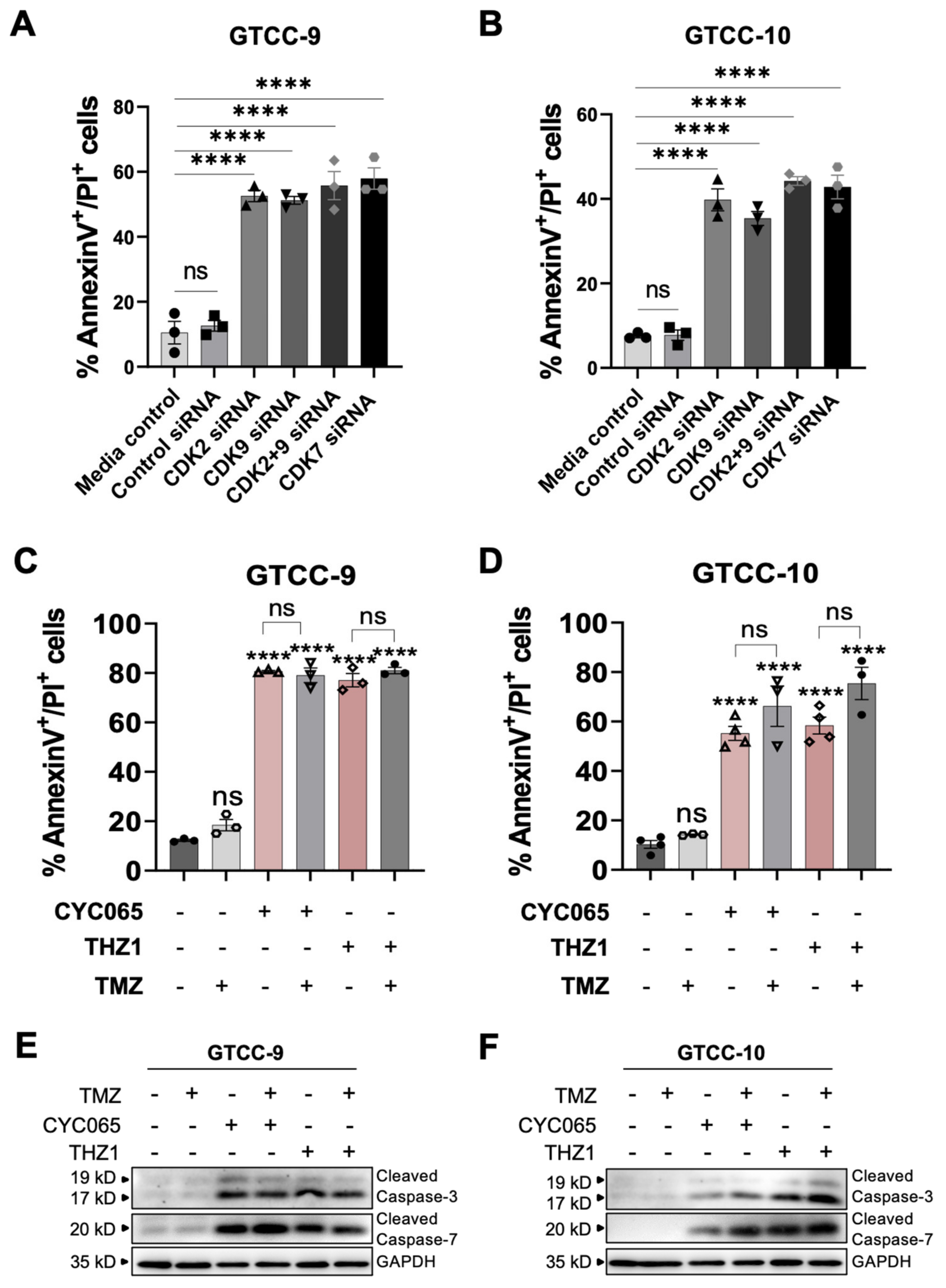
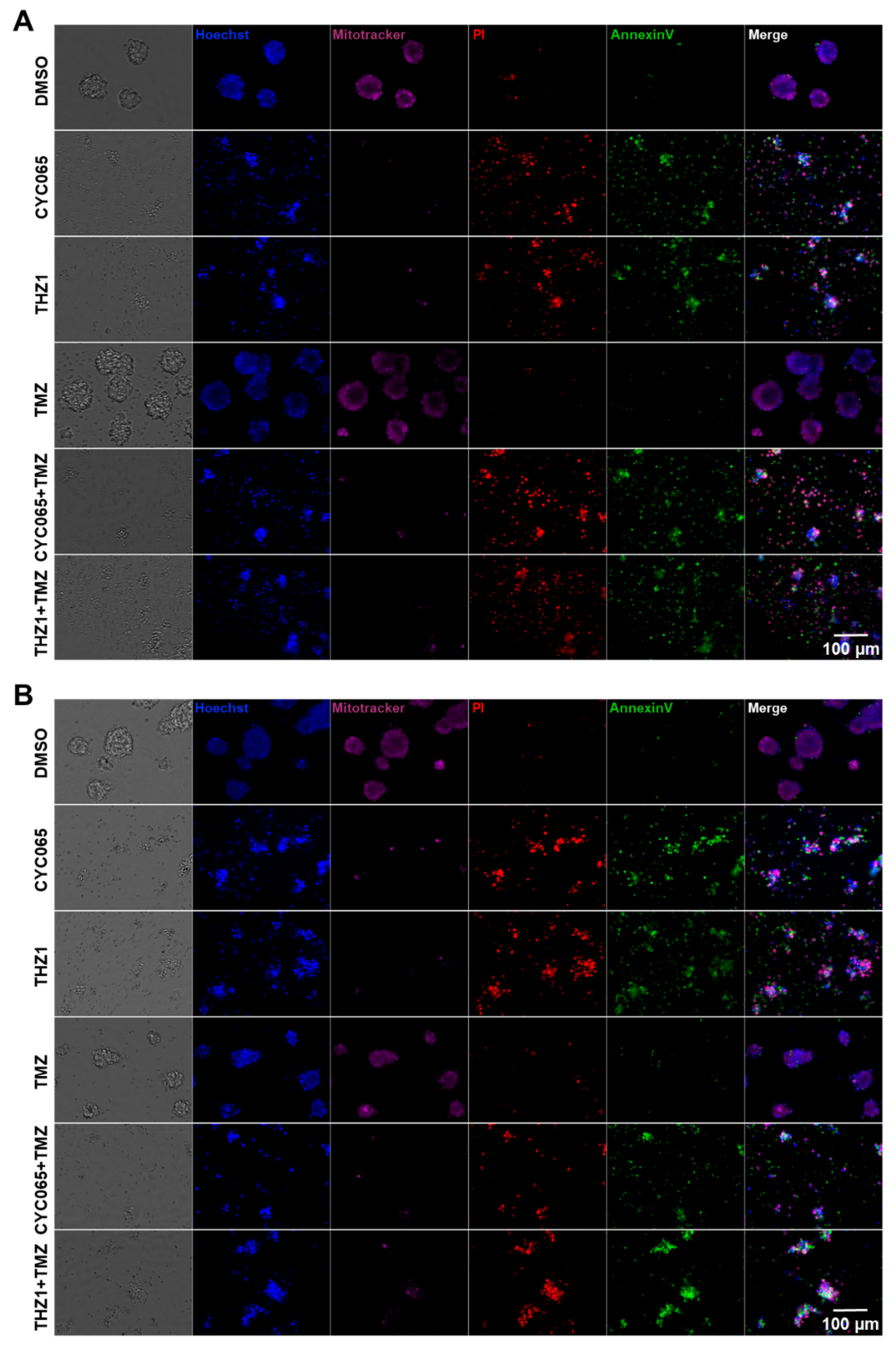
3.3. CDK2/9-Targeting, CYC065 and CDK7-Targeting, THZ1 Induce Apoptotic Cell Death in Recurrent Gliomaspheres
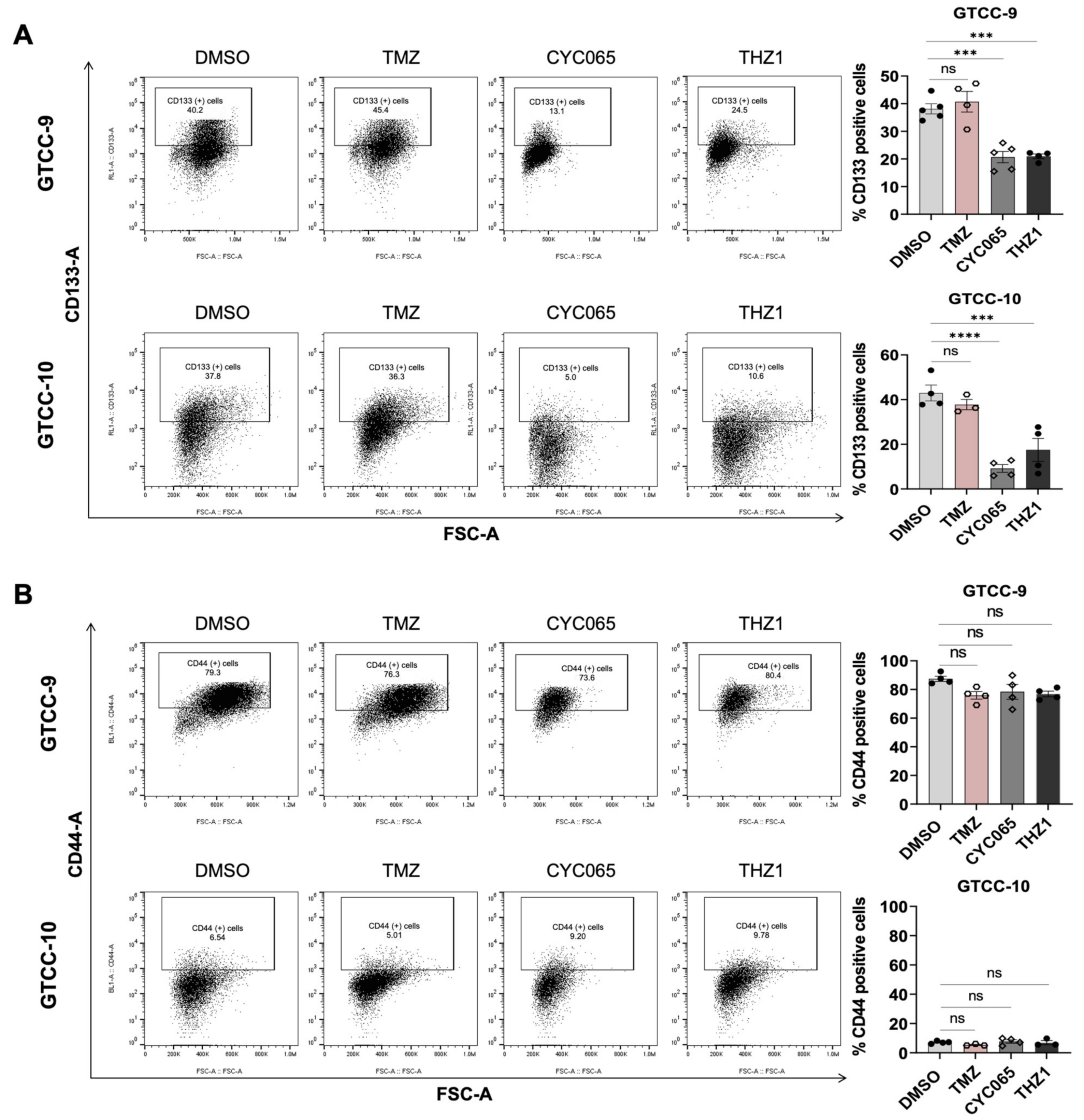
3.4. CYC065 and THZ1 Decrease the Expression of the CD133 Stem Cell Marker in Recurrent Gliomaspheres and Induce Apoptotic Cell Death in the Glioma Stem Cell Subpopulations
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stupp, R.; Mason, W.P.; Van Den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Sulman, E.; Aldape, K.; Colman, H. Brain tumor stem cells. Curr. Probl. Cancer 2008, 32, 124–142. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Yuan, X.; Zeng, Z.; Tunici, P.; Ng, H.; Abdulkadir, I.R.; Lu, L.; Irvin, D.; Black, K.L.; Yu, J.S. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol. Cancer 2006, 5, 67. [Google Scholar] [CrossRef]
- Zhou, H.-M.; Zhang, J.-G.; Zhang, X.; Li, Q. Targeting cancer stem cells for reversing therapy resistance: Mechanism, signaling, and prospective agents. Signal Transduct. Target. Ther. 2021, 6, 62. [Google Scholar] [CrossRef]
- Jamal, M.; Rath, B.H.; Tsang, P.S.; Camphausen, K.; Tofilon, P.J. The brain microenvironment preferentially enhances the radioresistance of CD133(+) glioblastoma stem-like cells. Neoplasia 2012, 14, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, K.; Troike, K.; Silver, D.J.; Lathia, J.D. The evolution of the cancer stem cell state in glioblastoma: Emerging insights into the next generation of functional interactions. Neuro Oncol. 2021, 23, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Beier, D.; Hau, P.; Proescholdt, M.; Lohmeier, A.; Wischhusen, J.; Oefner, P.J.; Aigner, L.; Brawanski, A.; Bogdahn, U.; Beier, C.P. CD133(+) and CD133(-) glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res. 2007, 67, 4010–4015. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; Sathornsumetee, S.; Hao, Y.; Li, Z.; Hjelmeland, A.B.; Shi, Q.; McLendon, R.E.; Bigner, D.D.; Rich, J.N. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006, 66, 7843–7848. [Google Scholar] [CrossRef]
- Wang, J.; Sakariassen, P.; Tsinkalovsky, O.; Immervoll, H.; Bøe, S.O.; Svendsen, A.; Prestegarden, L.; Røsland, G.; Thorsen, F.; Stuhr, L.; et al. CD133 negative glioma cells form tumors in nude rats and give rise to CD133 positive cells. Int. J. Cancer 2008, 122, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Chen, H.; Lv, S.; Yang, H. High CD133 Expression Is Associated with Worse Prognosis in Patients with Glioblastoma. Mol. Neurobiol. 2016, 53, 2354–2360. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.V.; Mantamadiotis, T. Insights into the next generation of cancer stem cell research. Front. Biosci. 2014, 19, 1015–1027. [Google Scholar] [CrossRef][Green Version]
- Zöller, M. CD44, Hyaluronan, the Hematopoietic Stem Cell, and Leukemia-Initiating Cells. Front. Immunol. 2015, 6, 235. [Google Scholar] [CrossRef]
- Nishikawa, M.; Inoue, A.; Ohnishi, T.; Kohno, S.; Ohue, S.; Matsumoto, S.; Suehiro, S.; Yamashita, D.; Ozaki, S.; Watanabe, H.; et al. Significance of Glioma Stem-Like Cells in the Tumor Periphery That Express High Levels of CD44 in Tumor Invasion, Early Progression, and Poor Prognosis in Glioblastoma. Stem Cells Int. 2018, 2018, 5387041. [Google Scholar] [CrossRef]
- Mooney, K.L.; Choy, W.; Sidhu, S.; Pelargos, P.; Bui, T.T.; Voth, B.; Barnette, N.; Yang, I. The role of CD44 in glioblastoma multiforme. J. Clin. Neurosci. 2016, 34, 1–5. [Google Scholar] [CrossRef]
- Wang, H.H.; Liao, C.C.; Chow, N.H.; Huang, L.L.; Chuang, J.I.; Wei, K.C.; Shin, J.W. Whether CD44 is an applicable marker for glioma stem cells. Am. J. Transl. Res. 2017, 9, 4785–4806. [Google Scholar]
- Grana, X.; Reddy, E.P. Cell cycle control in mammalian cells: Role of cyclins, cyclin dependent kinases (CDKs), growth suppressor genes and cyclin-dependent kinase inhibitors (CKIs). Oncogene 1995, 11, 211–219. [Google Scholar]
- Juric, V.; Murphy, B. Cyclin-dependent kinase inhibitors in brain cancer: Current state and future directions. Cancer Drug Resist. 2020, 3, 48–62. [Google Scholar] [CrossRef]
- Noonan, J.J.; Jarzabek, M.; Lincoln, F.A.; Cavanagh, B.L.; Pariag, A.R.; Juric, V.; Young, L.S.; Ligon, K.L.; Jahns, H.; Zheleva, D.; et al. Implementing Patient-Derived Xenografts to Assess the Effectiveness of Cyclin-Dependent Kinase Inhibitors in Glioblastoma. Cancers 2019, 11, 2005. [Google Scholar] [CrossRef]
- Frame, S.; Saladino, C.; MacKay, C.; Atrash, B.; Sheldrake, P.; McDonald, E.; Clarke, P.A.; Workman, P.; Blake, D.; Zheleva, D. Fadraciclib (CYC065), a novel CDK inhibitor, targets key pro-survival and oncogenic pathways in cancer. PLoS ONE 2020, 15, e0234103. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, N.; Zhang, T.; Rahl, P.B.; Abraham, B.J.; Reddy, J.; Ficarro, S.B.; Dastur, A.; Amzallag, A.; Ramaswamy, S.; Tesar, B.; et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 2014, 511, 616–620. [Google Scholar] [CrossRef]
- Chipumuro, E.; Marco, E.; Christensen, C.L.; Kwiatkowski, N.; Zhang, T.; Hatheway, C.M.; Abraham, B.J.; Sharma, B.; Yeung, C.; Altabef, A.; et al. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell 2014, 159, 1126–1139. [Google Scholar] [CrossRef]
- Christensen, C.L.; Kwiatkowski, N.; Abraham, B.J.; Carretero, J.; Al-Shahrour, F.; Zhang, T.; Chipumuro, E.; Herter-Sprie, G.S.; Akbay, E.A.; Altabef, A.; et al. Targeting transcriptional addictions in small cell lung cancer with a covalent CDK7 inhibitor. Cancer Cell 2014, 26, 909–922. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, T.; Kwiatkowski, N.; Abraham, B.J.; Lee, T.I.; Xie, S.; Yuzugullu, H.; Von, T.; Li, H.; Lin, Z.; et al. CDK7-dependent transcriptional addiction in triple-negative breast cancer. Cell 2015, 163, 174–186. [Google Scholar] [CrossRef]
- Greenall, S.A.; Lim, Y.C.; Mitchell, C.B.; Ensbey, K.S.; Stringer, B.W.; Wilding, A.L.; O’Neill, G.M.; McDonald, K.L.; Gough, D.J.; Day, B.W.; et al. Cyclin-dependent kinase 7 is a therapeutic target in high-grade glioma. Oncogenesis 2017, 6, e336. [Google Scholar] [CrossRef]
- Preibisch, S.; Saalfeld, S.; Schindelin, J.; Tomancak, P. Software for bead-based registration of selective plane illumination microscopy data. Nat. Methods 2010, 7, 418–419. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Bowman, R.L.; Wang, Q.; Carro, A.; Verhaak, R.G.; Squatrito, M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro Oncol. 2017, 19, 139–141. [Google Scholar] [CrossRef] [PubMed]
- Auffinger, B.; Spencer, D.; Pytel, P.; Ahmed, A.U.; Lesniak, M.S. The role of glioma stem cells in chemotherapy resistance and glioblastoma multiforme recurrence. Expert Rev. Neurother. 2015, 15, 741–752. [Google Scholar] [CrossRef]
- Vieira de Castro, J.; Gonçalves, C.S.; Hormigo, A.; Costa, B.M. Exploiting the Complexities of Glioblastoma Stem Cells: Insights for Cancer Initiation and Therapeutic Targeting. Int. J. Mol. Sci. 2020, 21, 5278. [Google Scholar] [CrossRef]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavaré, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef]
- Van Linde, M.E.; Brahm, C.G.; de Witt Hamer, P.C.; Reijneveld, J.C.; Bruynzeel, A.M.E.; Vandertop, W.P.; van de Ven, P.M.; Wagemakers, M.; van der Weide, H.L.; Enting, R.H.; et al. Treatment outcome of patients with recurrent glioblastoma multiforme: A retrospective multicenter analysis. J. Neuro-Oncol. 2017, 135, 183–192. [Google Scholar] [CrossRef]
- Kahana, S.; Finniss, S.; Cazacu, S.; Xiang, C.; Lee, H.K.; Brodie, S.; Goldstein, R.S.; Roitman, V.; Slavin, S.; Mikkelsen, T.; et al. Proteasome inhibitors sensitize glioma cells and glioma stem cells to TRAIL-induced apoptosis by PKCε-dependent downregulation of AKT and XIAP expressions. Cell Signal. 2011, 23, 1348–1357. [Google Scholar] [CrossRef]
- Bleau, A.M.; Hambardzumyan, D.; Ozawa, T.; Fomchenko, E.I.; Huse, J.T.; Brennan, C.W.; Holland, E.C. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell 2009, 4, 226–235. [Google Scholar] [CrossRef]
- Clement, V.; Sanchez, P.; de Tribolet, N.; Radovanovic, I.; Ruiz i Altaba, A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr. Biol. 2007, 17, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Metellus, P.; Nanni-Metellus, I.; Delfino, C.; Colin, C.; Coulibaly, B.; Fina, F.; Chinot, O.L.; Figarella-Branger, D.; Ouafik, L. Prognostic impact of stem cell marker CD133 in 61 glioblastoma patients treated with concomitant chemoradiation: A prospective study. J. Clin. Oncol. 2010, 28, 2027. [Google Scholar] [CrossRef]
- Ardebili, S.Y.; Zajc, I.; Gole, B.; Campos, B.; Herold-Mende, C.; Drmota, S.; Lah, T.T. CD133/prominin1 is prognostic for GBM patient’s survival, but inversely correlated with cysteine cathepsins’ expression in glioblastoma derived spheroids. Radiol. Oncol. 2011, 45, 102–115. [Google Scholar] [CrossRef]
- Zeppernick, F.; Ahmadi, R.; Campos, B.; Dictus, C.; Helmke, B.M.; Becker, N.; Lichter, P.; Unterberg, A.; Radlwimmer, B.; Herold-Mende, C.C. Stem Cell Marker CD133 Affects Clinical Outcome in Glioma Patients. Clin. Cancer Res. 2008, 14, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zheng, Z.; Guan, J.; Qi, D.; Zhou, S.; Shen, X.; Wang, F.; Wenkert, D.; Kirmani, B.; Solouki, T.; et al. Identification of a panel of genes as a prognostic biomarker for glioblastoma. EBioMedicine 2018, 37, 68–77. [Google Scholar] [CrossRef]
- Wei, K.C.; Huang, C.Y.; Chen, P.Y.; Feng, L.Y.; Wu, T.W.; Chen, S.M.; Tsai, H.C.; Lu, Y.J.; Tsang, N.M.; Tseng, C.K.; et al. Evaluation of the prognostic value of CD44 in glioblastoma multiforme. Anticancer Res. 2010, 30, 253–259. [Google Scholar] [PubMed]
- Edmondson, R.; Broglie, J.J.; Adcock, A.F.; Yang, L. Three-dimensional cell culture systems and their applications in drug discovery and cell-based biosensors. Assay Drug Dev. Technol. 2014, 12, 207–218. [Google Scholar] [CrossRef]
- Han, J.; Won, M.; Kim, J.H.; Jung, E.; Min, K.; Jangili, P.; Kim, J.S. Cancer stem cell-targeted bio-imaging and chemotherapeutic perspective. Chem. Soc. Rev. 2020, 49, 7856–7878. [Google Scholar] [CrossRef] [PubMed]
- Annovazzi, L.; Mellai, M.; Schiffer, D. Chemotherapeutic Drugs: DNA Damage and Repair in Glioblastoma. Cancers 2017, 9, 57. [Google Scholar] [CrossRef] [PubMed]
- Adamski, V.; Hempelmann, A.; Flüh, C.; Lucius, R.; Synowitz, M.; Hattermann, K.; Held-Feindt, J. Dormant glioblastoma cells acquire stem cell characteristics and are differentially affected by Temozolomide and AT101 treatment. Oncotarget 2017, 8, 108064–108078. [Google Scholar] [CrossRef] [PubMed]
- Garnier, D.; Meehan, B.; Kislinger, T.; Daniel, P.; Sinha, A.; Abdulkarim, B.; Nakano, I.; Rak, J. Divergent evolution of temozolomide resistance in glioblastoma stem cells is reflected in extracellular vesicles and coupled with radiosensitization. Neuro Oncol. 2018, 20, 236–248. [Google Scholar] [CrossRef] [PubMed]
- Murphy, A.C.; Weyhenmeyer, B.; Noonan, J.; Kilbride, S.M.; Schimansky, S.; Loh, K.P.; Kogel, D.; Letai, A.G.; Prehn, J.H.; Murphy, B.M. Modulation of Mcl-1 sensitizes glioblastoma to TRAIL-induced apoptosis. Apoptosis 2014, 19, 629–642. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Sex | Age at Diagnosis | Diagnosis | Alterations | MGMT Status |
|---|---|---|---|---|---|
| GTCC-9 | M | 44.00 | Recurrent | EGFR amp, PTEN loss | NA |
| GTCC-10 | M | 50.38 | Recurrent | NA | Methylated |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Juric, V.; Düssmann, H.; Lamfers, M.L.M.; Prehn, J.H.M.; Rehm, M.; Murphy, B.M. Transcriptional CDK Inhibitors CYC065 and THZ1 Induce Apoptosis in Glioma Stem Cells Derived from Recurrent GBM. Cells 2021, 10, 1182. https://doi.org/10.3390/cells10051182
Juric V, Düssmann H, Lamfers MLM, Prehn JHM, Rehm M, Murphy BM. Transcriptional CDK Inhibitors CYC065 and THZ1 Induce Apoptosis in Glioma Stem Cells Derived from Recurrent GBM. Cells. 2021; 10(5):1182. https://doi.org/10.3390/cells10051182
Chicago/Turabian StyleJuric, Viktorija, Heiko Düssmann, Martine L. M. Lamfers, Jochen H. M. Prehn, Markus Rehm, and Brona M. Murphy. 2021. "Transcriptional CDK Inhibitors CYC065 and THZ1 Induce Apoptosis in Glioma Stem Cells Derived from Recurrent GBM" Cells 10, no. 5: 1182. https://doi.org/10.3390/cells10051182
APA StyleJuric, V., Düssmann, H., Lamfers, M. L. M., Prehn, J. H. M., Rehm, M., & Murphy, B. M. (2021). Transcriptional CDK Inhibitors CYC065 and THZ1 Induce Apoptosis in Glioma Stem Cells Derived from Recurrent GBM. Cells, 10(5), 1182. https://doi.org/10.3390/cells10051182