
Anthracyclins Increase PUFAs: Potential Implications in ER Stress and Cell Death


 , ,
, ,  ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatment. Murine Model
2.2. Lipidomics Analysis and Data Pre-Treatment
2.3. Data Analysis and Biomedical Interpretation
2.4. Software and Graphical Material
3. Results
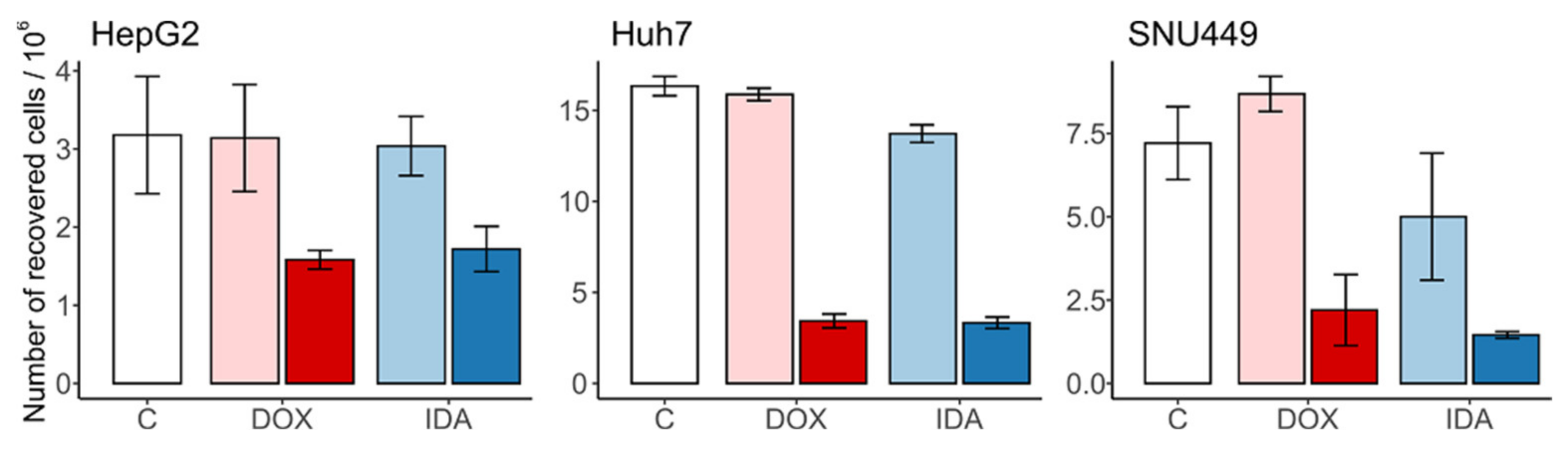
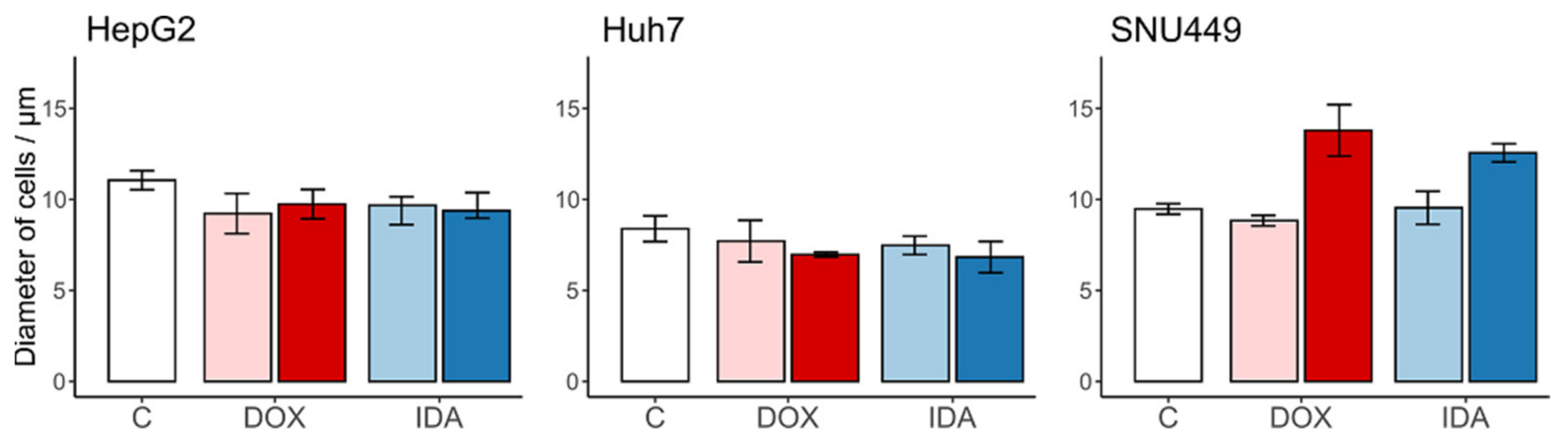
3.1. Number and Size of the Recovered Cells
3.2. Anthracyclins Increase etherPEs in All Cell Types
3.3. Anthracyclins Increase All etherPEs with PUFAs, but Not etherPC
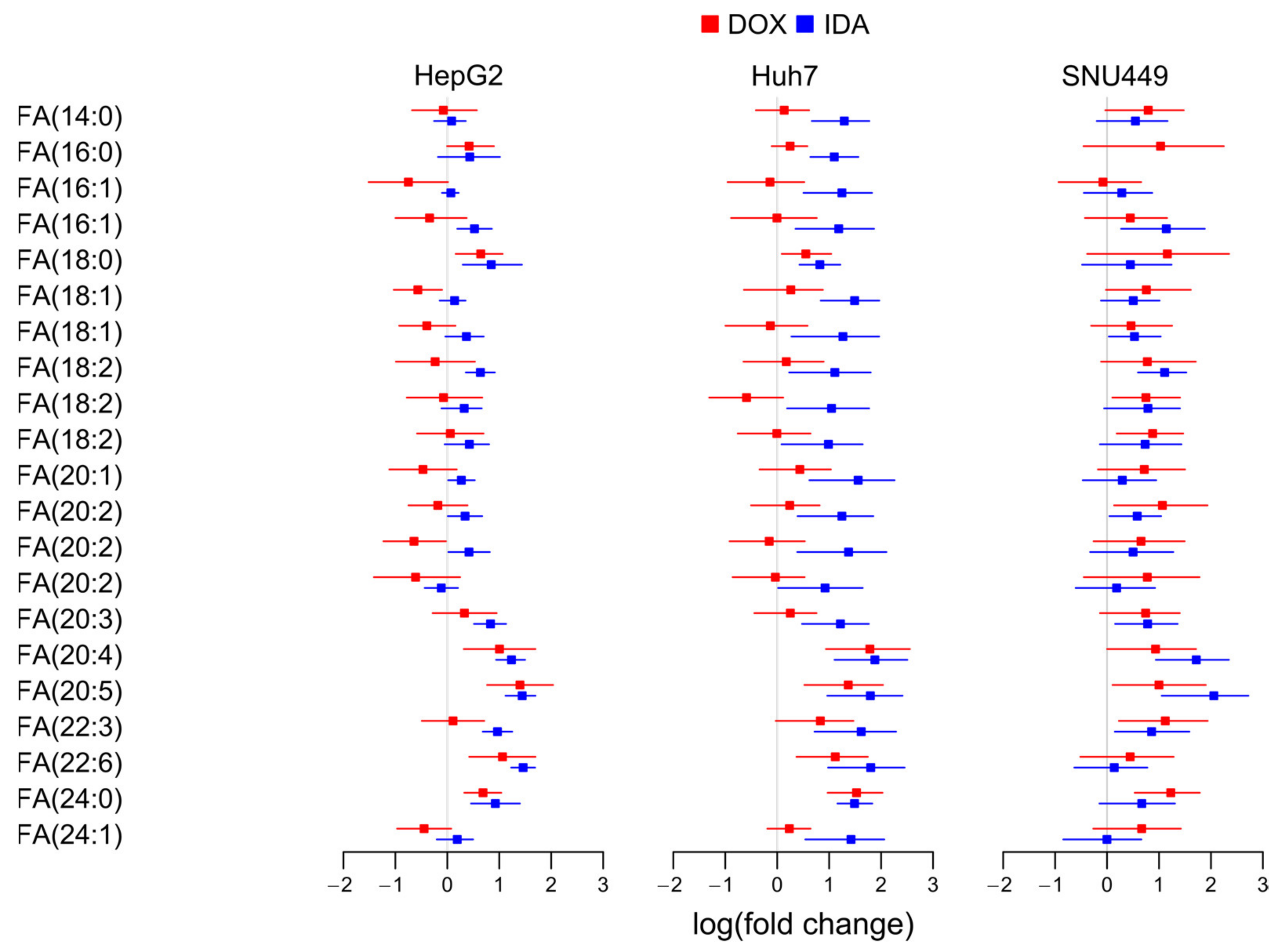
3.4. Anthracyclins Increase Free PUFAs
3.5. Anthracyclins Increase Other Glycerolipids with PUFAs
3.6. Regulation of Glycerolipids with Saturated and Monounsaturated Fatty Acids
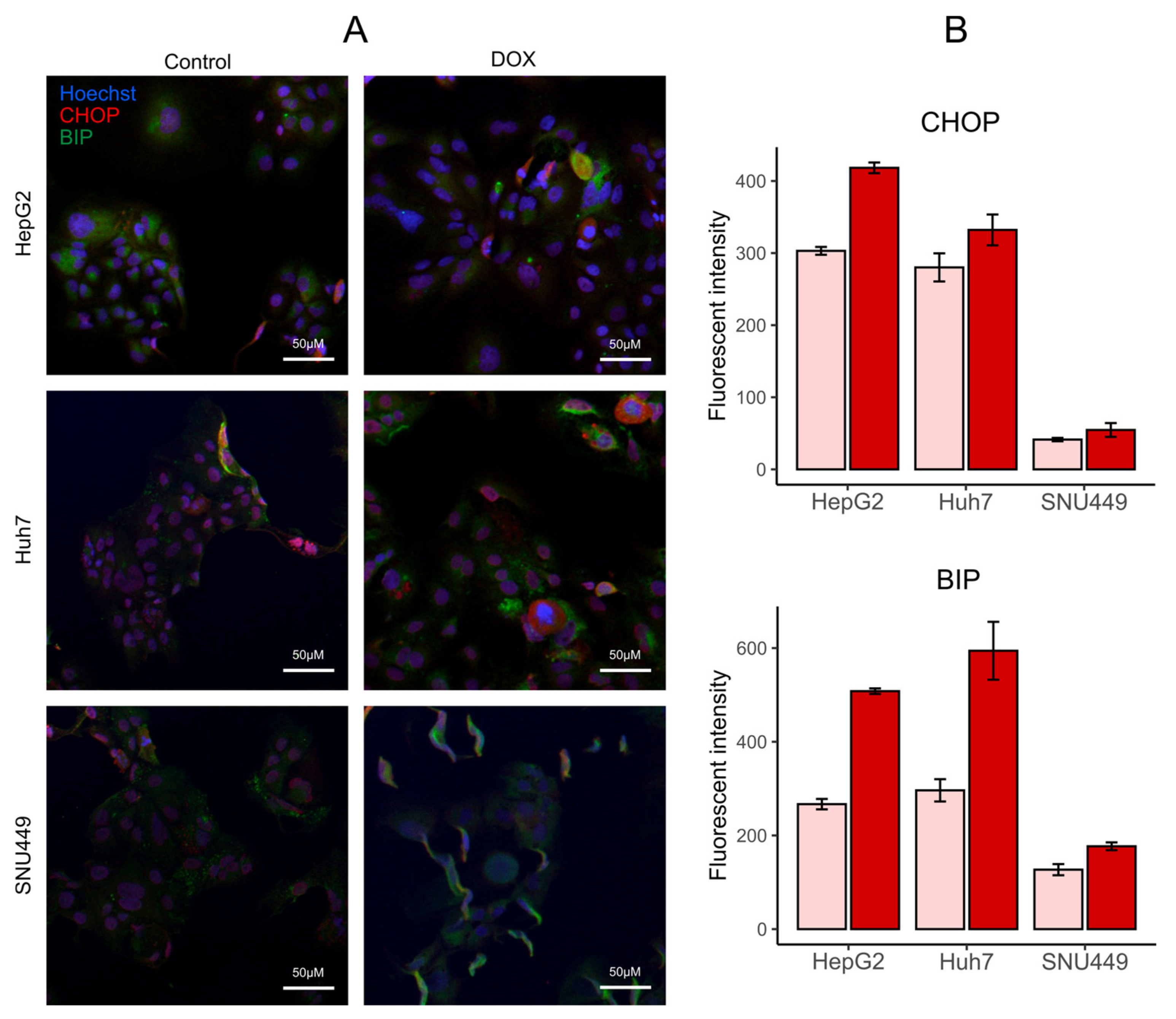
3.7. DOX Increases Markers of ER Stress
3.8. DOX Induces the Expression of Transferrin Receptor in the Liver of an HCC Murine Model
4. Discussion
4.1. Regulation of the De Novo Lipogenesis of Fatty Acids, Glycerolipids, and Cholesterol
4.2. Regulation of the Increase in etherPEs with PUFAs and PUFAs by Anthracyclins
4.3. Role of PUFAS and etherPEs with PUFAs: Potential Hallmark of Programmed Cell Death by Ferroptosis
4.4. Interplay Among the Lipidome, Anthracyclin Uptake, ER Stress, and Sensitivity to Anthracyclins
4.5. Perspectives in Metabochemotherapy and Anthracyclins
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of Cancer Cell Metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid Metabolic Reprogramming in Cancer Cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef]
- Kim, D.J.; Cho, E.J.; Yu, K.-S.; Jang, I.-J.; Yoon, J.-H.; Park, T.; Cho, J.-Y. Comprehensive Metabolomic Search for Biomarkers to Differentiate Early Stage Hepatocellular Carcinoma from Cirrhosis. Cancers 2019, 11, 1497. [Google Scholar] [CrossRef]
- Smith, M.R.; Chacko, B.K.; Johnson, M.S.; Benavides, G.A.; Uppal, K.; Go, Y.-M.; Jones, D.P.; Darley-Usmar, V.M. A Precision Medicine Approach to Defining the Impact of Doxorubicin on the Bioenergetic-Metabolite Interactome in Human Platelets. Redox Biol. 2020, 28, 101311. [Google Scholar] [CrossRef] [PubMed]
- Sangineto, M.; Villani, R.; Cavallone, F.; Romano, A.; Loizzi, D.; Serviddio, G. Lipid Metabolism in Development and Progression of Hepatocellular Carcinoma. Cancers 2020, 12, 1419. [Google Scholar] [CrossRef]
- Wang, W.; Bai, L.; Li, W.; Cui, J. The Lipid Metabolic Landscape of Cancers and New Therapeutic Perspectives. Front. Oncol. 2020, 10, 605154. [Google Scholar] [CrossRef] [PubMed]
- Van der Zanden, S.Y.; Qiao, X.; Neefjes, J. New Insights into the Activities and Toxicities of the Old Anticancer Drug Doxorubicin. FEBS J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Matyszewska, D.; Nazaruk, E.; Campbell, R.A. Interactions of Anticancer Drugs Doxorubicin and Idarubicin with Lipid Monolayers: New Insight into the Composition, Structure and Morphology. J. Colloid Interface Sci. 2021, 581, 403–416. [Google Scholar] [CrossRef]
- Todor, I.N.; Lukyanova, N.Y.; Chekhun, V.F. The Lipid Content of Cisplatin- and Doxorubicin-Resistant MCF-7 Human Breast Cancer Cells. Exp. Oncol. 2012, 34, 97–100. [Google Scholar]
- Gewirtz, D. A Critical Evaluation of the Mechanisms of Action Proposed for the Antitumor Effects of the Anthracycline Antibiotics Adriamycin and Daunorubicin. Biochem. Pharmacol. 1999, 57, 727–741. [Google Scholar] [CrossRef]
- Wander, D.P.A.; van der Zanden, S.Y.; van der Marel, G.A.; Overkleeft, H.S.; Neefjes, J.; Codée, J.D.C. Doxorubicin and Aclarubicin: Shuffling Anthracycline Glycans for Improved Anticancer Agents. J. Med. Chem. 2020, 63, 12814–12829. [Google Scholar] [CrossRef] [PubMed]
- Gil-de-Gómez, L.; Balgoma, D.; Montero, O. Lipidomic-Based Advances in Diagnosis and Modulation of Immune Response to Cancer. Metabolites 2020, 10, 332. [Google Scholar] [CrossRef]
- Riad, A.; Bien, S.; Westermann, D.; Becher, P.M.; Loya, K.; Landmesser, U.; Kroemer, H.K.; Schultheiss, H.P.; Tschöpe, C. Pretreatment with Statin Attenuates the Cardiotoxicity of Doxorubicin in Mice. Cancer Res. 2009, 69, 695–699. [Google Scholar] [CrossRef] [PubMed]
- Boslem, E.; Weir, J.M.; MacIntosh, G.; Sue, N.; Cantley, J.; Meikle, P.J.; Biden, T.J. Alteration of Endoplasmic Reticulum Lipid Rafts Contributes to Lipotoxicity in Pancreatic β-Cells. J. Biol. Chem. 2013, 288, 26569–26582. [Google Scholar] [CrossRef] [PubMed]
- Zugbi, S.; Winter, U.; Castañon, A.; Sampor, C.; Chantada, G.; Schaiquevich, P. Comparison of the Pharmacological Activity of Idarubicin and Doxorubicin for Retinoblastoma. Pediatr. Blood Cancer 2019, 66, e27441. [Google Scholar] [CrossRef]
- Capes-Davis, A.; Theodosopoulos, G.; Atkin, I.; Drexler, H.G.; Kohara, A.; MacLeod, R.A.F.; Masters, J.R.; Nakamura, Y.; Reid, Y.A.; Reddel, R.R.; et al. Check Your Cultures! A List of Cross-Contaminated or Misidentified Cell Lines. Int. J. Cancer 2010, 127, 1–8. [Google Scholar] [CrossRef]
- Kullenberg, F.; Degerstedt, O.; Calitz, C.; Pavlović, N.; Balgoma, D.; Gråsjö, J.; Sjögren, E.; Hedeland, M.; Heindryckx, F.; Lennernäs, H. In vitro cell toxicity and intracellular uptake of doxorubicin exposed as a solution or liposomes: Implications for treatment of hepatocellular carcinoma. Cells 2021. submitted. [Google Scholar]
- Heindryckx, F.; Mertens, K.; Charette, N.; Vandeghinste, B.; Casteleyn, C.; Van Steenkiste, C.; Slaets, D.; Libbrecht, L.; Staelens, S.; Starkel, P.; et al. Kinetics of Angiogenic Changes in a New Mouse Model for Hepatocellular Carcinoma. Mol. Cancer 2010, 9, 219. [Google Scholar] [CrossRef]
- Ruifrok, A.C.; Johnston, D.A. Quantification of Histochemical Staining by Color Deconvolution. Anal. Quant. Cytol. Histol. 2001, 23, 291–299. [Google Scholar]
- Balgoma, D.; Zelleroth, S.; Grönbladh, A.; Hallberg, M.; Pettersson, C.; Hedeland, M. Anabolic Androgenic Steroids Exert a Selective Remodeling of the Plasma Lipidome That Mirrors the Decrease of the de Novo Lipogenesis in the Liver. Metabolomics 2020, 16, 12. [Google Scholar] [CrossRef]
- Pulfer, M.; Murphy, R.C. Electrospray Mass Spectrometry of Phospholipids. Mass Spectrom. Rev. 2003, 22, 332–364. [Google Scholar] [CrossRef] [PubMed]
- McAnoy, A.M.; Wu, C.C.; Murphy, R.C. Direct Qualitative Analysis of Triacylglycerols by Electrospray Mass Spectrometry Using a Linear Ion Trap. J. Am. Soc. Mass Spectrom. 2005, 16, 1498–1509. [Google Scholar] [CrossRef] [PubMed]
- Betensky, R.A. The P-Value Requires Context, Not a Threshold. Am. Stat. 2019, 73, 115–117. [Google Scholar] [CrossRef]
- Wasserstein, R.L.; Lazar, N.A. The ASA’s Statement on p -Values: Context, Process, and Purpose. Am. Stat. 2016, 70, 129–133. [Google Scholar] [CrossRef]
- Wasserstein, R.L.; Schirm, A.L.; Lazar, N.A. Moving to a World Beyond “p < 0.05”. Am. Stat. 2019, 73, 1–19. [Google Scholar] [CrossRef]
- Gardner, M.J.; Altman, D.G. Confidence Intervals Rather than P Values: Estimation Rather than Hypothesis Testing. Br. Med. J. 1986, 292, 746–750. [Google Scholar] [CrossRef] [PubMed]
- Kell, D.B.; Oliver, S.G. Here Is the Evidence, Now What Is the Hypothesis? The Complementary Roles of Inductive and Hypothesis-Driven Science in the Post-Genomic Era. Bioessays 2004, 26, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Goodacre, R.; Vaidyanathan, S.; Dunn, W.B.; Harrigan, G.G.; Kell, D.B. Metabolomics by Numbers: Acquiring and Understanding Global Metabolite Data. Trends Biotechnol. 2004, 22, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Balsinde, J. Roles of Various Phospholipases A2 in Providing Lysophospholipid Acceptors for Fatty Acid Phospholipid Incorporation and Remodelling. Biochem. J. 2002, 364, 695–702. [Google Scholar] [CrossRef]
- Astudillo, A.M.; Balgoma, D.; Balboa, M.A.; Balsinde, J. Dynamics of Arachidonic Acid Mobilization by Inflammatory Cells. Biochim. Biophys. Acta 2012, 1821, 249–256. [Google Scholar] [CrossRef]
- Poirier, Y.; Antonenkov, V.D.; Glumoff, T.; Hiltunen, J.K. Peroxisomal Beta-Oxidation--a Metabolic Pathway with Multiple Functions. Biochim. Biophys. Acta 2006, 1763, 1413–1426. [Google Scholar] [CrossRef]
- Balgoma, D.; Pettersson, C.; Hedeland, M. Common Fatty Markers in Diseases with Dysregulated Lipogenesis. Trends Endocrinol. Metab. 2019, 30, 283–285. [Google Scholar] [CrossRef]
- Chen, X.; Comish, P.B.; Tang, D.; Kang, R. Characteristics and Biomarkers of Ferroptosis. Front. Cell Dev. Biol. 2021, 9, 637162. [Google Scholar] [CrossRef]
- Zou, Y.; Henry, W.S.; Ricq, E.L.; Graham, E.T.; Phadnis, V.V.; Maretich, P.; Paradkar, S.; Boehnke, N.; Deik, A.A.; Reinhardt, F.; et al. Plasticity of Ether Lipids Promotes Ferroptosis Susceptibility and Evasion. Nature 2020, 585, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Buranrat, B.; Suwannaloet, W.; Naowaboot, J. Simvastatin Potentiates Doxorubicin Activity against MCF-7 Breast Cancer Cells. Oncol. Lett. 2017. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Tontonoz, P. Liver X Receptors in Lipid Signalling and Membrane Homeostasis. Nat. Rev. Endocrinol. 2018, 14, 452–463. [Google Scholar] [CrossRef]
- Lin, Y.-C.; Wang, J.-C.; Wu, M.-S.; Lin, Y.-F.; Chen, C.-R.; Chen, C.-Y.; Chen, K.-C.; Peng, C.-C. Nifedipine Exacerbates Lipogenesis in the Kidney via KIM-1, CD36, and SREBP Upregulation: Implications from an Animal Model for Human Study. Int. J. Mol. Sci. 2020, 21, 4359. [Google Scholar] [CrossRef]
- Monzel, J.V.; Budde, T.; Meyer zu Schwabedissen, H.E.; Schwebe, M.; Bien-Möller, S.; Lütjohann, D.; Kroemer, H.K.; Jedlitschky, G.; Grube, M. Doxorubicin Enhances Oxysterol Levels Resulting in a LXR-Mediated Upregulation of Cardiac Cholesterol Transporters. Biochem. Pharmacol. 2017, 144, 108–119. [Google Scholar] [CrossRef]
- Yun, U.-J.; Lee, J.-H.; Shim, J.; Yoon, K.; Goh, S.-H.; Yi, E.H.; Ye, S.-K.; Lee, J.-S.; Lee, H.; Park, J.; et al. Anti-Cancer Effect of Doxorubicin Is Mediated by Downregulation of HMG-Co A Reductase via Inhibition of EGFR/Src Pathway. Lab. Investig. 2019, 99, 1157–1172. [Google Scholar] [CrossRef] [PubMed]
- Wanders, R.J.A. Peroxisomes, Lipid Metabolism, and Peroxisomal Disorders. Mol. Genet. Metab. 2004, 83, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Dean, J.M.; Lodhi, I.J. Structural and Functional Roles of Ether Lipids. Protein Cell 2018, 9, 196–206. [Google Scholar] [CrossRef]
- Braverman, N.E.; Moser, A.B. Functions of Plasmalogen Lipids in Health and Disease. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2012, 1822, 1442–1452. [Google Scholar] [CrossRef]
- Honsho, M.; Fujiki, Y. Plasmalogen Homeostasis—Regulation of Plasmalogen Biosynthesis and Its Physiological Consequence in Mammals. FEBS Lett. 2017, 591, 2720–2729. [Google Scholar] [CrossRef]
- Burdge, G. α-Linolenic Acid Metabolism in Men and Women: Nutritional and Biological Implications. Curr. Opin. Clin. Nutr. Metab. Care 2004, 7, 137–144. [Google Scholar] [CrossRef]
- Wallis, J.G.; Watts, J.L.; Browse, J. Polyunsaturated Fatty Acid Synthesis: What Will They Think of Next? Trends Biochem. Sci. 2002, 27, 467–473. [Google Scholar] [CrossRef]
- Sprecher, H. Metabolism of Highly Unsaturated N-3 and n-6 Fatty Acids. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2000, 1486, 219–231. [Google Scholar] [CrossRef]
- Balgoma, D.; Astudillo, A.M.; Pérez-Chacón, G.; Montero, O.; Balboa, M.A.; Balsinde, J. Markers of Monocyte Activation Revealed by Lipidomic Profiling of Arachidonic Acid-Containing Phospholipids. J. Immunol. 2010, 184, 3857–3865. [Google Scholar] [CrossRef]
- Pérez-Chacón, G.; Astudillo, A.M.; Balgoma, D.; Balboa, M.A.; Balsinde, J. Control of Free Arachidonic Acid Levels by Phospholipases A2 and Lysophospholipid Acyltransferases. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2009, 1791, 1103–1113. [Google Scholar] [CrossRef] [PubMed]
- Wanders, R.J.A.; Waterham, H.R.; Ferdinandusse, S. Metabolic Interplay between Peroxisomes and Other Subcellular Organelles Including Mitochondria and the Endoplasmic Reticulum. Front. Cell Dev. Biol. 2016, 3, 83. [Google Scholar] [CrossRef] [PubMed]
- Kinsella, J.E.; Black, J.M. Effects of Polyunsaturated Fatty Acids on the Efficacy of Antineoplastic Agents toward L5178Y Lymphoma Cells. Biochem. Pharm. 1993, 45, 1881–1887. [Google Scholar] [CrossRef]
- Koleini, N.; Nickel, B.E.; Edel, A.L.; Fandrich, R.R.; Ravandi, A.; Kardami, E. Oxidized Phospholipids in Doxorubicin-Induced Cardiotoxicity. Chem. Biol. Interact. 2019, 303, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Perez, M.A.; Magtanong, L.; Dixon, S.J.; Watts, J.L. Dietary Lipids Induce Ferroptosis in Caenorhabditiselegans and Human Cancer Cells. Dev. Cell 2020, 54, 447–454.e4. [Google Scholar] [CrossRef]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of Polyunsaturated Fatty Acids by Lipoxygenases Drives Ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef]
- Zheng, J.; Conrad, M. The Metabolic Underpinnings of Ferroptosis. Cell Metab. 2020, 32, 920–937. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Stoffel, W.; Holz, B.; Jenke, B.; Binczek, E.; Günter, R.H.; Kiss, C.; Karakesisoglou, I.; Thevis, M.; Weber, A.-A.; Arnhold, S.; et al. Δ6-Desaturase (FADS2) Deficiency Unveils the Role of Ω3- and Ω6-Polyunsaturated Fatty Acids. EMBO J. 2008, 27, 2281–2292. [Google Scholar] [CrossRef]
- Vriens, K.; Christen, S.; Parik, S.; Broekaert, D.; Yoshinaga, K.; Talebi, A.; Dehairs, J.; Escalona-Noguero, C.; Schmieder, R.; Cornfield, T.; et al. Evidence for an Alternative Fatty Acid Desaturation Pathway Increasing Cancer Plasticity. Nature 2019, 566, 403–406. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Tachtsis, B.; Whitfield, J.; Hawley, J.A.; Hoffman, N.J. Omega-3 Polyunsaturated Fatty Acids Mitigate Palmitate-Induced Impairments in Skeletal Muscle Cell Viability and Differentiation. Front. Physiol. 2020, 11, 563. [Google Scholar] [CrossRef] [PubMed]
- Ohta, M.; Kawano, H.; Notsu, T.; Naba, H.; Imada, K. Eicosapentaenoic Acid Attenuates Statin-Induced ER Stress and Toxicity in Myoblast. Biochem. Biophys. Res. Commun. 2012, 424, 301–307. [Google Scholar] [CrossRef]
- Chakraborty, S.; Doktorova, M.; Molugu, T.R.; Heberle, F.A.; Scott, H.L.; Dzikovski, B.; Nagao, M.; Stingaciu, L.-R.; Standaert, R.F.; Barrera, F.N.; et al. How Cholesterol Stiffens Unsaturated Lipid Membranes. Proc. Natl. Acad. Sci. USA 2020, 117, 21896–21905. [Google Scholar] [CrossRef] [PubMed]
- Speelmans, G.; Staffhorst, R.W.H.M.; de Kruijff, B. The Anionic Phospholipid-Mediated Membrane Interaction of the Anti-Cancer Drug Doxorubicin Is Enhanced by Phosphatidylethanolamine Compared to Other Zwitterionic Phospholipids. Biochemistry 1997, 36, 8657–8662. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-S.; Lee, D.-H.; Choudry, H.A.; Bartlett, D.L.; Lee, Y.J. Ferroptosis-Induced Endoplasmic Reticulum Stress: Cross-Talk between Ferroptosis and Apoptosis. Mol. Cancer Res. 2018, 16, 1073–1076. [Google Scholar] [CrossRef] [PubMed]
- Pavlović, N.; Calitz, C.; Thanapirom, K.; Mazza, G.; Rombouts, K.; Gerwins, P.; Heindryckx, F. Inhibiting IRE1α-Endonuclease Activity Decreases Tumor Burden in a Mouse Model for Hepatocellular Carcinoma. Elife 2020, 9. [Google Scholar] [CrossRef]
- Van Campenhout, S.; Tilleman, L.; Lefere, S.; Vandierendonck, A.; Raevens, S.; Verhelst, X.; Geerts, A.; Van Nieuwerburgh, F.; Van Vlierberghe, H.; Devisscher, L. Myeloid-Specific IRE1alpha Deletion Reduces Tumour Development in a Diabetic, Non-Alcoholic Steatohepatitis-Induced Hepatocellular Carcinoma Mouse Model. Metabolism 2020, 107, 154220. [Google Scholar] [CrossRef] [PubMed]
- Salaroglio, I.C.; Panada, E.; Moiso, E.; Buondonno, I.; Provero, P.; Rubinstein, M.; Kopecka, J.; Riganti, C. PERK Induces Resistance to Cell Death Elicited by Endoplasmic Reticulum Stress and Chemotherapy. Mol. Cancer 2017, 16, 91. [Google Scholar] [CrossRef]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luís, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic Reticulum Stress Signalling—From Basic Mechanisms to Clinical Applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef]
- Dubbelboer, I.R.; Pavlovic, N.; Heindryckx, F.; Sjögren, E.; Lennernäs, H. Liver Cancer Cell Lines Treated with Doxorubicin under Normoxia and Hypoxia: Cell Viability and Oncologic Protein Profile. Cancers 2019, 11, 1024. [Google Scholar] [CrossRef]
- Newell, M.; Patel, D.; Goruk, S.; Field, C.J. Docosahexaenoic Acid Incorporation Is Not Affected by Doxorubicin Chemotherapy in Either Whole Cell or Lipid Raft Phospholipids of Breast Cancer Cells in Vitro and Tumor Phospholipids in Vivo. Lipids 2020, 55, 549–565. [Google Scholar] [CrossRef]
- Lages, E.B.; Fernandes, R.S.; de Oliveira Silva, J.; de Souza, Â.M.; Cassali, G.D.; de Barros, A.L.B.; Ferreira, L.A.M. Co-Delivery of Doxorubicin, Docosahexaenoic Acid, and α-Tocopherol Succinate by Nanostructured Lipid Carriers Has a Synergistic Effect to Enhance Antitumor Activity and Reduce Toxicity. Biomed. Pharmacother. 2020, 132, 110876. [Google Scholar] [CrossRef]
- Tadokoro, T.; Ikeda, M.; Ide, T.; Deguchi, H.; Ikeda, S.; Okabe, K.; Ishikita, A.; Matsushima, S.; Koumura, T.; Yamada, K.-I.; et al. Mitochondria-Dependent Ferroptosis Plays a Pivotal Role in Doxorubicin Cardiotoxicity. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Serini, S.; Ottes Vasconcelos, R.; Nascimento Gomes, R.; Calviello, G. Protective Effects of ω-3 PUFA in Anthracycline-Induced Cardiotoxicity: A Critical Review. Int. J. Mol. Sci. 2017, 18, 2689. [Google Scholar] [CrossRef]
- Varela-López, A.; Battino, M.; Navarro-Hortal, M.D.; Giampieri, F.; Forbes-Hernández, T.Y.; Romero-Márquez, J.M.; Collado, R.; Quiles, J.L. An Update on the Mechanisms Related to Cell Death and Toxicity of Doxorubicin and the Protective Role of Nutrients. Food Chem. Toxicol. 2019, 134, 110834. [Google Scholar] [CrossRef] [PubMed]
- Jakobisiak, M.; Golab, J. Statins Can Modulate Effectiveness of Antitumor Therapeutic Modalities: Combinations of Statins with Other Drugs. Med. Res. Rev. 2010, 30, 102–135. [Google Scholar] [CrossRef]
- Werner, M.; Sacher, J.; Hohenegger, M. Mutual Amplification of Apoptosis by Statin-Induced Mitochondrial Stress and Doxorubicin Toxicity in Human Rhabdomyosarcoma Cells: Statin-Induced Apoptosis in Rhabdomyosarcoma Cells. Br. J. Pharmacol. 2004, 143, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Damrot, J.; Nübel, T.; Epe, B.; Roos, W.P.; Kaina, B.; Fritz, G. Lovastatin Protects Human Endothelial Cells from the Genotoxic and Cytotoxic Effects of the Anticancer Drugs Doxorubicin and Etoposide. Br. J. Pharmacol. 2006, 149, 988–997. [Google Scholar] [CrossRef]
- Henninger, C.; Huelsenbeck, J.; Huelsenbeck, S.; Grösch, S.; Schad, A.; Lackner, K.J.; Kaina, B.; Fritz, G. The Lipid Lowering Drug Lovastatin Protects against Doxorubicin-Induced Hepatotoxicity. Toxicol. Appl. Pharmacol. 2012, 261, 66–73. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | DOX Concentration (µM) | IDA Concentration (µM) |
|---|---|---|
| HepG2 | 0.5 | 0.1 |
| Huh7 | 5 | 0.1 |
| SNU449 | 30 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balgoma, D.; Kullenberg, F.; Calitz, C.; Kopsida, M.; Heindryckx, F.; Lennernäs, H.; Hedeland, M. Anthracyclins Increase PUFAs: Potential Implications in ER Stress and Cell Death. Cells 2021, 10, 1163. https://doi.org/10.3390/cells10051163
Balgoma D, Kullenberg F, Calitz C, Kopsida M, Heindryckx F, Lennernäs H, Hedeland M. Anthracyclins Increase PUFAs: Potential Implications in ER Stress and Cell Death. Cells. 2021; 10(5):1163. https://doi.org/10.3390/cells10051163
Chicago/Turabian StyleBalgoma, David, Fredrik Kullenberg, Carlemi Calitz, Maria Kopsida, Femke Heindryckx, Hans Lennernäs, and Mikael Hedeland. 2021. "Anthracyclins Increase PUFAs: Potential Implications in ER Stress and Cell Death" Cells 10, no. 5: 1163. https://doi.org/10.3390/cells10051163
APA StyleBalgoma, D., Kullenberg, F., Calitz, C., Kopsida, M., Heindryckx, F., Lennernäs, H., & Hedeland, M. (2021). Anthracyclins Increase PUFAs: Potential Implications in ER Stress and Cell Death. Cells, 10(5), 1163. https://doi.org/10.3390/cells10051163