
Metformin Dysregulates the Unfolded Protein Response and the WNT/β-Catenin Pathway in Endometrial Cancer Cells through an AMPK-Independent Mechanism

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Reagents
2.2. Cell Proliferation and Colony Formation Assay
2.3. Western Blot
2.4. RNA Isolation and Real-Time Reverse Transcription-PCR
2.5. Wound Healing Assay
2.6. Luciferase Assay
2.7. Immunofluorescence
2.8. Cell Fractionation
2.9. Statistical Analysis
3. Results
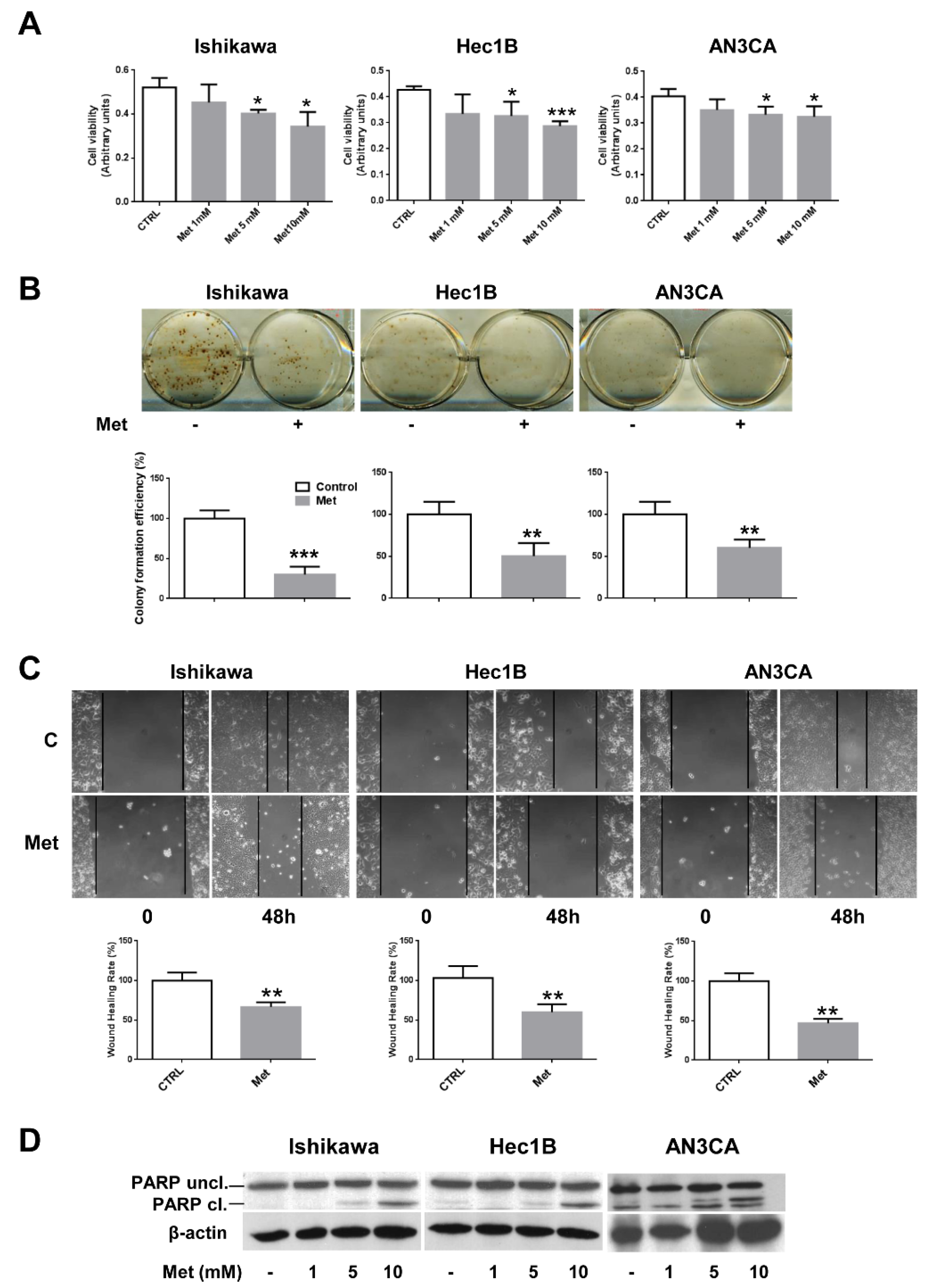
3.1. Metformin Affects Cell Growth and Viability of Endometrial Cancer Cells
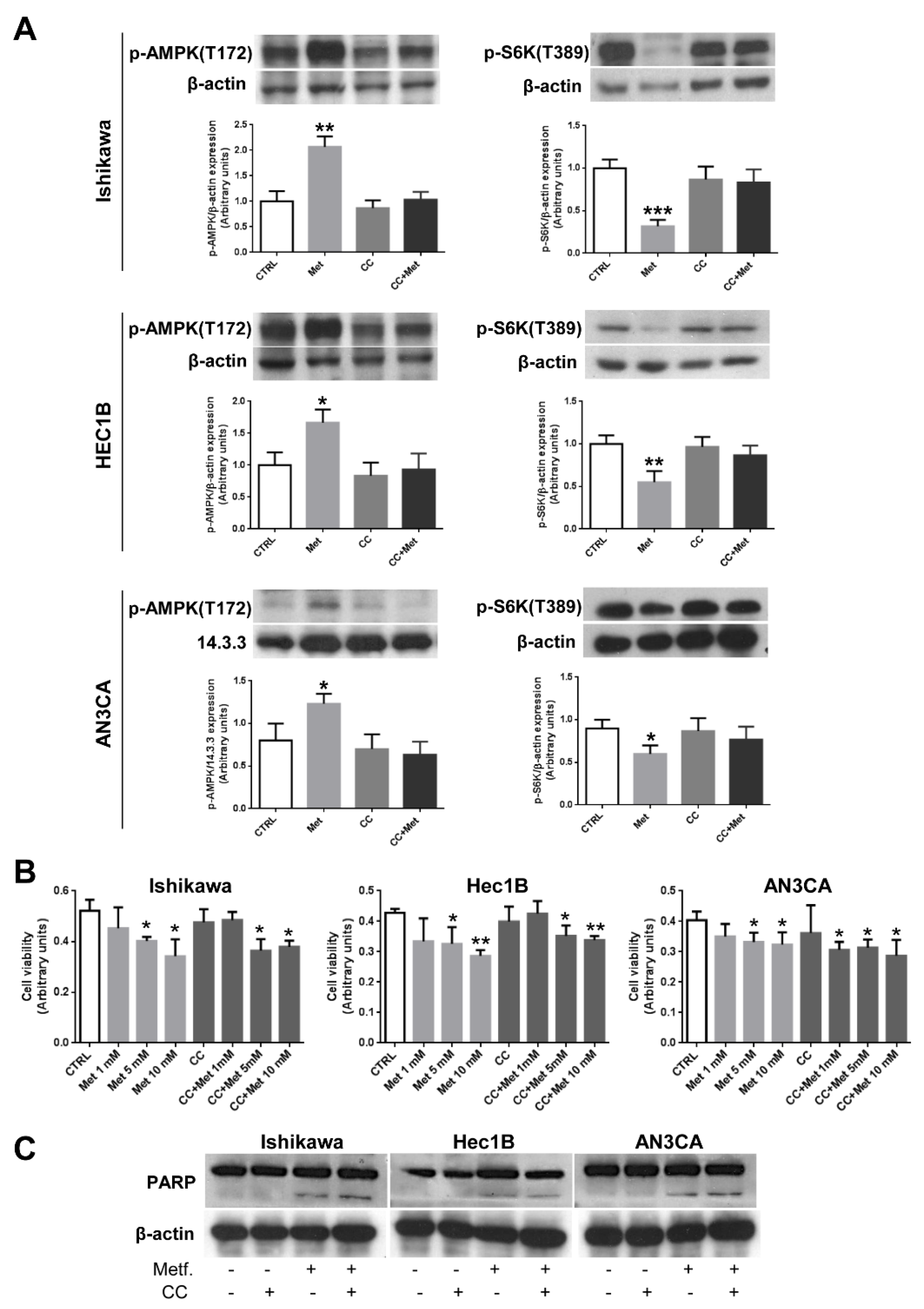
3.2. CC Does Not Prevent Metformin Effects on Endometrial Cancer Cells
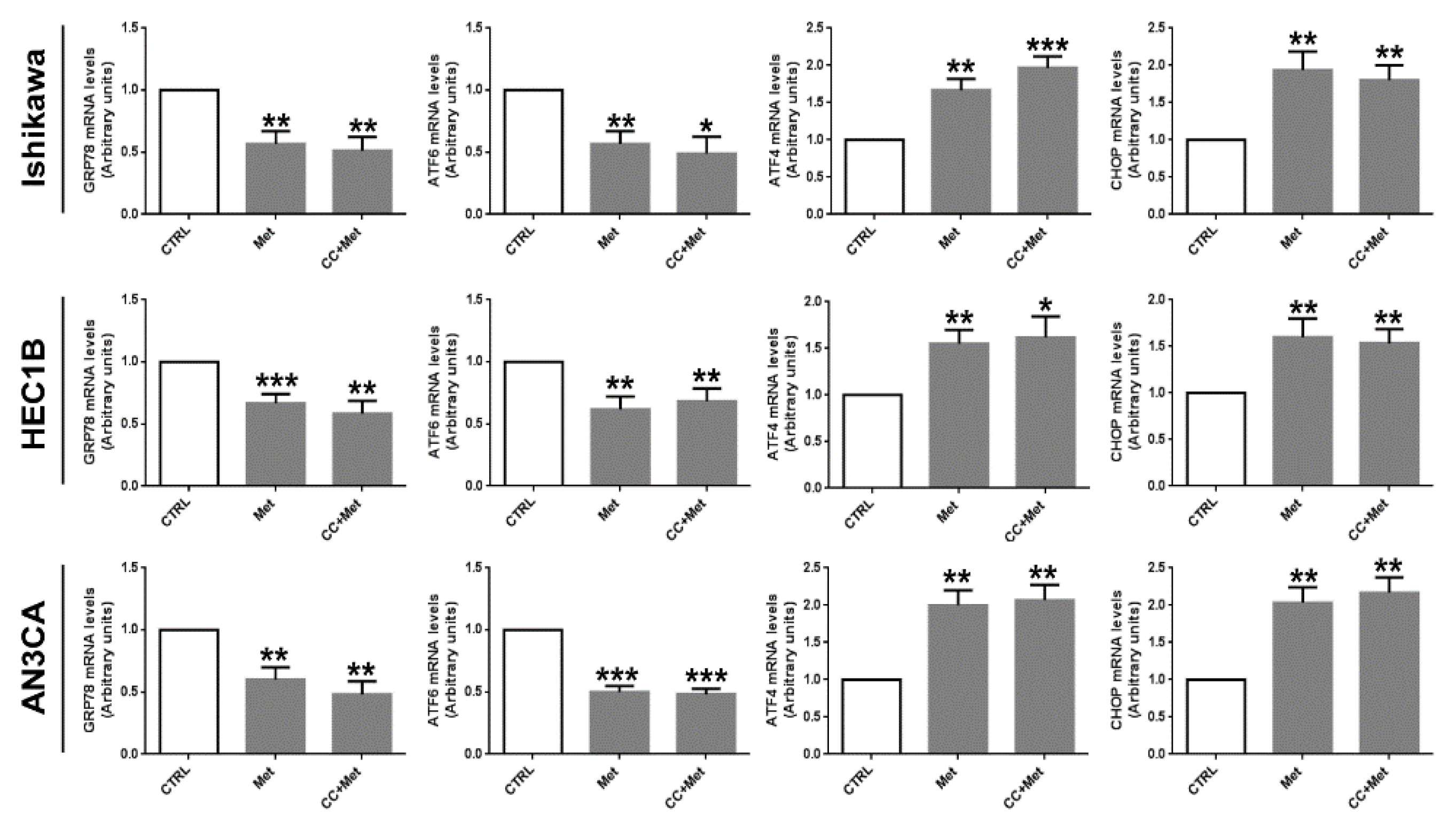
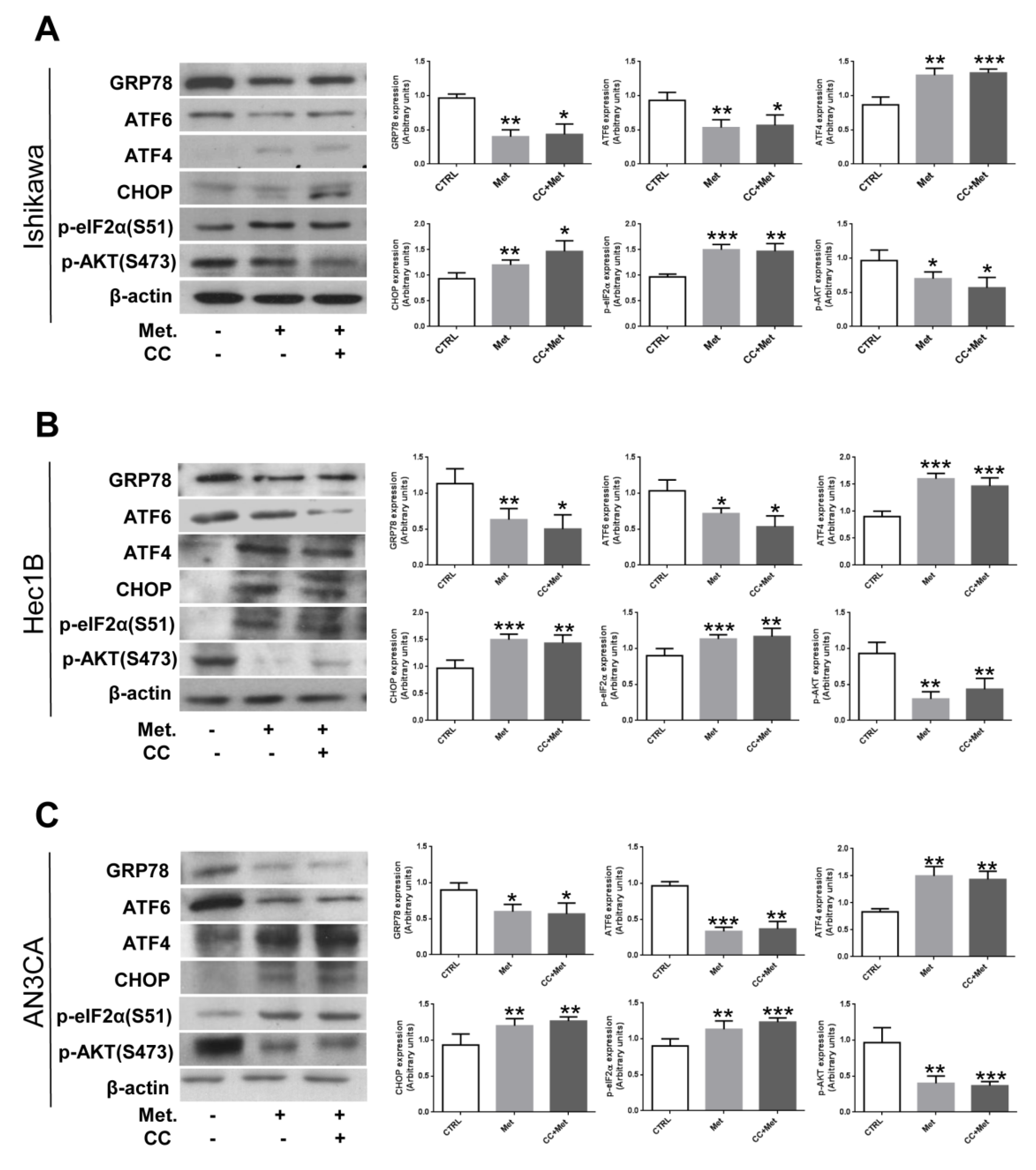
3.3. Metformin Modulates the Expression of UPR Key Players in Endometrial Cancer Cells
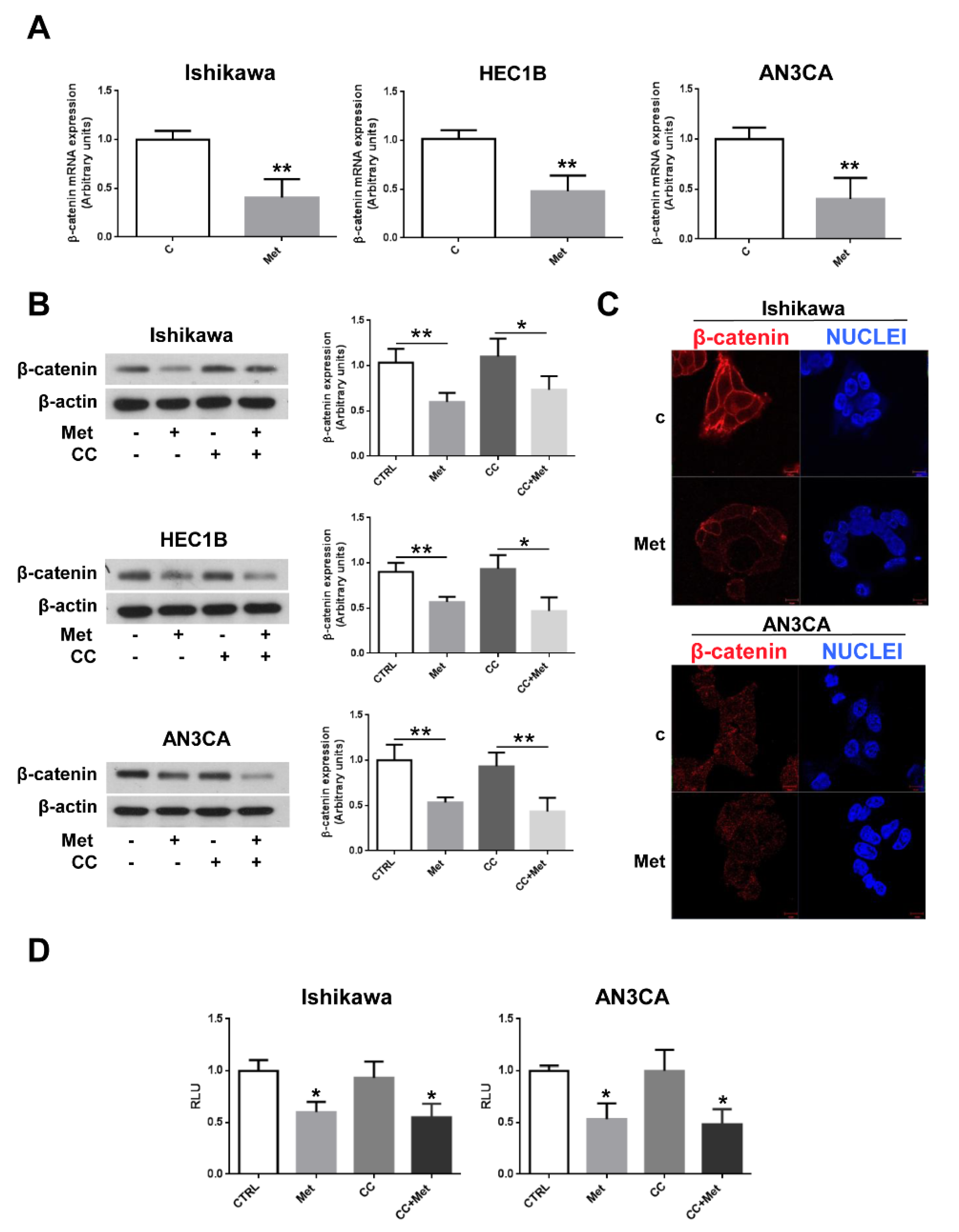
3.4. Metformin Inhibits β-Catenin Expression and Activity in Endometrial Cancer Cells
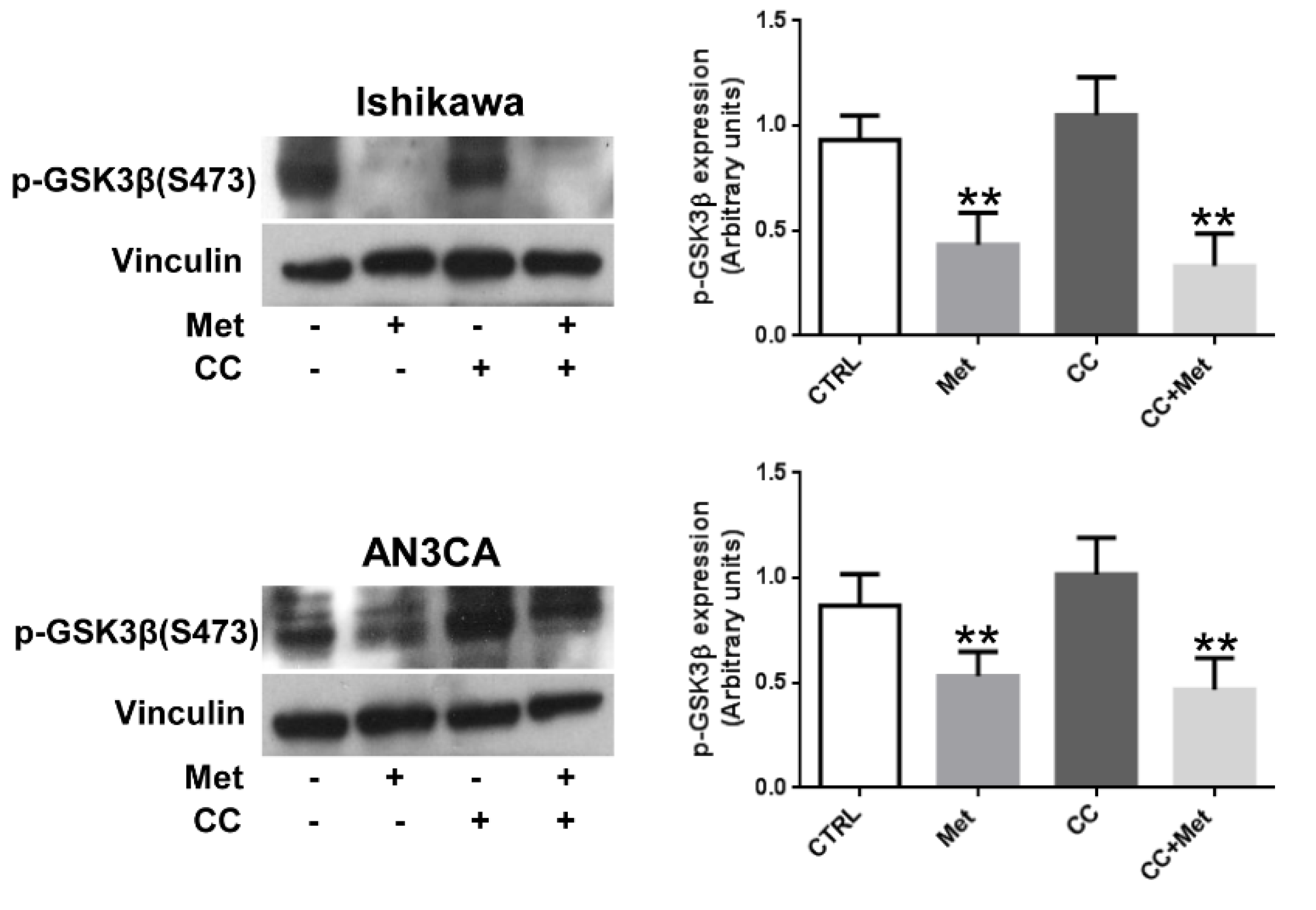
3.5. Metformin Inhibits GSK3β Phosphorylation in Endometrial Cancer Cells
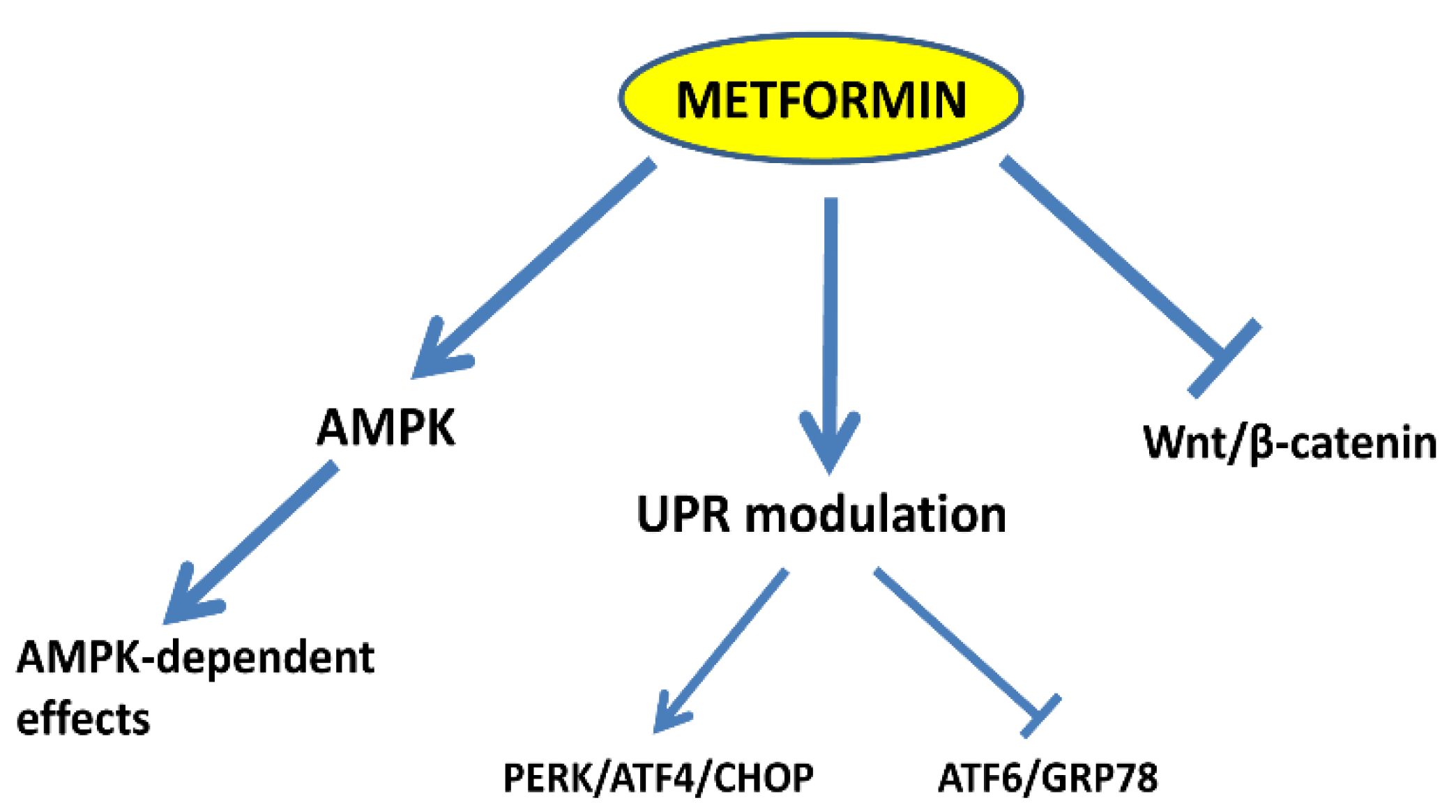
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pollak, M.N. Investigating metformin for cancer prevention and treatment: The end of the beginning. Cancer Discov. 2012, 2, 2778–2790. [Google Scholar] [CrossRef] [PubMed]
- Kourelis, T.V.; Siegel, R.D. Metformin and cancer: New applications for an old drug. Med. Oncol. 2012, 29, 1314–1327. [Google Scholar] [CrossRef]
- Evans, J.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and reduced risk of cancer in diabetic patients. BMJ 2005, 330, 1304–1305. [Google Scholar] [CrossRef]
- Bowker, S.L.; Majumdar, S.R.; Veugelers, P.; Johnson, J.A. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin. Diabetes Care 2006, 29, 254–258. [Google Scholar] [CrossRef]
- Ezewuiro, O.; Grushko, T.A.; Kocherginsky, M.; Habis, M.; Hurteau, J.A.; Mills, K.A.; Hunn, J.; Olopade, O.I.; Fleming, G.F.; Romero, I.L. Association of Metformin Use with Outcomes in Advanced Endometrial Cancer Treated with Chemotherapy. PLoS ONE 2016, 11, e0147145. [Google Scholar] [CrossRef] [PubMed]
- Mu, N.; Xu, T.; Gao, M.; Dong, M.; Tang, Q.; Hao, L.; Wang, G.; Li, Z.; Wang, W.; Yang, Y.; et al. Therapeutic effect of metformin in the treatment of endometrial cancer. Oncol. Lett. 2020, 20, 156. [Google Scholar] [CrossRef] [PubMed]
- Kitson, S.J.; Maskell, Z.; Sivalingam, V.N.; Allen, J.L.; Ali, S.; Burns, S.; Gilmour, K.; Latheef, R.; Slade, R.J.; Pemberton, P.W.; et al. PRE-surgical Metformin In Uterine Malignancy (PREMIUM): A Multi-Center, Randomized Double-Blind, Placebo-Controlled Phase III Trial. Clin. Cancer Res. 2019, 25, 2424–2432. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.L.; Yin, J.; Alimujiang, M.; Yu, X.Y.; Ai, L.G.; Bao, Y.Q.; Liu, F.; Jia, W.P. Inhibition of mitochondrial complex I improves glucose metabolism independently of AMPK activation. J. Cell Mol. Med. 2018, 22, 1316–1328. [Google Scholar] [CrossRef]
- Cameron, A.; Logie, L.; Patel, K.; Erhardt, S.; Bacon, S.; Middleton, P.; Harthill, J.; Forteath, C.; Coats, J.; Kerr, C.; et al. Metformin selectively targets redox control of complex I energy transduction. Redox Biol. 2018, 14, 187–197. [Google Scholar] [CrossRef]
- Stephenne, X.; Foretz, M.; Taleux, N.; van der Zon, G.C.; Sokal, E.; Hue, L.; Viollet, B.; Guigas, B. Metformin activates AMP-activated protein kinase in primary human hepatocytes by decreasing cellular energy status. Diabetologia 2011, 54, 3101–3110. [Google Scholar] [CrossRef]
- Bridges, H.R.; Jones, A.J.; Pollak, M.N.; Hirst, J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem. J. 2014, 462, 475–487. [Google Scholar] [CrossRef]
- Kuznetsov, J.N.; Leclerc, G.J.; Leclerc, G.M.; Barredo, J.C. AMPK and Akt determine apoptotic cell death following perturbations of one-carbon metabolism by regulating ER stress in acute lymphoblastic leukemia. Mol. Cancer Ther. 2011, 10, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Sha, H.; Davisson, R.L.; Qi, L. Phenformin Activates Unfolded Protein Response in an AMP-Activated Protein Kinase (AMPK)-Dependent Manner. J. Biol. Chem. 2013, 288, 13631–13638. [Google Scholar] [CrossRef]
- Yu-Chin, L.; Meng-Hsuan, W.; Tzu-Tang, W.; Yun-Chieh, L.; Wen-Chih, H.; Liang-Yu, H.; Yi-Ting, L.; Ching-Chow, C. Metformin sensitizes anticancer effect of dasatinib in head and neck squamous cell carcinoma cells through AMPK-dependent ER stress. Oncotarget 2014, 5, 298–308. [Google Scholar]
- Terai, K.; Hiramoto, Y.; Masaki, M.; Sugiyama, S.; Kuroda, T.; Hori, M.; Kawase, I.; Hirota, H. AMP-activated protein kinase protects cardiomyocytes against hypoxic injury through attenuation of endoplasmic reticulum stress. Mol. Cell Biol. 2005, 25, 9554–9575. [Google Scholar] [CrossRef]
- Yeh, C.H.; Chen, T.P.; Wang, Y.C.; Lin, Y.M.; Fang, S.W. AMP-activated protein kinase activation during cardioplegia-induced hypoxia/reoxygenation injury attenuates cardiomyocytic apoptosis via reduction of endoplasmic reticulum stress. Mediat. Inflamm. 2010, 2010, 130636. [Google Scholar] [CrossRef]
- Dong, Y.; Zhang, M.; Wang, S.; Liang, B.; Zhao, Z.; Liu, C.; Wu, M.; Chul Choi, H.; Lyons, T.J.; Zou, M.H. Activation of AMP-activated protein kinase inhibits oxidized LDL-triggered endoplasmic reticulum stress in vivo. Diabetes 2010, 59, 1386–1396. [Google Scholar] [CrossRef] [PubMed]
- DeSalvo, J.; Kuznetsov, J.N.; Du, J.; Leclerc, G.M.; Leclerc, G.J.; Lampidis, T.J.; Barredo, J.C. Inhibition of Akt potentiates 2-DG-induced apoptosis via downregulation of UPR in acute lymphoblastic leukemia. Mol. Cancer Res. 2012, 10, 969–978. [Google Scholar] [CrossRef]
- Schröder, M.; Kaufman, R. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Furuno, A.; Sakurai, J.; Sakamoto, A.; Park, H.R.; Shin-Ya, K.; Tsuruo, T.; Tomida, A. Chemical genomics identifies the unfolded protein response as a target for selective cancer cell killing during glucose deprivation. Cancer Res. 2009, 69, 4225–4234. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Tomida, A. Use of chemical genomics in assessment of the UPR. Methods Enzymol. 2011, 491, 327–341. [Google Scholar] [PubMed]
- Jagannathan, S.; Abdel-Malek, M.A.; Malek, E.; Vad, N.; Latif, T.; Anderson, K.C.; Driscoll, J.J. Pharmacologic screens reveal metformin that suppresses GRP78-dependent autophagy to enhance the anti-myeloma effect of bortezomib. Leukemia 2015, 29, 2184–2191. [Google Scholar] [CrossRef]
- Lee, A.S. The glucose-regulated proteins: Stress induction and clinical applications. Trends Biochem. Sci. 2001, 26, 504–510. [Google Scholar] [CrossRef]
- Leclerc, G.M.; Leclerc, G.J.; Kuznetsov, J.N.; de Salvo, J.; Barredo, J.C. Metformin induces apoptosis through AMPK-dependent inhibition of UPR signaling in ALL lymphoblasts. PLoS ONE 2013, 8, e74420. [Google Scholar] [CrossRef] [PubMed]
- Salis, O.; Bedir, A.; Ozdemir, T.; Okuyucu, A.; Alacam, H. The relationship between anticancer effect of metformin and the transcriptional regulation of certain genes (CHOP, CAV-1, HO-1, SGK-1 and Par-4) on MCF-7 cell line. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 1602–1609. [Google Scholar]
- Bifulco, G.; Miele, C.; di Jeso, B.; Beguinot, F.; Nappi, C.; di Carlo, C.; Capuozzo, S.; Terrazzano, G.; Insabato, L.; Ulianich, L. Endoplasmic reticulum stress is activated in endometrial adenocarcinoma. Gyn. Oncol. 2012, 125, 220–225. [Google Scholar] [CrossRef]
- Calì, G.; Insabato, L.; Conza, D.; Bifulco, G.; Parrillo, L.; Mirra, P.; Fiory, F.; Miele, C.; Raciti, G.A.; Di Jeso, B.; et al. GRP78 Mediates Cell Growth and Invasiveness in Endometrial Cancer. J. Cell Physiol. 2014, 229, 1417–1426. [Google Scholar] [CrossRef] [PubMed]
- Ulianich, L.; Insabato, L. Endoplasmic reticulum stress in endometrial cancer. Front. Med. 2014, 1, 55. [Google Scholar] [CrossRef]
- McMellen, A.; Woodruff, E.R.; Corr, B.R.; Bitler, B.G.; Moroney, M.R. Wnt Signaling in Gynecologic Malignancies. Int. J. Mol. Sci. 2020, 21, 4272. [Google Scholar] [CrossRef] [PubMed]
- Melnik, S.; Dvornikov, D.; Müller-Decker, K.; Depner, S.; Stannek, P.; Meister, M.; Warth, A.; Thomas, M.; Muley, T.; Risch, A.; et al. Cancer cell specific inhibition of Wnt/β-catenin signaling by forced intracellular acidification. Cell Discov. 2018, 4, 37. [Google Scholar] [CrossRef]
- Calì, G.; Gentile, F.; Mogavero, S.; Pallante, P.; Nitsch, R.; Ciancia, G.; Ferraro, A.; Fusco, A.; Nitsch, L. CDH16/Ksp-Cadherin Is Expressed in the Developing Thyroid Gland and Is Strongly Down-Regulated in Thyroid Carcinomas. Endocrinology 2012, 153, 522–534. [Google Scholar] [CrossRef]
- Xie, Y.; Wang, Y.L.; Yu, L.; Hu, Q.; Ji, L.; Zhang, Y.; Liao, Q.P. Metformin promotes progesterone receptor expression via inhibition of mammalian target of rapamycin (mTOR) in endometrial cancer cells. J. Steroid Biochem. Mol. Biol. 2011, 126, 113–120. [Google Scholar] [CrossRef]
- Cantrell, L.A.; Zhou, C.; Mendivil, A.; Malloy, K.M.; Gehrig, P.A.; Bae-Jump, V.L. Metformin is a potent inhibitor of endometrial cancer cell proliferation: Implications for a novel treatment strategy. Gynecol. Oncol. 2010, 116, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Kimura, F.; Yamanaka, A.; Takebayashi, A.; Kita, N.; Takahashi, K.; Murakami, T. Metformin impairs growth of endometrial cancer cells via cell cycle arrest and concomitant autophagy and apoptosis. Cancer Cell Int. 2014, 14, 53. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, S.H.; Desnoyers, S.; Ottaviano, Y.; Davidson, N.E.; Poirier, G.G. Specific proteolytic cleavage of poly(ADP-ribose) polymerase: An early marker of chemotherapy-induced apoptosis. Cancer Res. 1993, 53, 3976–3985. [Google Scholar]
- Gray, M.J.; Mhawech-Fauceglia, P.; Yoo, E.; Yang, W.; Wu, E.; Lee, A.S.; Lin, Y.G. AKT inhibition mitigates GRP78 (glucose-regulated protein) expression and contribution to chemoresistance in endometrial cancers. Int. J. Cancer 2013, 133, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Novellasdemunt, L.; Antas, P.; Li, V.S.W. Targeting Wnt signaling in colorectal cancer. A review in the theme: Cell signaling: Proteins, pathways and mechanisms. Am. J. Physiol. Cell Physiol. 2015, 309, C511–C521. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Kim, D.; Kee, S.H. Metformin-activated AMPK regulates β-catenin to reduce cell proliferation in colon carcinoma RKO cells. Oncol. Lett. 2019, 17, 2695–2702. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, D.A.; Yates, M.S.; van der Hoeven, D.; Rodkey, T.L.; Zhang, Q.; Co, N.N.; Burzawa, J.; Chigurupati, S.; Celestino, J.; Bowser, J.; et al. Another surprise from Metformin: Novel mechanism of action via K-Ras influences endometrial cancer response to therapy. Mol. Cancer Ther. 2013, 12, 2847–2856. [Google Scholar] [CrossRef] [PubMed]
- Sarfstein, R.; Friedman, Y.; Attias-Geva, Z.; Fishman, A.; Bruchim, I.; Werner, H. Metformin downregulates the insulin/IGF-I signaling pathway and inhibits different uterine serous carcinoma (USC) cells proliferation and migration in p53-dependent or -independent manners. PLoS ONE 2013, 8, e61537. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, M.X.; Wang, H.; Zeng, Z.; Li, X.M. Metformin down-regulates endometrial carcinoma cell secretion of IGF-1 and expression of IGF-1R. Asian Pac. J. Cancer Prev. 2015, 16, 221–225. [Google Scholar] [CrossRef]
- De Barros Machado, A.; Dos Reis, V.; Weber, S.; Jauckus, J.; Brum, I.S.; Von Eye Corleta, H.; Strowitzki, T.; Capp, E.; Germeyer, A. Proliferation and metastatic potential of endometrial cancer cells in response to metformin treatment in a high versus normal glucose environment. Oncol. Lett. 2016, 12, 3626–3632. [Google Scholar] [CrossRef]
- Zou, J.; Hong, L.; Luo, C.; Li, Z.; Zhu, Y.; Huang, T.; Zhang, Y.; Yuan, H.; Hu, Y.; Wen, T.; et al. Metformin inhibits estrogen-dependent endometrial cancer cell growth by activating the AMPK-FOXO1 signal pathway. Cancer Sci. 2016, 107, 1806–1817. [Google Scholar] [CrossRef] [PubMed]
- Wallbillich, J.J.; Josyula, S.; Saini, U.; Zingarelli, R.A.; Dorayappan, K.D.; Riley, M.K.; Wanner, R.A.; Cohn, D.E.; Selvendiran, K. High Glucose-mediated STAT3 activation in endometrial cancer is inhibited by metformin: Therapeutic implications for endometrial cancer. PLoS ONE 2017, 12, e0170318. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.I.; Lee, J.S.; Kwak, B.K.; Hwang, W.M.; Kim, M.J.; Kim, Y.B.; Chung, S.S.; Park, K.S. Metformin Ameliorates Lipotoxic beta-Cell Dysfunction through a Concentration-Dependent Dual Mechanism of Action. Diabetes Metab. J. 2019, 43, 854–866. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.Y.; Martinez-Outschoorn, U.E.; Schilder, R.J.; Kim, C.H.; Richard, S.D.; Rosenblum, N.G.; Johnson, J.M. Metformin as a Therapeutic Target in Endometrial Cancers. Front. Oncol. 2018, 8, 341. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.M.; Fiol, C.J.; DePaoli-Roach, A.A.; Roach, P.J. Glycogen synthase kinase-3 beta is a dual specificity kinase differentially regulated by tyrosine and serine/threonine phosphorylation. J. Biol. Chem. 1994, 269, 14566–14574. [Google Scholar] [CrossRef]
- King, T.D.; Song, L.; Jope, R.S. AMP-activated protein kinase (AMPK) activating agents cause dephosphorylation of Akt and glycogen synthase kinase-3. Biochem. Pharmacol. 2006, 71, 1637–1647. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Conza, D.; Mirra, P.; Calì, G.; Insabato, L.; Fiory, F.; Beguinot, F.; Ulianich, L. Metformin Dysregulates the Unfolded Protein Response and the WNT/β-Catenin Pathway in Endometrial Cancer Cells through an AMPK-Independent Mechanism. Cells 2021, 10, 1067. https://doi.org/10.3390/cells10051067
Conza D, Mirra P, Calì G, Insabato L, Fiory F, Beguinot F, Ulianich L. Metformin Dysregulates the Unfolded Protein Response and the WNT/β-Catenin Pathway in Endometrial Cancer Cells through an AMPK-Independent Mechanism. Cells. 2021; 10(5):1067. https://doi.org/10.3390/cells10051067
Chicago/Turabian StyleConza, Domenico, Paola Mirra, Gaetano Calì, Luigi Insabato, Francesca Fiory, Francesco Beguinot, and Luca Ulianich. 2021. "Metformin Dysregulates the Unfolded Protein Response and the WNT/β-Catenin Pathway in Endometrial Cancer Cells through an AMPK-Independent Mechanism" Cells 10, no. 5: 1067. https://doi.org/10.3390/cells10051067
APA StyleConza, D., Mirra, P., Calì, G., Insabato, L., Fiory, F., Beguinot, F., & Ulianich, L. (2021). Metformin Dysregulates the Unfolded Protein Response and the WNT/β-Catenin Pathway in Endometrial Cancer Cells through an AMPK-Independent Mechanism. Cells, 10(5), 1067. https://doi.org/10.3390/cells10051067