
Shaping of T Cell Functions by Trogocytosis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
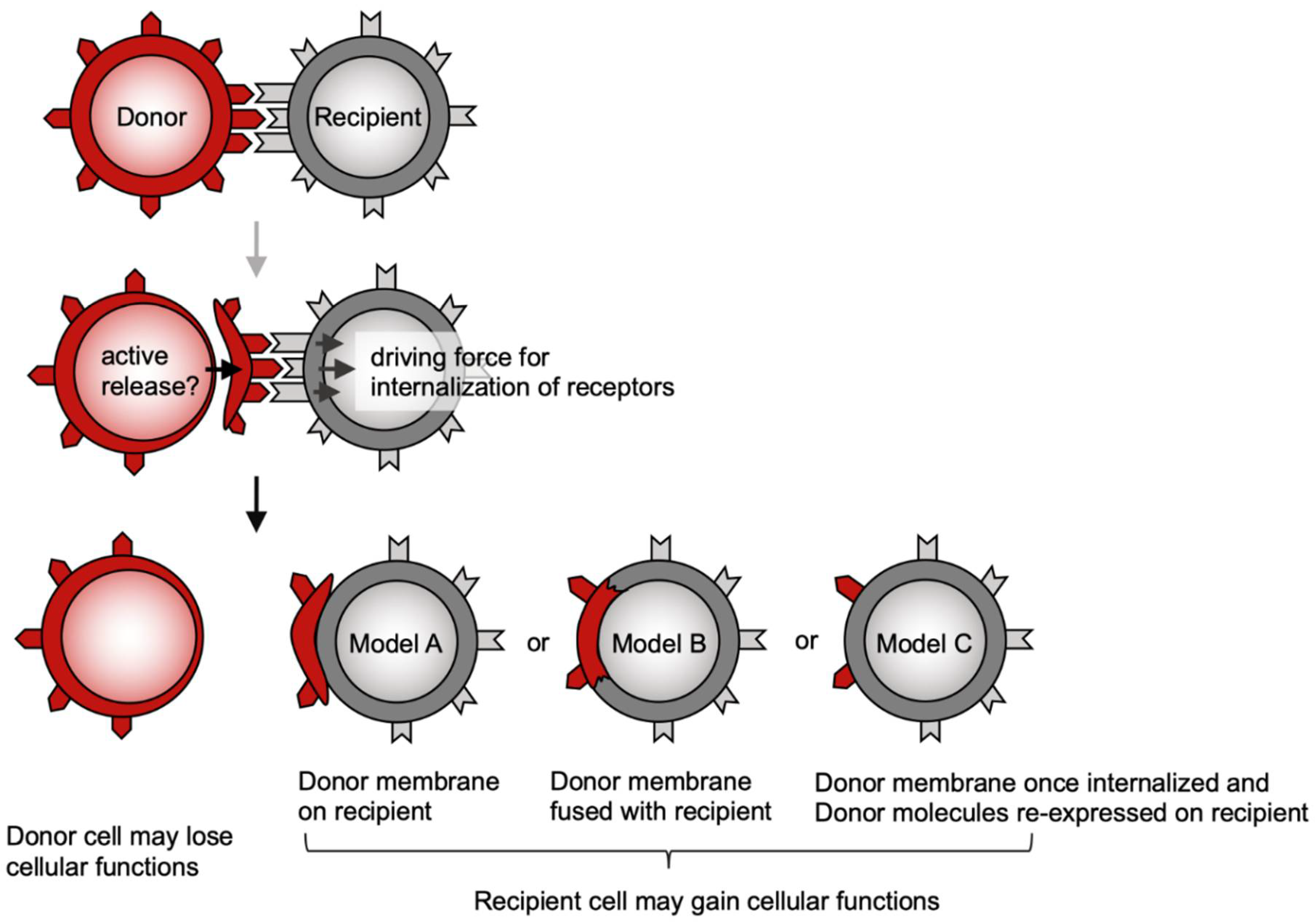
2. Possible Mechanisms Underlying Trogocytosis
3. Tumor
3.1. Priming of CD8+ T Cells by Cross-Presentation and Cross-Dressing
3.2. MHC Trogocytosis in the CTL Effector Phase
3.3. CAR-Mediated Trogocytosis
4. Transplantation
4.1. Allospecific T Cell Priming by Cross-Dressing
4.2. Induction of Allospecific T Cell Tolerance by Cross-Dressing
4.3. Induction of Allospecific T Cell Tolerance by Double-Negative T (DNT) Cell Trogocytosis
5. Infection
6. Th2 Diseases
7. Treg Trogocytosis
8. Application of Trogocytosis
9. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ahmed, K.A.; Munegowda, M.A.; Xie, Y.; Xiang, J. Intercellular trogocytosis plays an important role in modulation of immune responses. Cell. Mol. Immunol. 2008, 5, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.P.; Lindorfer, M.A. Fcgamma-receptor-mediated trogocytosis impacts mAb-based therapies: Historical precedence and recent developments. Blood 2015, 125, 762–766. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, M. Antigen presentation by MHC-dressed cells. Front. Immunol. 2015, 5, 672. [Google Scholar] [CrossRef] [PubMed]
- Dance, A. Core Concept: Cells nibble one another via the under-appreciated process of trogocytosis. Proc. Natl. Acad. Sci. USA 2019, 116, 17608–17610. [Google Scholar] [CrossRef] [PubMed]
- Joly, E.; Hudrisier, D. What is trogocytosis and what is its purpose? Nat. Immunol. 2003, 4, 815. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Bethune, M.T.; Wong, S.; Joglekar, A.V.; Leonard, M.T.; Wang, J.K.; Kim, J.T.; Cheng, D.; Peng, S.; Zaretsky, J.M.; et al. T cell antigen discovery via trogocytosis. Nat. Methods 2019, 16, 183–190. [Google Scholar] [CrossRef]
- Trambas, C.M.; Griffiths, G.M. Delivering the kiss of death. Nat. Immunol. 2003, 4, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Steele, S.; Radlinski, L.; Taft-Benz, S.; Brunton, J.; Kawula, T.H. Trogocytosis-associated cell to cell spread of intracellular bacterial pathogens. eLife 2016, 5, e10625. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, J.I.; Perez, F. Localized intercellular transfer of ephrin-as by trans-endocytosis enables long-term signaling. Dev. Cell 2020, 52, 104–117 e5. [Google Scholar] [CrossRef] [PubMed]
- Weinhard, L.; di Bartolomei, G.; Bolasco, G.; Machado, P.; Schieber, N.L.; Neniskyte, U.; Exiga, M.; Vadisiute, A.; Raggioli, A.; Schertel, A.; et al. Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nat. Commun. 2018, 9, 1228. [Google Scholar] [CrossRef]
- Andoh, M.; Shibata, K.; Okamoto, K.; Onodera, J.; Morishita, K.; Miura, Y.; Ikegaya, Y.; Koyama, R. Exercise reverses behavioral and synaptic abnormalities after maternal inflammation. Cell Rep. 2019, 27, 2817–2825 e5. [Google Scholar] [CrossRef] [PubMed]
- Ralston, K.S.; Solga, M.D.; Mackey-Lawrence, N.M.; Somlata; Bhattacharya, A.; Petri, W.A., Jr. Trogocytosis by Entamoeba histolytica contributes to cell killing and tissue invasion. Nature 2014, 508, 526–530. [Google Scholar] [CrossRef]
- Saito-Nakano, Y.; Wahyuni, R.; Nakada-Tsukui, K.; Tomii, K.; Nozaki, T. Rab7D small GTPase is involved in phago-, trogocytosis and cytoskeletal reorganization in the enteric protozoan Entamoeba histolytica. Cell. Microbiol. 2021, 23, e13267. [Google Scholar] [CrossRef] [PubMed]
- Bettadapur, A.; Miller, H.W.; Ralston, K.S. Biting off what can be chewed: Trogocytosis in health, infection, and disease. Infect. Immun. 2020, 88, e00930-19. [Google Scholar] [CrossRef] [PubMed]
- Thion, M.S.; Ginhoux, F.; Garel, S. Microglia and early brain development: An intimate journey. Science 2018, 362, 185–189. [Google Scholar] [CrossRef]
- Otto, G. Synaptic nibbling. Nat. Rev. Neurosci. 2018, 19, 322. [Google Scholar] [CrossRef] [PubMed]
- Li, K.J.; Wu, C.H.; Lu, C.H.; Shen, C.Y.; Kuo, Y.M.; Tsai, C.Y.; Hsieh, S.C.; Yu, C.L. Trogocytosis between non-immune cells for cell clearance, and among Immune-related cells for modulating immune responses and autoimmunity. Int. J. Mol. Sci. 2021, 22, 2236. [Google Scholar] [CrossRef]
- Karasuyama, H.; Miyake, K.; Yoshikawa, S.; Kawano, Y.; Yamanishi, Y. How do basophils contribute to Th2 cell differentiation and allergic responses? Int. Immunol. 2018, 30, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Martin, N.; Fernandez-Arenas, E.; Cemerski, S.; Delgado, P.; Turner, M.; Heuser, J.; Irvine, D.J.; Huang, B.; Bustelo, X.R.; Shaw, A.; et al. T cell receptor internalization from the immunological synapse is mediated by TC21 and RhoG GTPase-dependent phagocytosis. Immunity 2011, 35, 208–222. [Google Scholar] [CrossRef] [PubMed]
- Goodridge, H.S.; Underhill, D.M.; Touret, N. Mechanisms of Fc receptor and dectin-1 activation for phagocytosis. Traffic 2012, 13, 1062–1071. [Google Scholar] [CrossRef]
- Aucher, A.; Magdeleine, E.; Joly, E.; Hudrisier, D. Capture of plasma membrane fragments from target cells by trogocytosis requires signaling in T cells but not in B cells. Blood 2008, 111, 5621–5628. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Nakayama, M.; Kawano, M.; Amagai, R.; Ishii, T.; Harigae, H.; Ogasawara, K. Fratricide of natural killer cells dressed with tumor-derived NKG2D ligand. Proc. Natl. Acad. Sci. USA 2013, 110, 9421–9426. [Google Scholar] [CrossRef]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Thery, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Choudhuri, K.; Llodra, J.; Roth, E.W.; Tsai, J.; Gordo, S.; Wucherpfennig, K.W.; Kam, L.C.; Stokes, D.L.; Dustin, M.L. Polarized release of T-cell-receptor-enriched microvesicles at the immunological synapse. Nature 2014, 507, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Vietri, M.; Radulovic, M.; Stenmark, H. The many functions of ESCRTs. Nat. Rev. Mol. Cell Biol. 2020, 21, 25–42. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.R.; Mun, Y.; Lee, K.S.; Park, Y.J.; Park, J.S.; Park, J.H.; Jeon, B.N.; Kim, C.H.; Jun, Y.; Hyun, Y.M.; et al. T cell microvilli constitute immunological synaptosomes that carry messages to antigen-presenting cells. Nat. Commun. 2018, 9, 3630. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.R.; Jun, C.D. T cell microvilli: Sensors or senders? Front. Immunol. 2019, 10, 1753. [Google Scholar] [CrossRef]
- Balint, S.; Muller, S.; Fischer, R.; Kessler, B.M.; Harkiolaki, M.; Valitutti, S.; Dustin, M.L. Supramolecular attack particles are autonomous killing entities released from cytotoxic T cells. Science 2020, 368, 897–901. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yu, J.; Kadungure, T.; Beyene, J.; Zhang, H.; Lu, Q. ARMMs as a versatile platform for intracellular delivery of macromolecules. Nat. Commun. 2018, 9, 960. [Google Scholar] [CrossRef] [PubMed]
- Nabhan, J.F.; Hu, R.; Oh, R.S.; Cohen, S.N.; Lu, Q. Formation and release of arrestin domain-containing protein 1-mediated microvesicles (ARMMs) at plasma membrane by recruitment of TSG101 protein. Proc. Natl. Acad. Sci. USA 2012, 109, 4146–4151. [Google Scholar] [CrossRef] [PubMed]
- Norris, R.P. Transfer of mitochondria and endosomes between cells by gap junction internalization. Traffic 2021. Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Dopfer, E.P.; Minguet, S.; Schamel, W.W. A new vampire saga: The molecular mechanism of T cell trogocytosis. Immunity 2011, 35, 151–153. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.S.; Collinson, L.M.; Brzostek, J.; Eissmann, P.; Almeida, C.R.; McCann, F.E.; Burshtyn, D.; Davis, D.M. Membranous structures transfer cell surface proteins across NK cell immune synapses. Traffic 2007, 8, 1190–1204. [Google Scholar] [CrossRef] [PubMed]
- Hudrisier, D.; Clemenceau, B.; Balor, S.; Daubeuf, S.; Magdeleine, E.; Daeron, M.; Bruhns, P.; Vie, H. Ligand binding but undetected functional response of FcR after their capture by T cells via trogocytosis. J. Immunol. 2009, 183, 6102–6113. [Google Scholar] [CrossRef]
- Patel, D.M.; Dudek, R.W.; Mannie, M.D. Intercellular exchange of class II MHC complexes: Ultrastructural localization and functional presentation of adsorbed I-A/peptide complexes. Cell. Immunol. 2001, 214, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Chaudhri, G.; Quah, B.J.; Wang, Y.; Tan, A.H.; Zhou, J.; Karupiah, G.; Parish, C.R. T cell receptor sharing by cytotoxic T lymphocytes facilitates efficient virus control. Proc. Natl. Acad. Sci. USA 2009, 106, 14984–14989. [Google Scholar] [CrossRef]
- Somanchi, S.S.; Somanchi, A.; Cooper, L.J.; Lee, D.A. Engineering lymph node homing of ex vivo-expanded human natural killer cells via trogocytosis of the chemokine receptor CCR7. Blood 2012, 119, 5164–5172. [Google Scholar] [CrossRef]
- Palucka, K.; Banchereau, J. Cancer immunotherapy via dendritic cells. Nat. Rev. Cancer 2012, 12, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Cruz, F.M.; Colbert, J.D.; Merino, E.; Kriegsman, B.A.; Rock, K.L. The biology and underlying mechanisms of cross-presentation of exogenous antigens on MHC-I molecules. Annu. Rev. Immunol. 2017, 35, 149–176. [Google Scholar] [CrossRef] [PubMed]
- Blander, J.M. Regulation of the cell biology of antigen cross-presentation. Annu. Rev. Immunol. 2018, 36, 717–753. [Google Scholar] [CrossRef]
- Cabeza-Cabrerizo, M.; Cardoso, A.; Minutti, C.M.; Pereira da Costa, M.; Reis, E.S.C. Dendritic cells revisited. Annu. Rev. Immunol. 2021, 39, 131–166. [Google Scholar] [CrossRef] [PubMed]
- Veglia, F.; Gabrilovich, D.I. Dendritic cells in cancer: The role revisited. Curr. Opin. Immunol. 2017, 45, 43–51. [Google Scholar] [CrossRef]
- Reizis, B. Plasmacytoid dendritic cells: Development, regulation, and function. Immunity 2019, 50, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Joffre, O.P.; Segura, E.; Savina, A.; Amigorena, S. Cross-presentation by dendritic cells. Nat. Rev. Immunol. 2012, 12, 557–569. [Google Scholar] [CrossRef] [PubMed]
- Colbert, J.D.; Cruz, F.M.; Rock, K.L. Cross-presentation of exogenous antigens on MHC I molecules. Curr. Opin. Immunol. 2020, 64, 1–8. [Google Scholar] [CrossRef]
- Sancho, D.; Joffre, O.P.; Keller, A.M.; Rogers, N.C.; Martinez, D.; Hernanz-Falcon, P.; Rosewell, I.; Reis e Sousa, C. Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature 2009, 458, 899–903. [Google Scholar] [CrossRef] [PubMed]
- Ahrens, S.; Zelenay, S.; Sancho, D.; Hanc, P.; Kjaer, S.; Feest, C.; Fletcher, G.; Durkin, C.; Postigo, A.; Skehel, M.; et al. F-actin is an evolutionarily conserved damage-associated molecular pattern recognized by DNGR-1, a receptor for dead cells. Immunity 2012, 36, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Canton, J.; Blees, H.; Henry, C.M.; Buck, M.D.; Schulz, O.; Rogers, N.C.; Childs, E.; Zelenay, S.; Rhys, H.; Domart, M.C.; et al. The receptor DNGR-1 signals for phagosomal rupture to promote cross-presentation of dead-cell-associated antigens. Nat. Immunol. 2021, 22, 140–153. [Google Scholar] [CrossRef] [PubMed]
- Pitt, J.M.; Charrier, M.; Viaud, S.; Andre, F.; Besse, B.; Chaput, N.; Zitvogel, L. Dendritic cell-derived exosomes as immunotherapies in the fight against cancer. J. Immunol. 2014, 193, 1006–1011. [Google Scholar] [CrossRef]
- Campana, S.; De Pasquale, C.; Carrega, P.; Ferlazzo, G.; Bonaccorsi, I. Cross-dressing: An alternative mechanism for antigen presentation. Immunol. Lett. 2015, 168, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Dolan, B.P.; Gibbs, K.D., Jr.; Ostrand-Rosenberg, S. Dendritic cells cross-dressed with peptide MHC class I complexes prime CD8+ T cells. J. Immunol. 2006, 177, 6018–6024. [Google Scholar] [CrossRef]
- Wolfers, J.; Lozier, A.; Raposo, G.; Regnault, A.; Thery, C.; Masurier, C.; Flament, C.; Pouzieux, S.; Faure, F.; Tursz, T.; et al. Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat. Med. 2001, 7, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Andre, F.; Schartz, N.E.; Movassagh, M.; Flament, C.; Pautier, P.; Morice, P.; Pomel, C.; Lhomme, C.; Escudier, B.; Le Chevalier, T.; et al. Malignant effusions and immunogenic tumour-derived exosomes. Lancet 2002, 360, 295–305. [Google Scholar] [CrossRef]
- Helft, J.; Bottcher, J.; Chakravarty, P.; Zelenay, S.; Huotari, J.; Schraml, B.U.; Goubau, D.; Reis e Sousa, C. GM-CSF mouse bone marrow cultures comprise a heterogeneous population of CD11c+ MHCII+ macrophages and dendritic cells. Immunity 2015, 42, 1197–1211. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Unutmaz, D.; Wong, P.; Sano, G.; De los Santos, K.; Sparwasser, T.; Wu, S.; Vuthoori, S.; Ko, K.; Zavala, F.; et al. In vivo depletion of CD11c+ dendritic cells abrogates priming of CD8+ T cells by exogenous cell-associated antigens. Immunity 2002, 17, 211–220. [Google Scholar] [CrossRef]
- Zhang, Q.J.; Li, X.L.; Wang, D.; Huang, X.C.; Mathis, J.M.; Duan, W.M.; Knight, D.; Shi, R.; Glass, J.; Zhang, D.Q.; et al. Trogocytosis of MHC-I/peptide complexes derived from tumors and infected cells enhances dendritic cell cross-priming and promotes adaptive T cell responses. PLoS ONE 2008, 3, e3097. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, P.K.; Bollrath, J.; Pallangyo, C.K.; Matsutani, T.; Canli, O.; De Oliveira, T.; Diamanti, M.A.; Muller, N.; Gamrekelashvili, J.; Putoczki, T.; et al. Mitophagy in intestinal epithelial cells triggers adaptive immunity during tumorigenesis. Cell 2018, 174, 88–101.e16. [Google Scholar] [CrossRef] [PubMed]
- Das Mohapatra, A.; Tirrell, I.; Benechet, A.P.; Pattnayak, S.; Khanna, K.M.; Srivastava, P.K. Cross-dressing of CD8α+ dendritic cells with antigens from live mouse tumor cells is a major mechanism of cross-priming. Cancer Immunol. Res. 2020, 8, 1287–1299. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Kim, S.; Herndon, J.M.; Goedegebuure, P.; Belt, B.A.; Satpathy, A.T.; Fleming, T.P.; Hansen, T.H.; Murphy, K.M.; Gillanders, W.E. Cross-dressed CD8α+/CD103+ dendritic cells prime CD8+ T cells following vaccination. Proc. Natl. Acad. Sci. USA 2012, 109, 12716–12721. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Peng, P.; Loschko, J.; Feng, L.; Pham, P.; Cui, W.; Lee, K.P.; Krug, A.B.; Jiang, A. Plasmacytoid dendritic cells cross-prime naive CD8 T cells by transferring antigen to conventional dendritic cells through exosomes. Proc. Natl. Acad. Sci. USA 2020, 117, 23730–23741. [Google Scholar] [CrossRef]
- Smyth, L.A.; Harker, N.; Turnbull, W.; El-Doueik, H.; Klavinskis, L.; Kioussis, D.; Lombardi, G.; Lechler, R. The relative efficiency of acquisition of MHC:peptide complexes and cross-presentation depends on dendritic cell type. J. Immunol. 2008, 181, 3212–3220. [Google Scholar] [CrossRef]
- Wakim, L.M.; Bevan, M.J. Cross-dressed dendritic cells drive memory CD8+ T-cell activation after viral infection. Nature 2011, 471, 629–632. [Google Scholar] [CrossRef] [PubMed]
- Smyth, L.A.; Hervouet, C.; Hayday, T.; Becker, P.D.; Ellis, R.; Lechler, R.I.; Lombardi, G.; Klavinskis, L.S. Acquisition of MHC:peptide complexes by dendritic cells contributes to the generation of antiviral CD8+ T cell immunity in vivo. J. Immunol. 2012, 189, 2274–2282. [Google Scholar] [CrossRef]
- Bonaccorsi, I.; Morandi, B.; Antsiferova, O.; Costa, G.; Oliveri, D.; Conte, R.; Pezzino, G.; Vermiglio, G.; Anastasi, G.P.; Navarra, G.; et al. Membrane transfer from tumor cells overcomes deficient phagocytic ability of plasmacytoid dendritic cells for the acquisition and presentation of tumor antigens. J. Immunol. 2014, 192, 824–832. [Google Scholar] [CrossRef]
- Huang, J.F.; Yang, Y.; Sepulveda, H.; Shi, W.; Hwang, I.; Peterson, P.A.; Jackson, M.R.; Sprent, J.; Cai, Z. TCR-mediated internalization of peptide-MHC complexes acquired by T cells. Science 1999, 286, 952–954. [Google Scholar] [CrossRef] [PubMed]
- Daubeuf, S.; Puaux, A.L.; Joly, E.; Hudrisier, D. A simple trogocytosis-based method to detect, quantify, characterize and purify antigen-specific live lymphocytes by flow cytometry, via their capture of membrane fragments from antigen-presenting cells. Nat. Protoc. 2006, 1, 2536–2542. [Google Scholar] [CrossRef] [PubMed]
- Machlenkin, A.; Uzana, R.; Frankenburg, S.; Eisenberg, G.; Eisenbach, L.; Pitcovski, J.; Gorodetsky, R.; Nissan, A.; Peretz, T.; Lotem, M. Capture of tumor cell membranes by trogocytosis facilitates detection and isolation of tumor-specific functional CTLs. Cancer Res. 2008, 68, 2006–2013. [Google Scholar] [CrossRef] [PubMed]
- Wetzel, S.A.; McKeithan, T.W.; Parker, D.C. Peptide-specific intercellular transfer of MHC class II to CD4+ T cells directly from the immunological synapse upon cellular dissociation. J. Immunol. 2005, 174, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Osborne, D.G.; Wetzel, S.A. Trogocytosis results in sustained intracellular signaling in CD4+ T cells. J. Immunol. 2012, 189, 4728–4739. [Google Scholar] [CrossRef]
- Reed, J.; Wetzel, S.A. Trogocytosis-mediated intracellular signaling in CD4+ T cells drives TH2-associated effector cytokine production and differentiation. J. Immunol. 2019, 202, 2873–2887. [Google Scholar] [CrossRef] [PubMed]
- Stinchcombe, J.C.; Bossi, G.; Booth, S.; Griffiths, G.M. The immunological synapse of CTL contains a secretory domain and membrane bridges. Immunity 2001, 15, 751–761. [Google Scholar] [CrossRef]
- Hamieh, M.; Dobrin, A.; Cabriolu, A.; van der Stegen, S.J.C.; Giavridis, T.; Mansilla-Soto, J.; Eyquem, J.; Zhao, Z.; Whitlock, B.M.; Miele, M.M.; et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature 2019, 568, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Chung, B.; Stuge, T.B.; Murad, J.P.; Beilhack, G.; Andersen, E.; Armstrong, B.D.; Weber, J.S.; Lee, P.P. Antigen-specific inhibition of high-avidity T cell target lysis by low-avidity T cells via trogocytosis. Cell Rep. 2014, 8, 871–882. [Google Scholar] [CrossRef]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [PubMed]
- Salter, A.I.; Pont, M.J.; Riddell, S.R. Chimeric antigen receptor-modified T cells: CD19 and the road beyond. Blood 2018, 131, 2621–2629. [Google Scholar] [CrossRef]
- Sadelain, M.; Riviere, I.; Riddell, S. Therapeutic T cell engineering. Nature 2017, 545, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, O.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554. [Google Scholar] [CrossRef]
- Majzner, R.G.; Mackall, C.L. Tumor antigen escape from CAR T-cell therapy. Cancer Discov. 2018, 8, 1219–1226. [Google Scholar] [CrossRef]
- Lee, D.S.W.; Rojas, O.L.; Gommerman, J.L. B cell depletion therapies in autoimmune disease: Advances and mechanistic insights. Nat. Rev. Drug Discov. 2020, 20, 179–199. [Google Scholar] [CrossRef]
- Salles, G.; Barrett, M.; Foa, R.; Maurer, J.; O’Brien, S.; Valente, N.; Wenger, M.; Maloney, D.G. Rituximab in B-cell hematologic malignancies: A review of 20 years of clinical experience. Adv. Ther. 2017, 34, 2232–2273. [Google Scholar] [CrossRef] [PubMed]
- Sherman, L.A.; Chattopadhyay, S. The molecular basis of allorecognition. Ann. Rev. Immunol. 1993, 11, 385–402. [Google Scholar] [CrossRef]
- Auchincloss, H., Jr.; Lee, R.; Shea, S.; Markowitz, J.S.; Grusby, M.J.; Glimcher, L.H. The role of “indirect” recognition in initiating rejection of skin grafts from major histocompatibility complex class II-deficient mice. Proc. Natl. Acad. Sci. USA 1993, 90, 3373–3377. [Google Scholar] [CrossRef] [PubMed]
- Shoskes, D.A.; Wood, K.J. Indirect presentation of MHC antigens in transplantation. Immunol. Today 1994, 15, 32–38. [Google Scholar] [CrossRef]
- Herrera, O.B.; Golshayan, D.; Tibbott, R.; Salcido Ochoa, F.; James, M.J.; Marelli-Berg, F.M.; Lechler, R.I. A novel pathway of alloantigen presentation by dendritic cells. J. Immunol. 2004, 173, 4828–4837. [Google Scholar] [CrossRef] [PubMed]
- Smyth, L.A.; Afzali, B.; Tsang, J.; Lombardi, G.; Lechler, R.I. Intercellular transfer of MHC and immunological molecules: Molecular mechanisms and biological significance. Am. J. Transplant. 2007, 7, 1442–1449. [Google Scholar] [CrossRef] [PubMed]
- Siu, J.H.Y.; Surendrakumar, V.; Richards, J.A.; Pettigrew, G.J. T cell allorecognition pathways in solid organ transplantation. Front. Immunol. 2018, 9, 2548. [Google Scholar] [CrossRef]
- Russo, V.; Zhou, D.; Sartirana, C.; Rovere, P.; Villa, A.; Rossini, S.; Traversari, C.; Bordignon, C. Acquisition of intact allogeneic human leukocyte antigen molecules by human dendritic cells. Blood 2000, 95, 3473–3477. [Google Scholar] [CrossRef]
- Marino, J.; Babiker-Mohamed, M.H.; Crosby-Bertorini, P.; Paster, J.T.; LeGuern, C.; Germana, S.; Abdi, R.; Uehara, M.; Kim, J.I.; Markmann, J.F.; et al. Donor exosomes rather than passenger leukocytes initiate alloreactive T cell responses after transplantation. Sci. Immunol. 2016, 1, aaf8759. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Q.; Liu, Q.; Divito, S.J.; Zeng, Q.; Yatim, K.M.; Hughes, A.D.; Rojas-Canales, D.M.; Nakao, A.; Shufesky, W.J.; Williams, A.L.; et al. Graft-infiltrating host dendritic cells play a key role in organ transplant rejection. Nat. Commun. 2016, 7, 12623. [Google Scholar] [CrossRef] [PubMed]
- Hildner, K.; Edelson, B.T.; Purtha, W.E.; Diamond, M.; Matsushita, H.; Kohyama, M.; Calderon, B.; Schraml, B.U.; Unanue, E.R.; Diamond, M.S.; et al. Batf3 deficiency reveals a critical role for CD8α+ dendritic cells in cytotoxic T cell immunity. Science 2008, 322, 1097–1100. [Google Scholar] [CrossRef]
- Li, B.; Lu, C.; Oveissi, S.; Song, J.; Xiao, K.; Zanker, D.; Duan, M.; Chen, J.; Xu, H.; Zou, Q.; et al. Host CD8α+ and CD103+ dendritic cells prime transplant antigen-specific CD8+ T cells via cross-dressing. Immunol. Cell Biol. 2020, 98, 563–576. [Google Scholar] [CrossRef]
- Hughes, A.D.; Zhao, D.; Dai, H.; Abou-Daya, K.I.; Tieu, R.; Rammal, R.; Williams, A.L.; Landsittel, D.P.; Shlomchik, W.D.; Morelli, A.E.; et al. Cross-dressed dendritic cells sustain effector T cell responses in islet and kidney allografts. J. Clin. Investig. 2020, 130, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Harshyne, L.A.; Watkins, S.C.; Gambotto, A.; Barratt-Boyes, S.M. Dendritic cells acquire antigens from live cells for cross-presentation to CTL. J. Immunol. 2001, 166, 3717–3723. [Google Scholar] [CrossRef]
- Game, D.S.; Rogers, N.J.; Lechler, R.I. Acquisition of HLA-DR and costimulatory molecules by T cells from allogeneic antigen presenting cells. Am. J. Transplant. 2005, 5, 1614–1625. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Rojas-Canales, D.M.; Divito, S.J.; Shufesky, W.J.; Stolz, D.B.; Erdos, G.; Sullivan, M.L.; Gibson, G.A.; Watkins, S.C.; Larregina, A.T.; et al. Donor dendritic cell-derived exosomes promote allograft-targeting immune response. J. Clin. Investig. 2016, 126, 2805–2820. [Google Scholar] [CrossRef] [PubMed]
- Qian, S.; Demetris, A.J.; Murase, N.; Rao, A.S.; Fung, J.J.; Starzl, T.E. Murine liver allograft transplantation: Tolerance and donor cell chimerism. Hepatology 1994, 19, 916–924. [Google Scholar] [CrossRef] [PubMed]
- Londono, M.C.; Rimola, A.; O’Grady, J.; Sanchez-Fueyo, A. Immunosuppression minimization vs. complete drug withdrawal in liver transplantation. J. Hepatol. 2013, 59, 872–879. [Google Scholar] [CrossRef] [PubMed]
- Ono, Y.; Perez-Gutierrez, A.; Nakao, T.; Dai, H.; Camirand, G.; Yoshida, O.; Yokota, S.; Stolz, D.B.; Ross, M.A.; Morelli, A.E.; et al. Graft-infiltrating PD-L1hi cross-dressed dendritic cells regulate antidonor T cell responses in mouse liver transplant tolerance. Hepatology 2018, 67, 1499–1515. [Google Scholar] [CrossRef] [PubMed]
- Brandt, D.; Hedrich, C.M. TCRαβ+ CD3+ CD4- CD8- (double negative) T cells in autoimmunity. Autoimmun. Rev. 2018, 17, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.X.; Yang, L.; Young, K.J.; DuTemple, B.; Zhang, L. Identification of a previously unknown antigen-specific regulatory T cell and its mechanism of suppression. Nat. Med. 2000, 6, 782–789. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Yang, W.; Degauque, N.; Tian, Y.; Mikita, A.; Zheng, X.X. New differentiation pathway for double-negative regulatory T cells that regulates the magnitude of immune responses. Blood 2007, 109, 4071–4079. [Google Scholar] [CrossRef] [PubMed]
- Ford, M.S.; Young, K.J.; Zhang, Z.; Ohashi, P.S.; Zhang, L. The immune regulatory function of lymphoproliferative double negative T cells in vitro and in vivo. J. Exp. Med. 2002, 196, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Ford McIntyre, M.S.; Young, K.J.; Gao, J.; Joe, B.; Zhang, L. Cutting edge: In vivo trogocytosis as a mechanism of double negative regulatory T cell-mediated antigen-specific suppression. J. Immunol. 2008, 181, 2271–2275. [Google Scholar] [CrossRef] [PubMed]
- Tian, D.; Yang, L.; Wang, S.; Zhu, Y.; Shi, W.; Zhang, C.; Jin, H.; Tian, Y.; Xu, H.; Sun, G.; et al. Double negative T cells mediate Lag3-dependent antigen-specific protection in allergic asthma. Nat. Commun. 2019, 10, 4246. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, T.; Hirahara, K.; Onodera, A.; Endo, Y.; Hosokawa, H.; Shinoda, K.; Tumes, D.J.; Okamoto, Y. Th2 cells in health and disease. Annu. Rev. Immunol. 2017, 35, 53–84. [Google Scholar] [CrossRef] [PubMed]
- Tsang, J.Y.; Chai, J.G.; Lechler, R. Antigen presentation by mouse CD4+ T cells involving acquired MHC class II:peptide complexes: Another mechanism to limit clonal expansion? Blood 2003, 101, 2704–2710. [Google Scholar] [CrossRef] [PubMed]
- Helft, J.; Jacquet, A.; Joncker, N.T.; Grandjean, I.; Dorothee, G.; Kissenpfennig, A.; Malissen, B.; Matzinger, P.; Lantz, O. Antigen-specific T-T interactions regulate CD4 T-cell expansion. Blood 2008, 112, 1249–1258. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Tagaya, Y.; Tolouei-Semnani, R.; Schlom, J.; Sabzevari, H. Physiological relevance of antigen presentasome (APS), an acquired MHC/costimulatory complex, in the sustained activation of CD4+ T cells in the absence of APCs. Blood 2005, 105, 3238–3246. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.M.; Arnold, P.Y.; White, G.A.; Nardella, J.P.; Mannie, M.D. Class II MHC/peptide complexes are released from APC and are acquired by T cell responders during specific antigen recognition. J. Immunol. 1999, 163, 5201–5210. [Google Scholar] [PubMed]
- Arnold, P.Y.; Mannie, M.D. Vesicles bearing MHC class II molecules mediate transfer of antigen from antigen-presenting cells to CD4+ T cells. Eur. J. Immunol. 1999, 29, 1363–1373. [Google Scholar] [CrossRef]
- Tatari-Calderone, Z.; Semnani, R.T.; Nutman, T.B.; Schlom, J.; Sabzevari, H. Acquisition of CD80 by human T cells at early stages of activation: Functional involvement of CD80 acquisition in T cell to T cell interaction. J. Immunol. 2002, 169, 6162–6169. [Google Scholar] [CrossRef] [PubMed]
- Nolte-’t Hoen, E.N.; Buschow, S.I.; Anderton, S.M.; Stoorvogel, W.; Wauben, M.H. Activated T cells recruit exosomes secreted by dendritic cells via LFA-1. Blood 2009, 113, 1977–1981. [Google Scholar] [CrossRef] [PubMed]
- Karasuyama, H.; Miyake, K.; Yoshikawa, S.; Yamanishi, Y. Multifaceted roles of basophils in health and disease. J. Allergy Clin. Immunol. 2018, 142, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Miyake, K.; Shiozawa, N.; Nagao, T.; Yoshikawa, S.; Yamanishi, Y.; Karasuyama, H. Trogocytosis of peptide-MHC class II complexes from dendritic cells confers antigen-presenting ability on basophils. Proc. Natl. Acad. Sci. USA 2017, 114, 1111–1116. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate lymphoid cells: 10 years on. Cell 2018, 174, 1054–1066. [Google Scholar] [CrossRef]
- Oliphant, C.J.; Hwang, Y.Y.; Walker, J.A.; Salimi, M.; Wong, S.H.; Brewer, J.M.; Englezakis, A.; Barlow, J.L.; Hams, E.; Scanlon, S.T.; et al. MHCII-mediated dialog between group 2 innate lymphoid cells and CD4+ T cells potentiates type 2 immunity and promotes parasitic helminth expulsion. Immunity 2014, 41, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sanmamed, M.F.; Datar, I.; Su, T.T.; Ji, L.; Sun, J.; Chen, L.; Chen, Y.; Zhu, G.; Yin, W.; et al. Fibrinogen-like protein 1 is a major immune inhibitory ligand of LAG-3. Cell 2019, 176, 334–347 e12. [Google Scholar] [CrossRef] [PubMed]
- Akkaya, B.; Oya, Y.; Akkaya, M.; Al Souz, J.; Holstein, A.H.; Kamenyeva, O.; Kabat, J.; Matsumura, R.; Dorward, D.W.; Glass, D.D.; et al. Regulatory T cells mediate specific suppression by depleting peptide-MHC class II from dendritic cells. Nat. Immunol. 2019, 20, 218–231. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, M.; Takeda, K.; Kawano, M.; Takai, T.; Ishii, N.; Ogasawara, K. Natural killer (NK)-dendritic cell interactions generate MHC class II-dressed NK cells that regulate CD4+ T cells. Proc. Natl. Acad. Sci. USA 2011, 108, 18360–18365. [Google Scholar] [CrossRef] [PubMed]
- Dubrot, J.; Duraes, F.V.; Potin, L.; Capotosti, F.; Brighouse, D.; Suter, T.; LeibundGut-Landmann, S.; Garbi, N.; Reith, W.; Swartz, M.A.; et al. Lymph node stromal cells acquire peptide-MHCII complexes from dendritic cells and induce antigen-specific CD4+ T cell tolerance. J. Exp. Med. 2014, 211, 1153–1166. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Mikami, N.; Wing, J.B.; Tanaka, A.; Ichiyama, K.; Ohkura, N. Regulatory T cells and human disease. Annu. Rev. Immunol. 2020, 38, 541–566. [Google Scholar] [CrossRef] [PubMed]
- Raffin, C.; Vo, L.T.; Bluestone, J.A. Treg cell-based therapies: Challenges and perspectives. Nat. Rev. Immunol. 2020, 20, 158–172. [Google Scholar] [CrossRef] [PubMed]
- Wing, K.; Onishi, Y.; Prieto-Martin, P.; Yamaguchi, T.; Miyara, M.; Fehervari, Z.; Nomura, T.; Sakaguchi, S. CTLA-4 control over Foxp3+ regulatory T cell function. Science 2008, 322, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, O.S.; Zheng, Y.; Nakamura, K.; Attridge, K.; Manzotti, C.; Schmidt, E.M.; Baker, J.; Jeffery, L.E.; Kaur, S.; Briggs, Z.; et al. Trans-endocytosis of CD80 and CD86: A molecular basis for the cell-extrinsic function of CTLA-4. Science 2011, 332, 600–603. [Google Scholar] [CrossRef] [PubMed]
- Gu, P.; Gao, J.F.; D’Souza, C.A.; Kowalczyk, A.; Chou, K.Y.; Zhang, L. Trogocytosis of CD80 and CD86 by induced regulatory T cells. Cell. Mol. Immunol. 2012, 9, 136–146. [Google Scholar] [CrossRef]
- Zanin-Zhorov, A.; Ding, Y.; Kumari, S.; Attur, M.; Hippen, K.L.; Brown, M.; Blazar, B.R.; Abramson, S.B.; Lafaille, J.J.; Dustin, M.L. Protein kinase C-theta mediates negative feedback on regulatory T cell function. Science 2010, 328, 372–376. [Google Scholar] [CrossRef]
- Sims, T.N.; Soos, T.J.; Xenias, H.S.; Dubin-Thaler, B.; Hofman, J.M.; Waite, J.C.; Cameron, T.O.; Thomas, V.K.; Varma, R.; Wiggins, C.H.; et al. Opposing effects of PKCtheta and WASp on symmetry breaking and relocation of the immunological synapse. Cell 2007, 129, 773–785. [Google Scholar] [CrossRef]
- Gros, A.; Parkhurst, M.R.; Tran, E.; Pasetto, A.; Robbins, P.F.; Ilyas, S.; Prickett, T.D.; Gartner, J.J.; Crystal, J.S.; Roberts, I.M.; et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat. Med. 2016, 22, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Stronen, E.; Toebes, M.; Kelderman, S.; van Buuren, M.M.; Yang, W.; van Rooij, N.; Donia, M.; Boschen, M.L.; Lund-Johansen, F.; Olweus, J.; et al. Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science 2016, 352, 1337–1341. [Google Scholar] [CrossRef]
- Gee, M.H.; Han, A.; Lofgren, S.M.; Beausang, J.F.; Mendoza, J.L.; Birnbaum, M.E.; Bethune, M.T.; Fischer, S.; Yang, X.; Gomez-Eerland, R.; et al. Antigen identification for orphan T cell receptors expressed on tumor-infiltrating lymphocytes. Cell 2018, 172, 549–563 e16. [Google Scholar] [CrossRef]
- Tomaru, U.; Yamano, Y.; Nagai, M.; Maric, D.; Kaumaya, P.T.; Biddison, W.; Jacobson, S. Detection of virus-specific T cells and CD8+ T-cell epitopes by acquisition of peptide-HLA-GFP complexes: Analysis of T-cell phenotype and function in chronic viral infections. Nat. Med. 2003, 9, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Beadling, C.; Slifka, M.K. Quantifying viable virus-specific T cells without a priori knowledge of fine epitope specificity. Nat. Med. 2006, 12, 1208–1212. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakayama, M.; Hori, A.; Toyoura, S.; Yamaguchi, S.-I. Shaping of T Cell Functions by Trogocytosis. Cells 2021, 10, 1155. https://doi.org/10.3390/cells10051155
Nakayama M, Hori A, Toyoura S, Yamaguchi S-I. Shaping of T Cell Functions by Trogocytosis. Cells. 2021; 10(5):1155. https://doi.org/10.3390/cells10051155
Chicago/Turabian StyleNakayama, Masafumi, Arisa Hori, Saori Toyoura, and Shin-Ichiro Yamaguchi. 2021. "Shaping of T Cell Functions by Trogocytosis" Cells 10, no. 5: 1155. https://doi.org/10.3390/cells10051155
APA StyleNakayama, M., Hori, A., Toyoura, S., & Yamaguchi, S.-I. (2021). Shaping of T Cell Functions by Trogocytosis. Cells, 10(5), 1155. https://doi.org/10.3390/cells10051155