
Emerging Roles of SKP2 in Cancer Drug Resistance

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Expression of Skp2 Associated with Tumor Malignancy and Drug Resistance
3. The Molecular Mechanism of Skp2 Involved in Cancer Drug Resistance
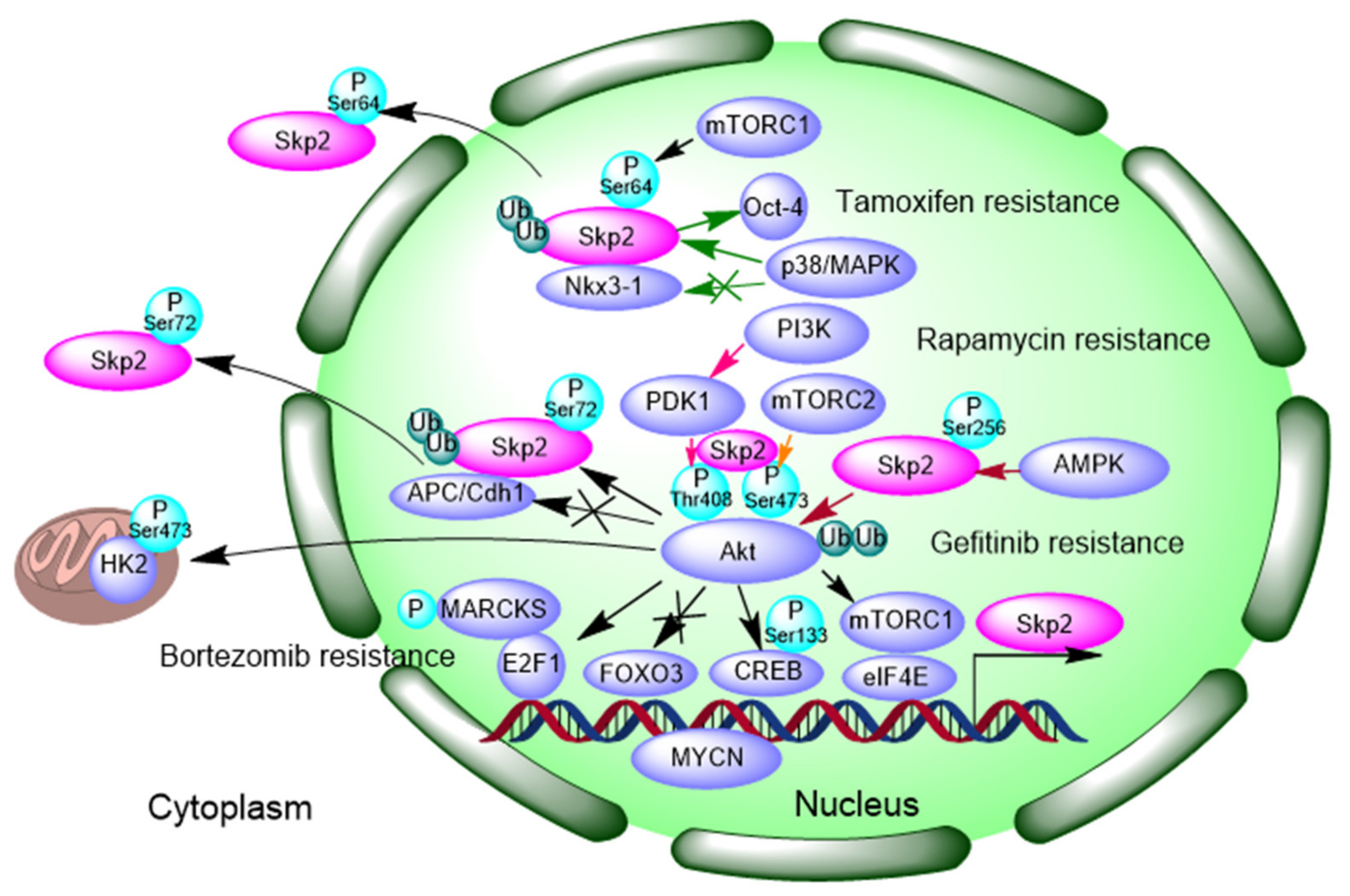
3.1. Signaling Feedback Loop between Akt-Skp2
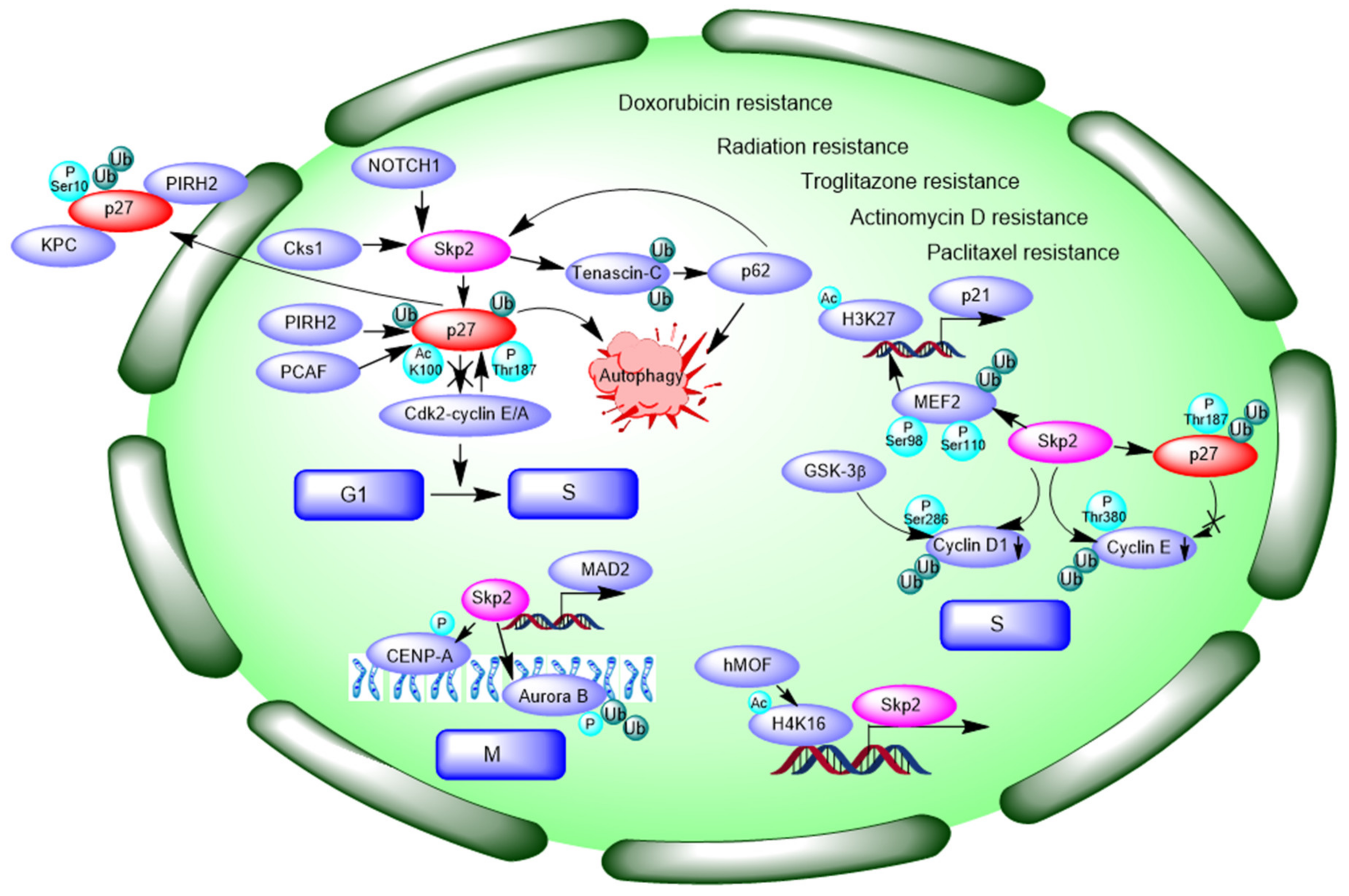
3.2. Negative Modulating Substrates p27 and Contribution to Autophagy
3.3. Interaction and Ubiquitination of Cyclin Proteins and Control of Cell Cycle
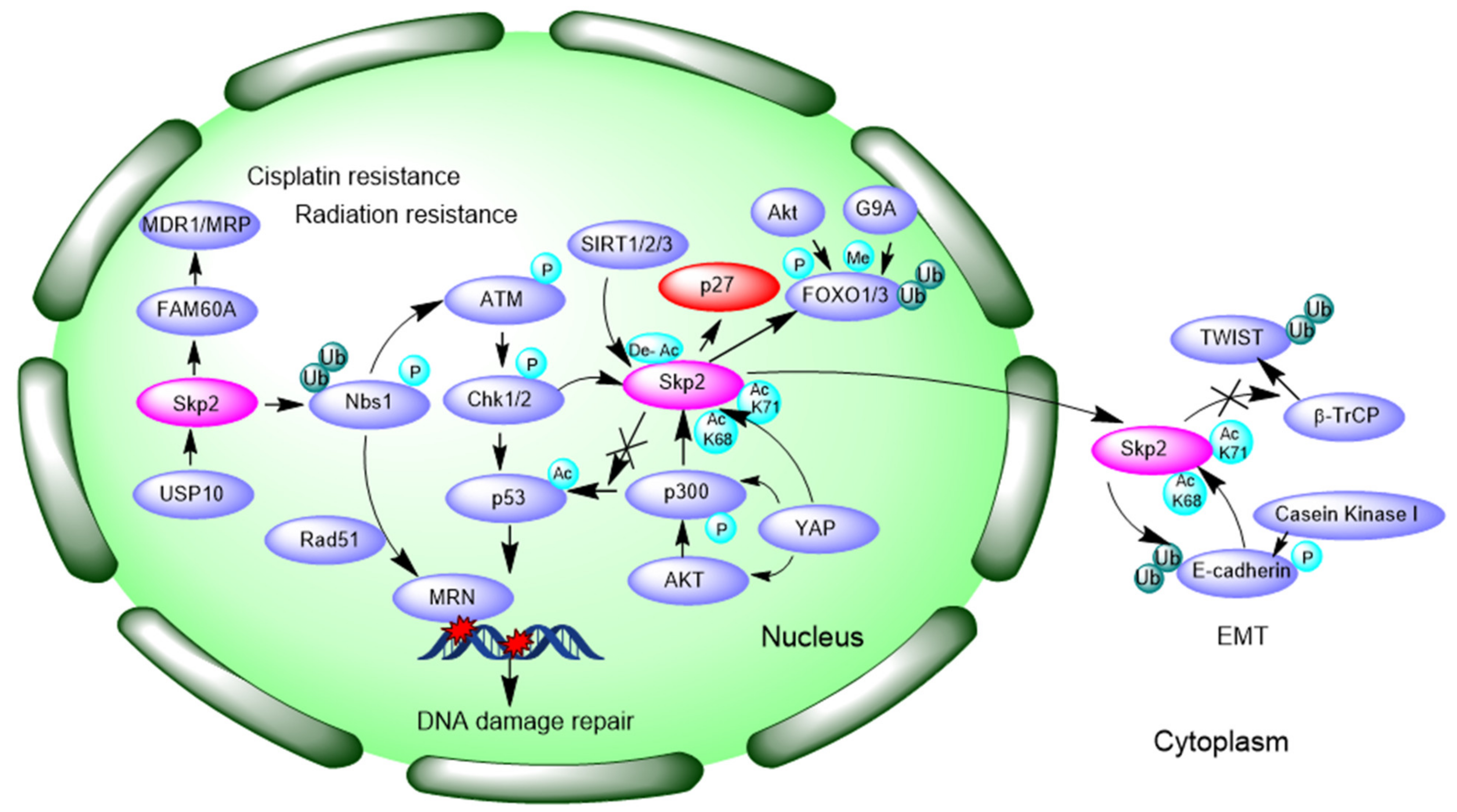
3.4. Promotion of DNA Damage Response and Repair
3.5. Inducing Epithelial–Mesenchymal Transition (EMT)
3.6. MYC Regulates Skp2 mRNA Transcription
3.7. FOXO1 and FOXO3 Interact with Skp2
3.8. DUB Enzymes Facilitate Skp2 Function
3.9. Other Mechanisms
4. Inhibitors Targeting Skp2 Overcome Resistance
4.1. Targeting Skp2 Expression
4.2. Targeting Skp2-SKP1 Interaction and SCF Formation
4.3. Targeting the Interactions between Skp2 and Cks1
4.4. Targeting the Binding Interface for p27
4.5. Targeting the Binding between Skp2 with p300
5. Conclusion and Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Buckley, A.M.; Lynam-Lennon, N.; O’Neill, H.; O’Sullivan, J. Targeting hallmarks of cancer to enhance radiosensitivity in gastrointestinal cancers. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 298–313. [Google Scholar] [CrossRef] [PubMed]
- Torres-Collado, A.X.; Knott, J.; Jazirehi, A.R. Reversal of Resistance in Targeted Therapy of Metastatic Melanoma: Lessons Learned from Vemurafenib (BRAF(V600E)-Specific Inhibitor). Cancers 2018, 10, 157. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Arnst, K.; Miller, D.D.; Li, W. Recent Advances in Elucidating Paclitaxel Resistance Mechanisms in Non-small Cell Lung Cancer and Strategies to Overcome Drug Resistance. Curr. Med. Chem. 2020, 27, 6573–6595. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.J.; Bjorklund, C.C.; Baladandayuthapani, V.; Kuhn, D.J.; Orlowski, R.Z. Drug resistance to inhibitors of the human double minute-2 E3 ligase is mediated by point mutations of p53, but can be overcome with the p53 targeting agent RITA. Mol. Cancer Ther. 2012, 11, 2243–2253. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Meng, Y.; Xu, L.; Qiu, L.; Wei, M.; Su, D.; Qi, X.; Wang, Z.; Yang, S.; Liu, C.; et al. Cul4 E3 ubiquitin ligase regulates ovarian cancer drug resistance by targeting the antiapoptotic protein BIRC3. Cell Death Dis. 2019, 10, 104. [Google Scholar] [CrossRef]
- Benanti, J.A. Coordination of cell growth and division by the ubiquitin-proteasome system. Semin. Cell Dev. Biol. 2012, 23, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Zheng, N.; Wang, Z.; Wei, W. Ubiquitination-mediated degradation of cell cycle-related proteins by F-box proteins. Int. J. Biochem. Cell Biol. 2016, 73, 99–110. [Google Scholar] [CrossRef]
- Liu, Y.; Mallampalli, R.K. Small molecule therapeutics targeting F-box proteins in cancer. Semin. Cancer Biol. 2016, 36, 105–119. [Google Scholar] [CrossRef]
- Liu, J.; Shaik, S.; Dai, X.; Wu, Q.; Zhou, X.; Wang, Z.; Wei, W. Targeting the ubiquitin pathway for cancer treatment. Biochim. Biophys. Acta 2015, 1855, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Kobayashi, R.; Galaktionov, K.; Beach, D. p19Skp1 and p45Skp2 are essential elements of the cyclin A-CDK2 S phase kinase. Cell 1995, 82, 915–925. [Google Scholar] [CrossRef]
- Demetrick, D.J.; Zhang, H.; Beach, D.H. Chromosomal mapping of the genes for the human CDK2/cyclin A-associated proteins p19 (SKP1A and SKP1B) and p45 (SKP2). Cytogenet. Cell Genet. 1996, 73, 104–107. [Google Scholar] [CrossRef]
- Hnit, S.S.T.; Xie, C.; Yao, M.; Holst, J.; Bensoussan, A.; de Souza, P.; Li, Z.; Dong, Q. p27Kip1 signaling: Transcriptional and post-translational regulation. Int. J. Biochem. Cell Biol. 2015, 68, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gao, D.; Fukushima, H.; Inuzuka, H.; Liu, P.; Wan, L.; Sarkar, F.H.; Wei, W. Skp2: A novel potential therapeutic target for prostate cancer. Biochim. Biophys. Acta 2012, 1825, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Zheng, N.; Schulman, B.A.; Song, L.; Miller, J.J.; Jeffrey, P.D.; Wang, P.; Chu, C.; Koepp, D.M.; Elledge, S.J.; Pagano, M.; et al. Structure of the Cul1-Rbx1-Skp1-F boxSkp2 SCF ubiquitin ligase complex. Nature 2002, 416, 703–709. [Google Scholar] [CrossRef]
- Hao, Z.; Huang, S. E3 ubiquitin ligase Skp2 as an attractive target in cancer therapy. Front. Biosci. 2015, 20, 474–490. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Moten, A.; Peng, D.; Hsu, C.C.; Pan, B.S.; Manne, R.; Li, H.Y.; Lin, H.K. The Skp2 Pathway: A Critical Target for Cancer Therapy. Semin. Cancer Biol. 2020, 67, 16–33. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Chan, C.H.; Gao, Y.; Lin, H.K. Novel roles of Skp2 E3 ligase in cellular senescence, cancer progression, and metastasis. Chin. J. Cancer 2012, 31, 169–177. [Google Scholar] [CrossRef]
- Chan, C.H.; Morrow, J.K.; Zhang, S.; Lin, H.K. Skp2: A dream target in the coming age of cancer therapy. Cell Cycle 2014, 13, 679–680. [Google Scholar] [CrossRef]
- Bu, W.; Luo, T. miR-1297 Promotes Cell Proliferation of Non-Small Cell Lung Cancer Cells: Involving in PTEN/Akt/Skp2 Signaling Pathway. DNA Cell Biol. 2017, 36, 976–982. [Google Scholar] [CrossRef]
- Su, L.; Han, D.; Wu, J.; Huo, X. Skp2 regulates non-small cell lung cancer cell growth by Meg3 and miR-3163. Tumour Biol. 2016, 37, 3925–3931. [Google Scholar] [CrossRef]
- Arbini, A.A.; Greco, M.; Yao, J.L.; Bourne, P.; Marra, E.; Hsieh, J.T.; di Sant’agnese, P.A.; Moro, L. Skp2 overexpression is associated with loss of BRCA2 protein in human prostate cancer. Am. J. Pathol. 2011, 178, 2367–2376. [Google Scholar] [CrossRef]
- Šimečková, Š.; Kahounová, Z.; Fedr, R.; Remšík, J.; Slabáková, E.; Suchánková, T.; Procházková, J.; Bouchal, J.; Kharaishvili, G.; Král, M.; et al. High Skp2 expression is associated with a mesenchymal phenotype and increased tumorigenic potential of prostate cancer cells. Sci. Rep. 2019, 9, 5695. [Google Scholar] [CrossRef]
- Su, J.; Zhou, X.; Wang, L.; Yin, X.; Wang, Z. Curcumin inhibits cell growth and invasion and induces apoptosis through down-regulation of Skp2 in pancreatic cancer cells. Am. J. Cancer Res. 2016, 6, 1949–1962. [Google Scholar]
- Yang, Y.; Yan, W.; Liu, Z.; Wei, M. Skp2 inhibitor SKPin C1 decreased viability and proliferation of multiple myeloma cells and induced apoptosis. Braz. J. Med. Biol. Res. 2019, 52, e8412. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Wang, K.; Yang, K. Inhibiting the role of Skp2 suppresses cell proliferation and tumorigenesis of human gastric cancer cells via the upregulation of p27kip1. Mol. Med. Rep. 2016, 14, 3917–3924. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Jiang, X.; Liu, F.; Qiao, H.; Zhou, B.; Zhai, B.; Zhang, L.; Zhang, X.; Han, L.; Jiang, H.; et al. Downregulation of Skp2 inhibits the growth and metastasis of gastric cancer cells in vitro and in vivo. Tumour Biol. 2013, 34, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Woenckhaus, C.; Maile, S.; Uffmann, S.; Bansemir, M.; Dittberner, T.; Poetsch, M.; Giebel, J. Expression of Skp2 and p27KIP1 in naevi and malignant melanoma of the skin and its relation to clinical outcome. Histol. Histopathol. 2005, 20, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Murphy, M.; Ross, J.; Sheehan, C.; Carlson, J.A. Skp2 and p27kip1 expression in melanocytic nevi and melanoma: An inverse relationship. J. Cutan. Pathol. 2004, 31, 633–642. [Google Scholar] [CrossRef]
- Zhao, H.; Pan, H.; Wang, H.; Chai, P.; Ge, S.; Jia, R.; Fan, X. SKP2 targeted inhibition suppresses human uveal melanoma progression by blocking ubiquitylation of p27. Onco. Targets Ther. 2019, 12, 4297–4308. [Google Scholar] [CrossRef]
- Latres, E.; Chiarle, R.; Schulman, B.A.; Pavletich, N.P.; Pellicer, A.; Inghirami, G.; Pagano, M. Role of the F-box protein Skp2 in lymphomagenesis. Proc. Natl. Acad. Sci. USA 2001, 98, 2515–2520. [Google Scholar] [CrossRef]
- Yu, W.N.; Nogueira, V.; Sobhakumari, A.; Patra, K.C.; Bhaskar, P.T.; Hay, N. Systemic Akt1 Deletion after Tumor Onset in p53(-/-) Mice Increases Lifespan and Regresses Thymic Lymphoma Emulating p53 Restoration. Cell Rep. 2015, 12, 610–621. [Google Scholar] [CrossRef][Green Version]
- Yan, W.; Yang, Y.; Yang, W. Inhibition of SKP2 Activity Impaired ATM-Mediated DNA Repair and Enhanced Sensitivity of Cisplatin-Resistant Mantle Cell Lymphoma Cells. Cancer Biother. Radiopharm. 2019, 34, 451–458. [Google Scholar] [CrossRef]
- Wang, J.; Huang, Y.; Guan, Z.; Zhang, J.L.; Su, H.K.; Zhang, W.; Yue, C.F.; Yan, M.; Guan, S.; Liu, Q.Q. E3-ligase Skp2 predicts poor prognosis and maintains cancer stem cell pool in nasopharyngeal carcinoma. Oncotarget 2014, 5, 5591–5601. [Google Scholar] [CrossRef]
- Fang, F.M.; Chien, C.Y.; Li, C.F.; Shiu, W.Y.; Chen, C.H.; Huang, H.Y. Effect of S-phase kinase-associated protein 2 expression on distant metastasis and survival in nasopharyngeal carcinoma patients. Int. J. Radiat. Oncol. Biol. Phys. 2009, 73, 202–207. [Google Scholar] [CrossRef]
- Zhang, Y.; Zvi, Y.S.; Batko, B.; Zaphiros, N.; O’Donnell, E.F.; Wang, J.; Sato, K.; Yang, R.; Geller, D.S.; Koirala, P.; et al. Down-regulation of Skp2 expression inhibits invasion and lung metastasis in osteosarcoma. Sci. Rep. 2018, 8, 14294. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Li, R.; Han, X.; Zhou, Y.; Zhang, H.; Cui, Y.; Wang, W.; Bai, J. Inhibition of Skp2 suppresses the proliferation and invasion of osteosarcoma cells. Oncol. Rep. 2017, 38, 933–940. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Nan, H.; Ma, J.; Jiang, L.; Guo, Q.; Han, L.; Zhang, Y.; Nan, K.; Guo, H. High Skp2/Low p57(Kip2) Expression is Associated with Poor Prognosis in Human Breast Carcinoma. Breast Cancer 2015, 9, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Cao, L.; Sun, Z.; Xu, J.; Tang, L.; Chen, W.; Luo, J.; Yang, F.; Wang, Y.; Guan, X. Skp2 is over-expressed in breast cancer and promotes breast cancer cell proliferation. Cell Cycle 2016, 15, 1344–1351. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.F.; Chen, T.J.; Lin, C.Y.; Chen, L.T.; Lin, L.C.; Hsing, C.H.; Lee, S.W.; Sheu, M.J.; Lee, H.H.; Shiue, Y.L.; et al. SKP2 overexpression is associated with a poor prognosis of rectal cancer treated with chemoradiotherapy and represents a therapeutic target with high potential. Tumour Biol. 2013, 34, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Shintani, S.; Li, C.; Mihara, M.; Hino, S.; Nakashiro, K.; Hamakawa, H. Skp2 and Jab1 expression are associated with inverse expression of p27(KIP1) and poor prognosis in oral squamous cell carcinomas. Oncology 2003, 65, 355–362. [Google Scholar] [CrossRef]
- Yokoi, S.; Yasui, K.; Mori, M.; Iizasa, T.; Fujisawa, T.; Inazawa, J. Amplification and overexpression of SKP2 are associated with metastasis of non-small-cell lung cancers to lymph nodes. Am. J. Pathol. 2004, 165, 175–180. [Google Scholar] [CrossRef][Green Version]
- Yang, Q.; Huang, J.; Wu, Q.; Cai, Y.; Zhu, L.; Lu, X.; Chen, S.; Chen, C.; Wang, Z. Acquisition of epithelial-mesenchymal transition is associated with Skp2 expression in paclitaxel-resistant breast cancer cells. Br. J. Cancer 2014, 110, 1958–1967. [Google Scholar] [CrossRef]
- Yang, Y.; Lu, Y.; Wang, L.; Mizokami, A.; Keller, E.T.; Zhang, J.; Fu, J. Skp2 is associated with paclitaxel resistance in prostate cancer cells. Oncol. Rep. 2016, 36, 559–566. [Google Scholar] [CrossRef]
- Huang, T.; Yang, L.; Wang, G.; Ding, G.; Peng, B.; Wen, Y.; Wang, Z. Inhibition of Skp2 sensitizes lung cancer cells to paclitaxel. Onco. Targets Ther. 2017, 10, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Byun, W.S.; Jin, M.; Yu, J.; Kim, W.K.; Song, J.; Chung, H.J.; Jeong, L.S.; Lee, S.K. A novel selenonucleoside suppresses tumor growth by targeting Skp2 degradation in paclitaxel-resistant prostate cancer. Biochem. Pharmacol. 2018, 158, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Wang, R.; Zhang, Y.; Zhou, L.; Wang, W.; Liu, H.; Li, W. Skp2-mediated ubiquitination and mitochondrial localization of Akt drive tumor growth and chemoresistance to cisplatin. Oncogene 2019, 38, 7457–7472. [Google Scholar] [CrossRef] [PubMed]
- Hou, Q.; Jiang, Z.; Li, Y.; Wu, H.; Yu, J.; Jiang, M. FAM60A promotes cisplatin resistance in lung cancer cells by activating SKP2 expression. Anticancer. Drugs 2020, 31, 776–784. [Google Scholar] [CrossRef]
- Zeng, L.; Nikolaev, A.; Xing, C.; Della Manna, D.L.; Yang, E.S. CHK1/2 Inhibitor Prexasertib Suppresses NOTCH Signaling and Enhances Cytotoxicity of Cisplatin and Radiation in Head and Neck Squamous Cell Carcinoma. Mol. Cancer Ther. 2020, 19, 1279–1288. [Google Scholar] [CrossRef] [PubMed]
- Davidovich, S.; Ben-Izhak, O.; Shapira, M.; Futerman, B.; Hershko, D.D. Over-expression of Skp2 is associated with resistance to preoperative doxorubicin-based chemotherapy in primary breast cancer. Breast Cancer Res. 2008, 10, R63. [Google Scholar] [CrossRef]
- Bhatt, S.; Stender, J.D.; Joshi, S.; Wu, G.; Katzenellenbogen, B.S. OCT-4: A novel estrogen receptor-α collaborator that promotes tamoxifen resistance in breast cancer cells. Oncogene 2016, 35, 5722–5734. [Google Scholar] [CrossRef]
- Lin, Y.S.; Lin, Y.Y.; Yang, Y.H.; Lin, C.L.; Kuan, F.C.; Lu, C.N.; Chang, G.H.; Tsai, M.S.; Hsu, C.M.; Yeh, R.A.; et al. Antrodia cinnamomea extract inhibits the proliferation of tamoxifen-resistant breast cancer cells through apoptosis and skp2/microRNAs pathway. BMC Complement. Altern. Med. 2018, 18, 152. [Google Scholar] [CrossRef]
- Malek, E.; Abdel-Malek, M.A.; Jagannathan, S.; Vad, N.; Karns, R.; Jegga, A.G.; Broyl, A.; van Duin, M.; Sonneveld, P.; Cottini, F.; et al. Pharmacogenomics and chemical library screens reveal a novel SCF(SKP2) inhibitor that overcomes Bortezomib resistance in multiple myeloma. Leukemia 2017, 31, 645–653. [Google Scholar] [CrossRef]
- Liu, X.; Wang, H.; Ma, J.; Xu, J.; Sheng, C.; Yang, S.; Sun, L.; Ni, Q. The expression and prognosis of Emi1 and Skp2 in breast carcinoma: Associated with PI3K/Akt pathway and cell proliferation. Med. Oncol. 2013, 30, 735. [Google Scholar] [CrossRef]
- Jia, T.; Zhang, L.; Duan, Y.; Zhang, M.; Wang, G.; Zhang, J.; Zhao, Z. The differential susceptibilities of MCF-7 and MDA-MB-231 cells to the cytotoxic effects of curcumin are associated with the PI3K/Akt-SKP2-Cip/Kips pathway. Cancer Cell Int. 2014, 14, 126. [Google Scholar] [CrossRef] [PubMed]
- Clement, E.; Inuzuka, H.; Nihira, N.T.; Wei, W.; Toker, A. Skp2-dependent reactivation of AKT drives resistance to PI3K inhibitors. Sci. Signal 2018, 11. [Google Scholar] [CrossRef]
- Tian, Y.F.; Wang, H.C.; Luo, C.W.; Hung, W.C.; Lin, Y.H.; Chen, T.Y.; Li, C.F.; Lin, C.Y.; Pan, M.R. Preprogramming therapeutic response of PI3K/mTOR dual inhibitor via the regulation of EHMT2 and p27 in pancreatic cancer. Am. J. Cancer Res. 2018, 8, 1812–1822. [Google Scholar] [PubMed]
- Han, F.; Li, C.F.; Cai, Z.; Zhang, X.; Jin, G.; Zhang, W.N.; Xu, C.; Wang, C.Y.; Morrow, J.; Zhang, S.; et al. The critical role of AMPK in driving Akt activation under stress, tumorigenesis and drug resistance. Nat. Commun. 2018, 9, 4728. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zi, X.; Pollak, M. Molecular mechanisms underlying IGF-I-induced attenuation of the growth-inhibitory activity of trastuzumab (Herceptin) on SKBR3 breast cancer cells. Int. J. Cancer 2004, 108, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Ling, Y.H.; Li, T.; Yuan, Z.; Haigentz, M.; Weber, T.K., Jr.; Perez-Soler, R. Erlotinib, an effective epidermal growth factor receptor tyrosine kinase inhibitor, induces p27KIP1 up-regulation and nuclear translocation in association with cell growth inhibition and G1/S phase arrest in human non-small-cell lung cancer cell lines. Mol. Pharmacol. 2007, 72, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Perdreau, S.A.; Chatterjee, P.; Wang, L.; Kuan, S.F.; Duensing, A. Imatinib mesylate induces quiescence in gastrointestinal stromal tumor cells through the CDH1-SKP2-p27Kip1 signaling axis. Cancer Res. 2008, 68, 9015–9023. [Google Scholar] [CrossRef]
- Liao, Y.; Liu, N.; Xia, X.; Guo, Z.; Li, Y.; Jiang, L.; Zhou, R.; Tang, D.; Huang, H.; Liu, J. USP10 modulates the SKP2/Bcr-Abl axis via stabilizing SKP2 in chronic myeloid leukemia. Cell Discov. 2019, 5, 24. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Huang, Z.; Wu, W.; Xia, R. Inhibition of Skp2 Sensitizes Chronic Myeloid Leukemia Cells to Imatinib. Cancer Manag. Res. 2020, 12, 4777–4787. [Google Scholar] [CrossRef]
- Titus, A.S.; Kailasam, S. Coordinated regulation of cell survival and cell cycle pathways by DDR2-dependent SRF transcription factor in cardiac fibroblasts. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H1538–H1558. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Wang, C.; Cui, Y.; Han, X.; Zhou, Y.; Bai, J.; Li, R. S-phase kinase-associated protein 2 is involved in epithelial-mesenchymal transition in methotrexate-resistant osteosarcoma cells. Int. J. Oncol. 2018, 52, 1841–1852. [Google Scholar] [CrossRef] [PubMed]
- Panaccione, A.; Chang, M.T.; Carbone, B.E.; Guo, Y.; Moskaluk, C.A.; Virk, R.K.; Chiriboga, L.; Prasad, M.L.; Judson, B.; Mehra, S.; et al. NOTCH1 and SOX10 are Essential for Proliferation and Radiation Resistance of Cancer Stem-Like Cells in Adenoid Cystic Carcinoma. Clin. Cancer Res. 2016, 22, 2083–2095. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.C.; Tian, L.L.; Tian, J.; Jiang, X.Y. Overexpression of SKP2 promotes the radiation resistance of esophageal squamous cell carcinoma. Radiat. Res. 2012, 177, 52–58. [Google Scholar] [CrossRef]
- Wang, X.C.; Tian, L.L.; Tian, J.; Li, D.; Wang, Y.; Wu, H.; Zheng, H.; Meng, A.M. Overexpression of Cks1 increases the radiotherapy resistance of esophageal squamous cell carcinoma. J. Radiat. Res. 2012, 53, 72–78. [Google Scholar] [CrossRef]
- Uehara, N.; Yoshizawa, K.; Tsubura, A. Vorinostat enhances protein stability of p27 and p21 through negative regulation of Skp2 and Cks1 in human breast cancer cells. Oncol. Rep. 2012, 28, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Fenner, A. Prostate cancer: Twist and Skp2 castration resistance. Nat. Rev. Urol. 2017, 14, 387. [Google Scholar] [CrossRef] [PubMed]
- Totary-Jain, H.; Sanoudou, D.; Dautriche, C.N.; Schneller, H.; Zambrana, L.; Marks, A.R. Rapamycin resistance is linked to defective regulation of Skp2. Cancer Res. 2012, 72, 1836–1843. [Google Scholar] [CrossRef]
- Reichert, M.; Saur, D.; Hamacher, R.; Schmid, R.M.; Schneider, G. Phosphoinositide-3-kinase signaling controls S-phase kinase-associated protein 2 transcription via E2F1 in pancreatic ductal adenocarcinoma cells. Cancer Res. 2007, 67, 4149–4156. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Lee, S.W.; Zhang, X.; Han, F.; Kwan, S.Y.; Yuan, X.; Yang, W.L.; Jeong, Y.S.; Rezaeian, A.H.; Gao, Y.; et al. Foxo3a transcription factor is a negative regulator of Skp2 and Skp2 SCF complex. Oncogene 2013, 32, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, V.; Sundararajan, D.; Kwan, J.M.; Peng, X.D.; Sarvepalli, N.; Sonenberg, N.; Hay, N. Akt-dependent Skp2 mRNA translation is required for exiting contact inhibition, oncogenesis, and adipogenesis. Embo J. 2012, 31, 1134–1146. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Chen, Y.; Saha, M.N.; Chen, J.; Evans, K.; Qiu, L.; Reece, D.; Chen, G.A.; Chang, H. Targeting phospho-MARCKS overcomes drug-resistance and induces antitumor activity in preclinical models of multiple myeloma. Leukemia 2015, 29, 715–726. [Google Scholar] [CrossRef]
- Iskandarani, A.; Bhat, A.A.; Siveen, K.S.; Prabhu, K.S.; Kuttikrishnan, S.; Khan, M.A.; Krishnankutty, R.; Kulinski, M.; Nasr, R.R.; Mohammad, R.M.; et al. Bortezomib-mediated downregulation of S-phase kinase protein-2 (SKP2) causes apoptotic cell death in chronic myelogenous leukemia cells. J. Transl. Med. 2016, 14, 69. [Google Scholar] [CrossRef]
- Gao, D.; Inuzuka, H.; Tseng, A.; Chin, R.Y.; Toker, A.; Wei, W. Phosphorylation by Akt1 promotes cytoplasmic localization of Skp2 and impairs APCCdh1-mediated Skp2 destruction. Nat. Cell Biol. 2009, 11, 397–408. [Google Scholar] [CrossRef]
- Geng, Q.; Liu, J.; Gong, Z.; Chen, S.; Chen, S.; Li, X.; Lu, Y.; Zhu, X.; Lin, H.K.; Xu, D. Phosphorylation by mTORC1 stablizes Skp2 and regulates its oncogenic function in gastric cancer. Mol. Cancer 2017, 16, 83. [Google Scholar] [CrossRef] [PubMed]
- Shintani, S.; Li, C.; Mihara, M.; Yano, J.; Terakado, N.; Nakashiro, K.; Hamakawa, H. Gefitinib (‘Iressa’, ZD1839), an epidermal growth factor receptor tyrosine kinase inhibitor, up-regulates p27KIP1 and induces G1 arrest in oral squamous cell carcinoma cell lines. Oral Oncol. 2004, 40, 43–51. [Google Scholar] [CrossRef]
- Chan, C.H.; Li, C.F.; Yang, W.L.; Gao, Y.; Lee, S.W.; Feng, Z.; Huang, H.Y.; Tsai, K.K.; Flores, L.G.; Shao, Y.; et al. The Skp2-SCF E3 ligase regulates Akt ubiquitination, glycolysis, herceptin sensitivity, and tumorigenesis. Cell 2012, 149, 1098–1111. [Google Scholar] [CrossRef]
- Slingerland, J.; Pagano, M. Regulation of the cdk inhibitor p27 and its deregulation in cancer. J. Cell Physiol. 2000, 183, 10–17. [Google Scholar] [CrossRef]
- Frescas, D.; Pagano, M. Deregulated proteolysis by the F-box proteins SKP2 and beta-TrCP: Tipping the scales of cancer. Nat. Rev. Cancer 2008, 8, 438–449. [Google Scholar] [CrossRef]
- Timmerbeul, I.; Garrett-Engele, C.M.; Kossatz, U.; Chen, X.; Firpo, E.; Grünwald, V.; Kamino, K.; Wilkens, L.; Lehmann, U.; Buer, J.; et al. Testing the importance of p27 degradation by the SCFskp2 pathway in murine models of lung and colon cancer. Proc. Natl. Acad. Sci. USA 2006, 103, 14009–14014. [Google Scholar] [CrossRef]
- Hill, S.; Reichermeier, K.; Scott, D.C.; Samentar, L.; Coulombe-Huntington, J.; Izzi, L.; Tang, X.; Ibarra, R.; Bertomeu, T.; Moradian, A.; et al. Robust cullin-RING ligase function is established by a multiplicity of poly-ubiquitylation pathways. Elife 2019, 8. [Google Scholar] [CrossRef]
- Koga, H.; Harada, M.; Ohtsubo, M.; Shishido, S.; Kumemura, H.; Hanada, S.; Taniguchi, E.; Yamashita, K.; Kumashiro, R.; Ueno, T.; et al. Troglitazone induces p27Kip1-associated cell-cycle arrest through down-regulating Skp2 in human hepatoma cells. Hepatology 2003, 37, 1086–1096. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.A.; Inoue, H.; Sonoda, H.; Mine, S.; Yoshikawa, Y.; Nakayama, K.; Nakayama, K.; Mori, M. Clinical and biological significance of S-phase kinase-associated protein 2 (Skp2) gene expression in gastric carcinoma: Modulation of malignant phenotype by Skp2 overexpression, possibly via p27 proteolysis. Cancer Res. 2002, 62, 3819–3825. [Google Scholar] [PubMed]
- Hao, B.; Zheng, N.; Schulman, B.A.; Wu, G.; Miller, J.J.; Pagano, M.; Pavletich, N.P. Structural basis of the Cks1-dependent recognition of p27(Kip1) by the SCF(Skp2) ubiquitin ligase. Mol. Cell 2005, 20, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Cai, L.; Yu, Y.; Meng, Q.; Cheng, X.; Zhao, Y.; Sui, G.; Zhang, F. Knockdown of S-phase kinase-associated protein-2 expression in MCF-7 inhibits cell growth and enhances the cytotoxic effects of epirubicin. Acta Biochim. Biophys. Sin. 2007, 39, 999–1007. [Google Scholar] [CrossRef][Green Version]
- Chen, Q.; Xie, W.; Kuhn, D.J.; Voorhees, P.M.; Lopez-Girona, A.; Mendy, D.; Corral, L.G.; Krenitsky, V.P.; Xu, W.; Moutouh-de Parseval, L.; et al. Targeting the p27 E3 ligase SCF(Skp2) results in p27- and Skp2-mediated cell-cycle arrest and activation of autophagy. Blood 2008, 111, 4690–4699. [Google Scholar] [CrossRef]
- Li, Z.L.; Zhang, H.L.; Huang, Y.; Huang, J.H.; Sun, P.; Zhou, N.N.; Chen, Y.H.; Mai, J.; Wang, Y.; Yu, Y.; et al. Autophagy deficiency promotes triple-negative breast cancer resistance to T cell-mediated cytotoxicity by blocking tenascin-C degradation. Nat. Commun. 2020, 11, 3806. [Google Scholar] [CrossRef]
- Jung, D.; Khurana, A.; Roy, D.; Kalogera, E.; Bakkum-Gamez, J.; Chien, J.; Shridhar, V. Quinacrine upregulates p21/p27 independent of p53 through autophagy-mediated downregulation of p62-Skp2 axis in ovarian cancer. Sci. Rep. 2018, 8, 2487. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Han, F.; Wu, J.; Lee, S.W.; Chan, C.H.; Wu, C.Y.; Yang, W.L.; Gao, Y.; Zhang, X.; Jeong, Y.S.; et al. The role of Skp2 in hematopoietic stem cell quiescence, pool size, and self-renewal. Blood 2011, 118, 5429–5438. [Google Scholar] [CrossRef] [PubMed]
- Schüler, S.; Diersch, S.; Hamacher, R.; Schmid, R.M.; Saur, D.; Schneider, G. SKP2 confers resistance of pancreatic cancer cells towards TRAIL-induced apoptosis. Int. J. Oncol. 2011, 38, 219–225. [Google Scholar] [CrossRef]
- Harada, K.; Supriatno; Kawashima, Y.; Itashiki, Y.; Yoshida, H.; Sato, M. Down-regulation of S-phase kinase associated protein 2 (Skp2) induces apoptosis in oral cancer cells. Oral Oncol. 2005, 41, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Luna, M.; Aguasca, M.; Perearnau, A.; Serratosa, J.; Martínez-Balbas, M.; Jesús Pujol, M.; Bachs, O. PCAF regulates the stability of the transcriptional regulator and cyclin-dependent kinase inhibitor p27 Kip1. Nucleic Acids Res. 2012, 40, 6520–6533. [Google Scholar] [CrossRef] [PubMed]
- Yeh, K.H.; Kondo, T.; Zheng, J.; Tsvetkov, L.M.; Blair, J.; Zhang, H. The F-box protein SKP2 binds to the phosphorylated threonine 380 in cyclin E and regulates ubiquitin-dependent degradation of cyclin E. Biochem. Biophys. Res. Commun. 2001, 281, 884–890. [Google Scholar] [CrossRef]
- Alao, J.P.; Lam, E.W.; Ali, S.; Buluwela, L.; Bordogna, W.; Lockey, P.; Varshochi, R.; Stavropoulou, A.V.; Coombes, R.C.; Vigushin, D.M. Histone deacetylase inhibitor trichostatin A represses estrogen receptor alpha-dependent transcription and promotes proteasomal degradation of cyclin D1 in human breast carcinoma cell lines. Clin. Cancer Res. 2004, 10, 8094–8104. [Google Scholar] [CrossRef]
- Wu, J.; Huang, Y.F.; Zhou, X.K.; Zhang, W.; Lian, Y.F.; Lv, X.B.; Gao, X.R.; Lin, H.K.; Zeng, Y.X.; Huang, J.Q. Skp2 is required for Aurora B activation in cell mitosis and spindle checkpoint. Cell Cycle 2015, 14, 3877–3884. [Google Scholar] [CrossRef][Green Version]
- Zhao, L.; Wang, D.L.; Liu, Y.; Chen, S.; Sun, F.L. Histone acetyltransferase hMOF promotes S phase entry and tumorigenesis in lung cancer. Cell Signal 2013, 25, 1689–1698. [Google Scholar] [CrossRef]
- Di Giorgio, E.; Gagliostro, E.; Clocchiatti, A.; Brancolini, C. The control operated by the cell cycle machinery on MEF2 stability contributes to the downregulation of CDKN1A and entry into S phase. Mol. Cell Biol. 2015, 35, 1633–1647. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhang, X.; Zhang, L.; Wu, C.Y.; Rezaeian, A.H.; Chan, C.H.; Li, J.M.; Wang, J.; Gao, Y.; Han, F.; et al. Skp2 E3 ligase integrates ATM activation and homologous recombination repair by ubiquitinating NBS1. Mol. Cell 2012, 46, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Inuzuka, H.; Zhong, J.; Liu, P.; Sarkar, F.H.; Sun, Y.; Wei, W. Identification of acetylation-dependent regulatory mechanisms that govern the oncogenic functions of Skp2. Oncotarget 2012, 3, 1294–1300. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, M.; Lee, S.H.; McCormick, F. Skp2 suppresses p53-dependent apoptosis by inhibiting p300. Mol. Cell 2008, 29, 217–231. [Google Scholar] [CrossRef]
- Zhang, S.; Chen, Q.; Liu, Q.; Li, Y.; Sun, X.; Hong, L.; Ji, S.; Liu, C.; Geng, J.; Zhang, W.; et al. Hippo Signaling Suppresses Cell Ploidy and Tumorigenesis through Skp2. Cancer Cell 2017, 31, 669–684. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Huang, J.; Yuan, H.; Chen, Z.; Luo, Q.; Lu, S. SIRT2 inhibits non-small cell lung cancer cell growth through impairing Skp2-mediated p27 degradation. Oncotarget 2016, 7, 18927–18939. [Google Scholar] [CrossRef]
- De Craene, B.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef]
- Kajiyama, H.; Shibata, K.; Terauchi, M.; Yamashita, M.; Ino, K.; Nawa, A.; Kikkawa, F. Chemoresistance to paclitaxel induces epithelial-mesenchymal transition and enhances metastatic potential for epithelial ovarian carcinoma cells. Int. J. Oncol. 2007, 31, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Shen, L.; Zheng, Y.; Cui, Y.; Feng, Z.; Liu, F.; Liu, J. A signal transduction pathway from TGF-β1 to SKP2 via Akt1 and c-Myc and its correlation with progression in human melanoma. J. Invest. Dermatol. 2014, 134, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Inuzuka, H.; Gao, D.; Finley, L.W.; Yang, W.; Wan, L.; Fukushima, H.; Chin, Y.R.; Zhai, B.; Shaik, S.; Lau, A.W.; et al. Acetylation-dependent regulation of Skp2 function. Cell 2012, 150, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Ruan, D.; He, J.; Li, C.F.; Lee, H.J.; Liu, J.; Lin, H.K.; Chan, C.H. Skp2 deficiency restricts the progression and stem cell features of castration-resistant prostate cancer by destabilizing Twist. Oncogene 2017, 36, 4299–4310. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kim, W.; Ko, C.; Ryu, W.S. Hepatitis B virus X protein enhances Myc stability by inhibiting SCF(Skp2) ubiquitin E3 ligase-mediated Myc ubiquitination and contributes to oncogenesis. Oncogene 2016, 35, 1857–1867. [Google Scholar] [CrossRef]
- Feng, L.; Li, J.; Bu, X.; Zuo, Y.; Shen, L.; Qu, X. BRAF(V600E) dictates cell survival via c-Myc-dependent induction of Skp2 in human melanoma. Biochem. Biophys. Res. Commun. 2020, 524, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Evans, L.; Chen, L.; Milazzo, G.; Gherardi, S.; Perini, G.; Willmore, E.; Newell, D.R.; Tweddle, D.A. SKP2 is a direct transcriptional target of MYCN and a potential therapeutic target in neuroblastoma. Cancer Lett. 2015, 363, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Regan, K.M.; Wang, F.; Wang, D.; Smith, D.I.; van Deursen, J.M.; Tindall, D.J. Skp2 inhibits FOXO1 in tumor suppression through ubiquitin-mediated degradation. Proc. Natl. Acad. Sci. USA 2005, 102, 1649–1654. [Google Scholar] [CrossRef]
- Huang, H.; Tindall, D.J. Regulation of FOXO protein stability via ubiquitination and proteasome degradation. Biochim. Biophys. Acta 2011, 1813, 1961–1964. [Google Scholar] [CrossRef]
- Chae, Y.C.; Kim, J.Y.; Park, J.W.; Kim, K.B.; Oh, H.; Lee, K.H.; Seo, S.B. FOXO1 degradation via G9a-mediated methylation promotes cell proliferation in colon cancer. Nucleic Acids Res. 2019, 47, 1692–1705. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Chan, C.H.; Chen, K.; Guan, X.; Lin, H.K.; Tong, Q. Deacetylation of FOXO3 by SIRT1 or SIRT2 leads to Skp2-mediated FOXO3 ubiquitination and degradation. Oncogene 2012, 31, 1546–1557. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Zhou, G.; Wang, X.; Chen, W.; Gao, H. USP18 promotes breast cancer growth by upregulating EGFR and activating the AKT/Skp2 pathway. Int. J. Oncol. 2018, 53, 371–383. [Google Scholar] [CrossRef]
- Ren, H.; Zhang, Y.; Zhu, H. MiR-339 depresses cell proliferation via directly targeting S-phase kinase-associated protein 2 mRNA in lung cancer. Thorac. Cancer 2018, 9, 408–414. [Google Scholar] [CrossRef]
- Lee, Y.; Lim, H.S. Skp2 Inhibitors: Novel Anticancer Strategies. Curr. Med. Chem. 2016, 23, 2363–2379. [Google Scholar] [CrossRef]
- Li, X.; Pham, V.; Tippin, M.; Fu, D.; Rendon, R.; Song, L.; Uchio, E.; Hoang, B.H.; Zi, X. Flavokawain B targets protein neddylation for enhancing the anti-prostate cancer effect of Bortezomib via Skp2 degradation. Cell Commun. Signal 2019, 17, 25. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yokoyama, N.N.; Zhang, S.; Ding, L.; Liu, H.M.; Lilly, M.B.; Mercola, D.; Zi, X. Flavokawain A induces deNEDDylation and Skp2 degradation leading to inhibition of tumorigenesis and cancer progression in the TRAMP transgenic mouse model. Oncotarget 2015, 6, 41809–41824. [Google Scholar] [CrossRef]
- Pham, V.; Rendon, R.; Le, V.X.; Tippin, M.; Fu, D.J.; Le, T.H.; Miller, M.; Agredano, E.; Cedano, J.; Zi, X. Gartanin is a novel NEDDylation inhibitor for induction of Skp2 degradation, FBXW2 expression, and autophagy. Mol. Carcinog. 2020, 59, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Dow, R.; Hendley, J.; Pirkmaier, A.; Musgrove, E.A.; Germain, D. Retinoic acid-mediated growth arrest requires ubiquitylation and degradation of the F-box protein Skp2. J. Biol. Chem. 2001, 276, 45945–45951. [Google Scholar] [CrossRef] [PubMed]
- Rico-Bautista, E.; Yang, C.C.; Lu, L.; Roth, G.P.; Wolf, D.A. Chemical genetics approach to restoring p27Kip1 reveals novel compounds with antiproliferative activity in prostate cancer cells. BMC Biol. 2010, 8, 153. [Google Scholar] [CrossRef]
- Chan, C.H.; Morrow, J.K.; Li, C.F.; Gao, Y.; Jin, G.; Moten, A.; Stagg, L.J.; Ladbury, J.E.; Cai, Z.; Xu, D.; et al. Pharmacological inactivation of Skp2 SCF ubiquitin ligase restricts cancer stem cell traits and cancer progression. Cell 2013, 154, 556–568. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhang, F.; Jiang, L.; Hu, G.; Sun, W.; Zhang, C.; Ding, X. Inhibition of SKP2 Sensitizes Bromocriptine-Induced Apoptosis in Human Prolactinoma Cells. Cancer Res. Treat. 2017, 49, 358–373. [Google Scholar] [CrossRef]
- Driscoll, J.J.; Malek, E. Pharmacogenomics of Bortezomib in Multiple Myeloma Patients Reveals that the Ubiquitin Ligase SCF-Skp2 Promotes Drug Resistance. Clin. Lymphoma Myeloma Leuk. 2015, 15, S54. [Google Scholar] [CrossRef]
- Ungermannova, D.; Lee, J.; Zhang, G.; Dallmann, H.G.; McHenry, C.S.; Liu, X. High-throughput screening AlphaScreen assay for identification of small-molecule inhibitors of ubiquitin E3 ligase SCFSkp2-Cks1. J. Biomol. Screen 2013, 18, 910–920. [Google Scholar] [CrossRef] [PubMed]
- Ooi, L.C.; Watanabe, N.; Futamura, Y.; Sulaiman, S.F.; Darah, I.; Osada, H. Identification of small molecule inhibitors of p27(Kip1) ubiquitination by high-throughput screening. Cancer Sci. 2013, 104, 1461–1467. [Google Scholar] [CrossRef]
- Wu, L.; Grigoryan, A.V.; Li, Y.; Hao, B.; Pagano, M.; Cardozo, T.J. Specific small molecule inhibitors of Skp2-mediated p27 degradation. Chem. Biol. 2012, 19, 1515–1524. [Google Scholar] [CrossRef]
- Yang, E.S.; Burnstein, K.L. Vitamin D inhibits G1 to S progression in LNCaP prostate cancer cells through p27Kip1 stabilization and Cdk2 mislocalization to the cytoplasm. J. Biol. Chem. 2003, 278, 46862–46868. [Google Scholar] [CrossRef] [PubMed]
- Oh, M.; Lee, J.H.; Moon, H.; Hyun, Y.J.; Lim, H.S. A Chemical Inhibitor of the Skp2/p300 Interaction that Promotes p53-Mediated Apoptosis. Angew Chem. Int. Ed. Engl. 2016, 55, 602–606. [Google Scholar] [CrossRef]
- Huang, H.C.; Lin, C.L.; Lin, J.K. 1,2,3,4,6-penta-O-galloyl-β-D-glucose, quercetin, curcumin and lycopene induce cell-cycle arrest in MDA-MB-231 and BT474 cells through downregulation of Skp2 protein. J. Agric. Food Chem. 2011, 59, 6765–6775. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.C.; Way, T.D.; Lin, C.L.; Lin, J.K. EGCG stabilizes p27kip1 in E2-stimulated MCF-7 cells through down-regulation of the Skp2 protein. Endocrinology 2008, 149, 5972–5983. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Zhang, X.; Tang, J.; Kasiappan, R.; Jinwal, U.; Li, P.; Hann, S.; Nicosia, S.V.; Wu, J.; Zhang, X.; et al. The coupling of epidermal growth factor receptor down regulation by 1alpha,25-dihydroxyvitamin D3 to the hormone-induced cell cycle arrest at the G1-S checkpoint in ovarian cancer cells. Mol. Cell Endocrinol. 2011, 338, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Yu, X.; Li, M.; Gong, G.; Liu, W.; Li, T.; Zuo, H.; Li, W.; Gao, F.; Liu, H. Cdh1-mediated Skp2 degradation by dioscin reprogrammes aerobic glycolysis and inhibits colorectal cancer cells growth. EBioMedicine 2020, 51, 102570. [Google Scholar] [CrossRef]
- Jandial, D.D.; Krill, L.S.; Chen, L.; Wu, C.; Ke, Y.; Xie, J.; Hoang, B.H.; Zi, X. Induction of G2M Arrest by Flavokawain A, a Kava Chalcone, Increases the Responsiveness of HER2-Overexpressing Breast Cancer Cells to Herceptin. Molecules 2017, 22, 462. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Simoneau, A.R.; Xie, J.; Shahandeh, B.; Zi, X. Effects of the kava chalcone flavokawain A differ in bladder cancer cells with wild-type versus mutant p53. Cancer Prev. Res. 2008, 1, 439–451. [Google Scholar] [CrossRef]
- Wang, J.; Sato, K.; O’Donnell, E.; Singla, A.; Yaguare, S.; Aldahamsheh, O.; Batko, B.; Borjihan, H.; Tingling, J.; Zhang, J.; et al. Skp2 depletion reduces tumor-initiating properties and promotes apoptosis in synovial sarcoma. Transl. Oncol. 2020, 13, 100809. [Google Scholar] [CrossRef]
- Sun, X.; Gao, H.; Yang, Y.; He, M.; Wu, Y.; Song, Y.; Tong, Y.; Rao, Y. PROTACs: Great opportunities for academia and industry. Signal Transduct. Target Ther. 2019, 4, 64. [Google Scholar] [CrossRef]
- Fulcher, L.J.; Macartney, T.; Bozatzi, P.; Hornberger, A.; Rojas-Fernandez, A.; Sapkota, G.P. An affinity-directed protein missile system for targeted proteolysis. Open Biol. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Khoo, R.; Peh, K.M.; Teo, J.; Chang, S.C.; Ng, S.; Beilhartz, G.L.; Melnyk, R.A.; Johannes, C.W.; Brown, C.J.; et al. bioPROTACs as versatile modulators of intracellular therapeutic targets including proliferating cell nuclear antigen (PCNA). Proc. Natl. Acad. Sci. USA 2020, 117, 5791–5800. [Google Scholar] [CrossRef] [PubMed]
- Mayor-Ruiz, C.; Bauer, S.; Brand, M.; Kozicka, Z.; Siklos, M.; Imrichova, H.; Kaltheuner, I.H.; Hahn, E.; Seiler, K.; Koren, A.; et al. Rational discovery of molecular glue degraders via scalable chemical profiling. Nat. Chem. Biol. 2020, 16, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Slabicki, M.; Kozicka, Z.; Petzold, G.; Li, Y.D.; Manojkumar, M.; Bunker, R.D.; Donovan, K.A.; Sievers, Q.L.; Koeppel, J.; Suchyta, D.; et al. The CDK inhibitor CR8 acts as a molecular glue degrader that depletes cyclin K. Nature 2020, 585, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.K.; Bihani, T. Selective estrogen receptor modulators (SERMs) and selective estrogen receptor degraders (SERDs) in cancer treatment. Pharmacol. Ther. 2018, 186, 1–24. [Google Scholar] [CrossRef]
- Nepali, K.; Liou, J.P. Recent developments in epigenetic cancer therapeutics: Clinical advancement and emerging trends. J. Biomed. Sci. 2021, 28, 27. [Google Scholar] [CrossRef]
- Lu, G.; Middleton, R.E.; Sun, H.; Naniong, M.; Ott, C.J.; Mitsiades, C.S.; Wong, K.K.; Bradner, J.E.; Kaelin, W.G., Jr. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 2014, 343, 305–309. [Google Scholar] [CrossRef]
- Krönke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.; et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 2014, 343, 301–305. [Google Scholar] [CrossRef]
- Sievers, Q.L.; Petzold, G.; Bunker, R.D.; Renneville, A.; Słabicki, M.; Liddicoat, B.J.; Abdulrahman, W.; Mikkelsen, T.; Ebert, B.L.; Thomä, N.H. Defining the human C2H2 zinc finger degrome targeted by thalidomide analogs through CRBN. Science 2018, 362. [Google Scholar] [CrossRef]
- Gandhi, A.K.; Kang, J.; Havens, C.G.; Conklin, T.; Ning, Y.; Wu, L.; Ito, T.; Ando, H.; Waldman, M.F.; Thakurta, A.; et al. Immunomodulatory agents lenalidomide and pomalidomide co-stimulate T cells by inducing degradation of T cell repressors Ikaros and Aiolos via modulation of the E3 ubiquitin ligase complex CRL4(CRBN.). Br. J. Haematol. 2014, 164, 811–821. [Google Scholar] [CrossRef]
- Yao, S.; He, Z.; Chen, C. CRISPR/Cas9-Mediated Genome Editing of Epigenetic Factors for Cancer Therapy. Hum. Gene Ther. 2015, 26, 463–471. [Google Scholar] [CrossRef]
- Thakore, P.I.; D’Ippolito, A.M.; Song, L.; Safi, A.; Shivakumar, N.K.; Kabadi, A.M.; Reddy, T.E.; Crawford, G.E.; Gersbach, C.A. Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat. Methods 2015, 12, 1143–1149. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.K.; Hatanaka, F.; Araoka, T.; Reddy, P.; Wu, M.Z.; Sui, Y.; Yamauchi, T.; Sakurai, M.; O’Keefe, D.D.; Núñez-Delicado, E.; et al. In Vivo Target Gene Activation via CRISPR/Cas9-Mediated Trans-epigenetic Modulation. Cell 2017, 171, 1495–1507.e15. [Google Scholar] [CrossRef] [PubMed]
- Chavez, A.; Scheiman, J.; Vora, S.; Pruitt, B.W.; Tuttle, M.; E, P.R.I.; Lin, S.; Kiani, S.; Guzman, C.D.; Wiegand, D.J.; et al. Highly efficient Cas9-mediated transcriptional programming. Nat. Methods 2015, 12, 326–328. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, T.; Gu, X.; Cui, H. Emerging Roles of SKP2 in Cancer Drug Resistance. Cells 2021, 10, 1147. https://doi.org/10.3390/cells10051147
Wu T, Gu X, Cui H. Emerging Roles of SKP2 in Cancer Drug Resistance. Cells. 2021; 10(5):1147. https://doi.org/10.3390/cells10051147
Chicago/Turabian StyleWu, Ting, Xinsheng Gu, and Hongmei Cui. 2021. "Emerging Roles of SKP2 in Cancer Drug Resistance" Cells 10, no. 5: 1147. https://doi.org/10.3390/cells10051147
APA StyleWu, T., Gu, X., & Cui, H. (2021). Emerging Roles of SKP2 in Cancer Drug Resistance. Cells, 10(5), 1147. https://doi.org/10.3390/cells10051147