
Moderately Inducing Autophagy Reduces Tertiary Brain Injury after Perinatal Hypoxia-Ischemia

 , , and
, , and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Rodents
2.2. Neonatal Hypoxic-Ischemic Brain Injury
2.3. SB505124 Drug Delivery
2.4. Tat-Beclin1 Administration
2.5. Organotypic Slice Culture
2.6. Sensorimotor Tests
2.7. Western Blot Analyses
2.8. Brain Histology and Immunofluorescence
2.9. Data Analyses and Statistics
3. Results
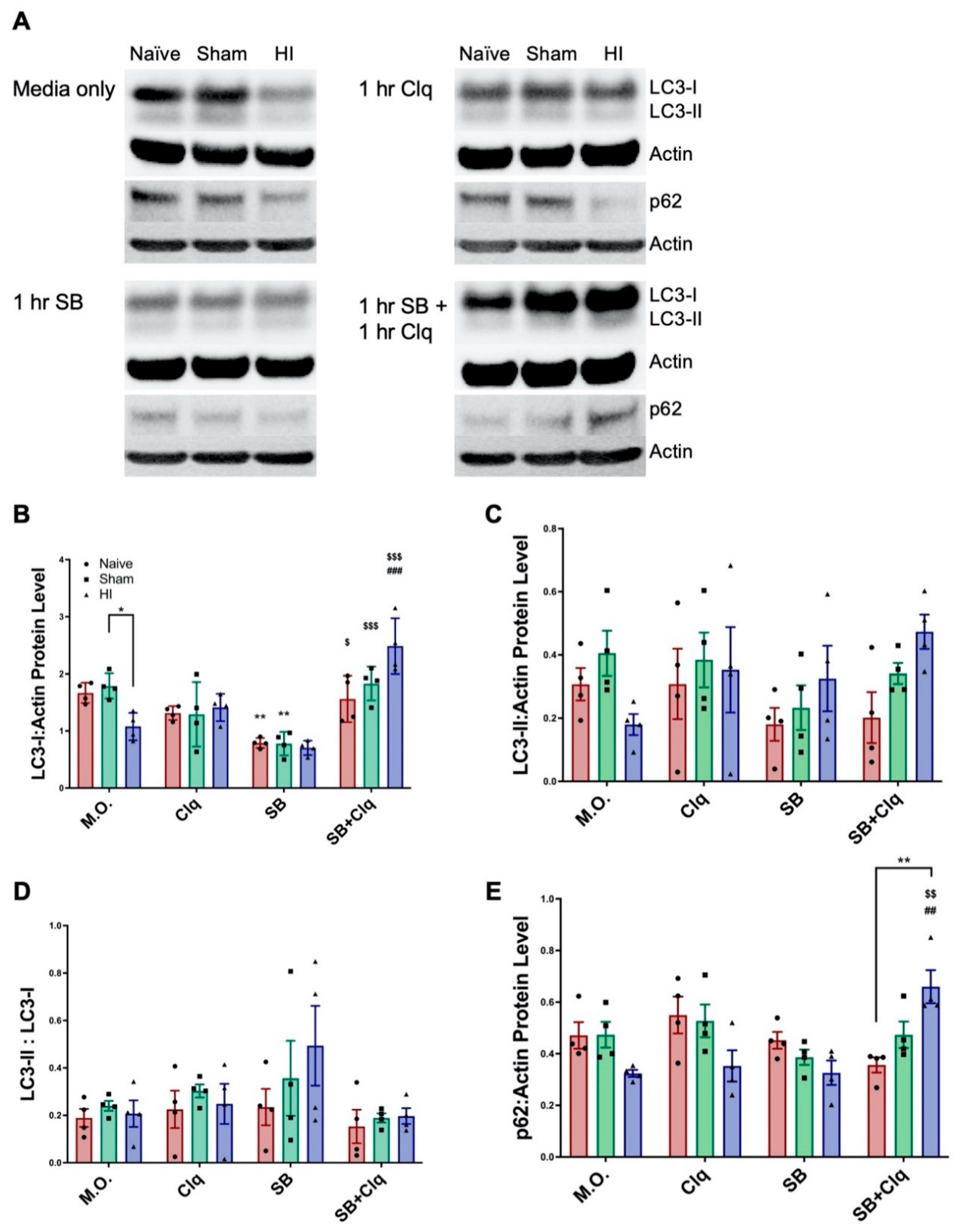
3.1. Delayed SB505124 Reduces the Number of Apoptotic Cells in the Neocortex after HI Injury
3.2. Delayed SB505124 Induces Autophagic Flux after HI Injury
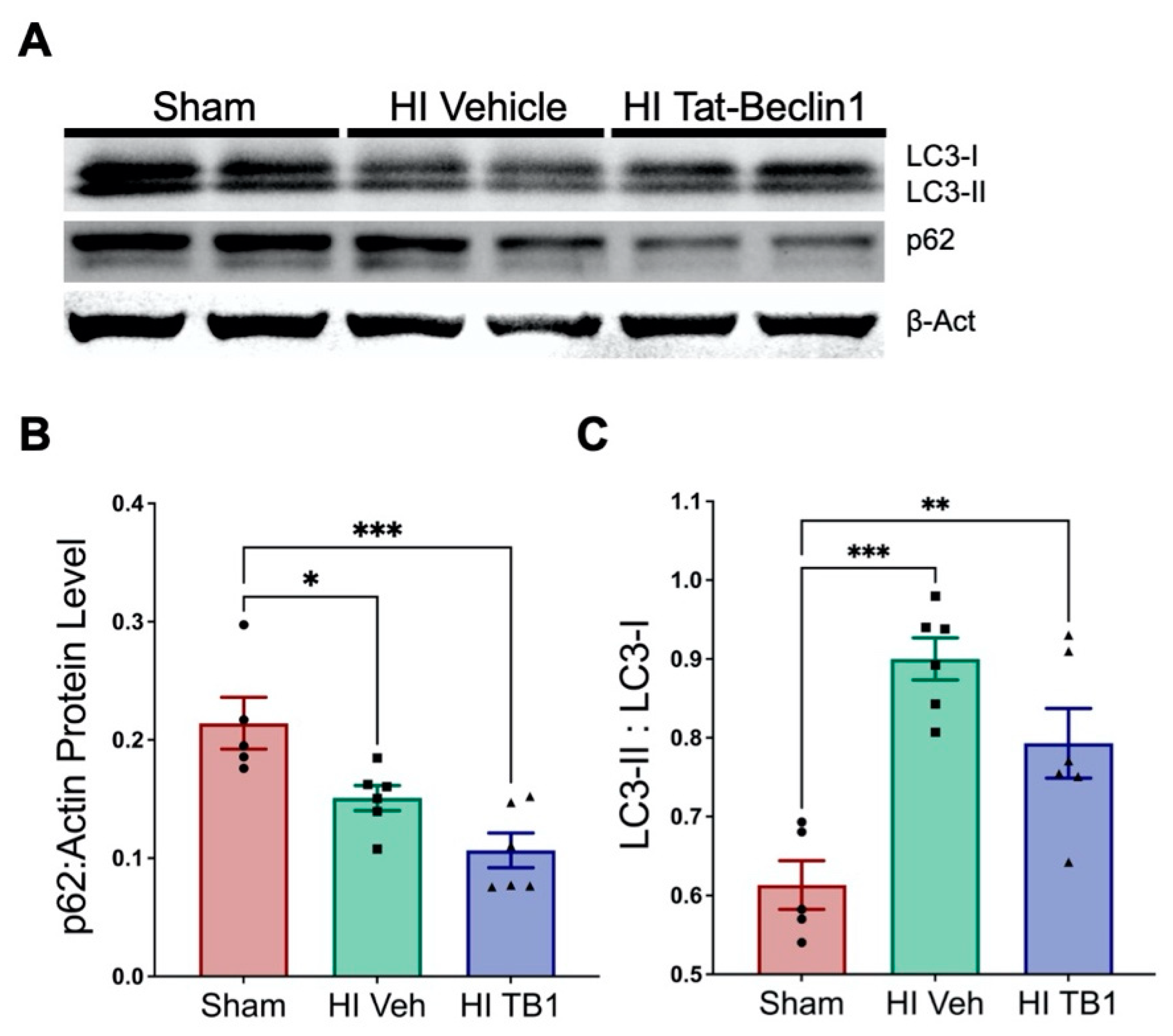
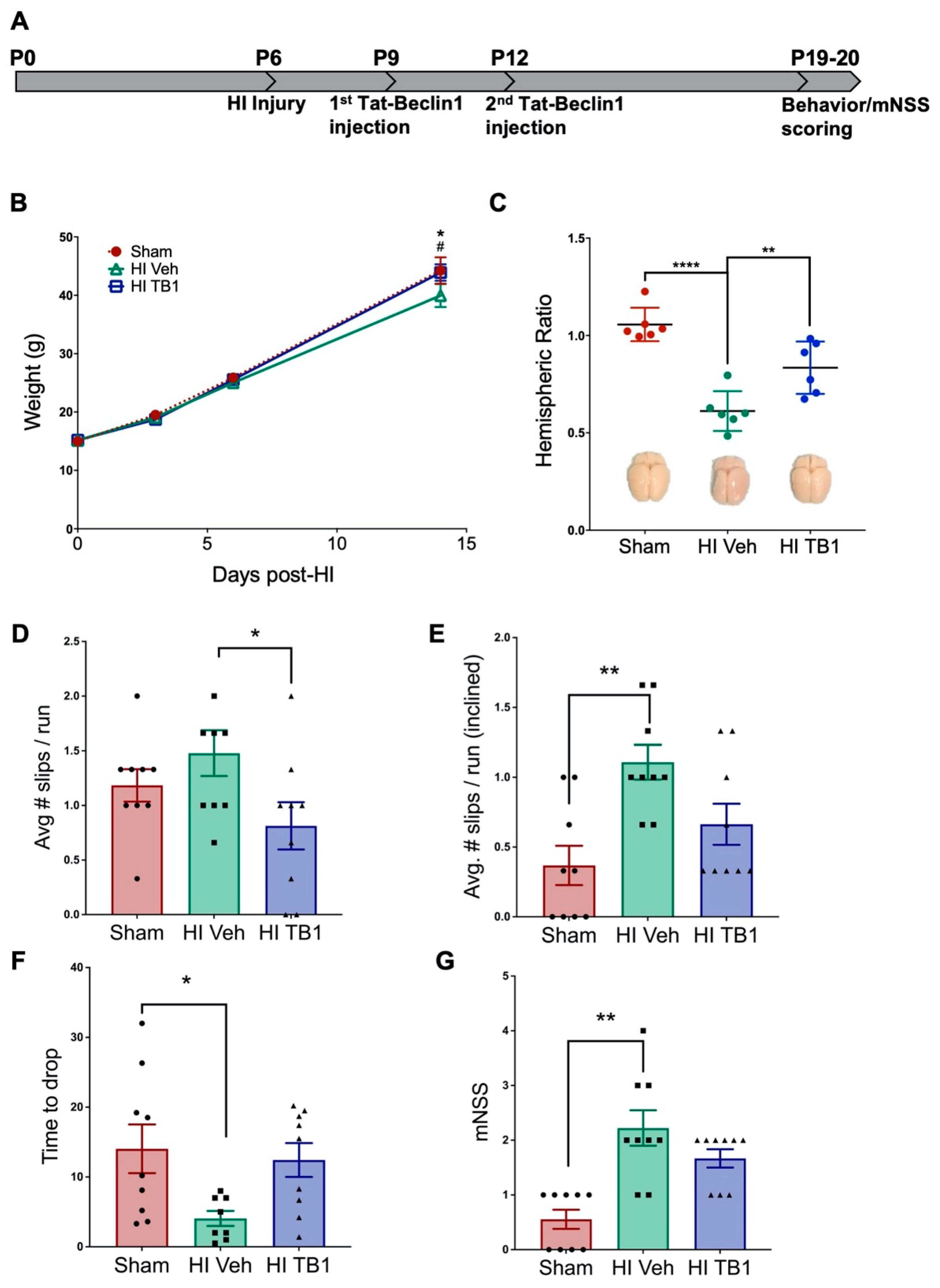
3.3. Independently Augmenting Autophagy after HI Injury Improves Sensorimotor Performance and Limits Long-Term Neurodegeneration
4. Discussion
4.1. Investigating Autophagic Flux Ex Vivo
4.2. Inducing Autophagy with Tat-Beclin1
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lloyd-Jones, D.; Adams, R.; Carnethon, M.; de Simone, G.; Ferguson, T.B.; Flegal, K.; Ford, E.; Furie, K.; Go, A.; Greenlund, K.; et al. Heart disease and stroke statistics—2009 update: A report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2009, 119, 480–486. [Google Scholar] [CrossRef]
- Ferriero, D.M. Neonatal brain injury. N. Engl. J. Med. 2004, 351, 1985–1995. [Google Scholar] [CrossRef] [PubMed]
- Graham, E.M.; Ruis, K.A.; Hartman, A.L.; Northington, F.J.; Fox, H.E. A systematic review of the role of intrapartum hypoxia-ischemia in the causation of neonatal encephalopathy. Am. J. Obstet. Gynecol. 2008, 199, 587–595. [Google Scholar] [CrossRef]
- Levison, S.; Rocha-Ferreira, E.; Kim, B.; Hagberg, H.; Fleiss, B.; Gressens, P.; Dobrowolski, R. Mechanisms of Tertiary Neurodegeneration after Neonatal Hypoxic-Ischemic Brain Damage. Pediatr. Med. 2021, in press. [Google Scholar] [CrossRef]
- Takeoka, M.; Soman, T.B.; Yoshii, A.; Caviness, V.S., Jr.; Gonzalez, R.G.; Grant, P.E.; Krishnamoorthy, K.S. Diffusion-weighted images in neonatal cerebral hypoxic-ischemic injury. Pediatr. Neurol. 2002, 26, 274–281. [Google Scholar] [CrossRef]
- Hagberg, H.; Mallard, C.; Ferriero, D.M.; Vannucci, S.J.; Levison, S.W.; Vexler, Z.S.; Gressens, P. The role of inflammation in perinatal brain injury. Nat. Rev. Neurol. 2015, 11, 192–208. [Google Scholar] [CrossRef]
- Qin, X.; Cheng, J.; Zhong, Y.; Mahgoub, O.K.; Akter, F.; Fan, Y.; Aldughaim, M.; Xie, Q.; Qin, L.; Gu, L.; et al. Mechanism and treatment related to oxidative stress in neonatal hypoxic-ischemic encephalopathy. Front. Mol. Neurosci. 2019, 12, 88. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.C.; Yang, S.N. Perinatal hypoxic-ischemic encephalopathy. J. Biomed. Biotechnol. 2011, 2011, 609813. [Google Scholar] [CrossRef] [PubMed]
- Askalan, R.; Gabarin, N.; Armstrong, E.A.; Fang Liu, Y.; Couchman, D.; Yager, J.Y. Mechanisms of neurodegeneration after severe hypoxic-ischemic injury in the neonatal rat brain. Brain Res. 2015, 1629, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.H.; Levison, S.W. TGFβ1: Friend or foe during recovery in encephalopathy. Neuroscientist 2019, 25, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Puyal, J.; Vaslin, A.; Mottier, V.; Clarke, P.G. Postischemic treatment of neonatal cerebral ischemia should target autophagy. Ann. Neurol. 2009, 66, 378–389. [Google Scholar] [CrossRef]
- Wang, Q.; Lv, H.; Lu, L.; Ren, P.; Li, L. Neonatal hypoxic-ischemic encephalopathy: Emerging therapeutic strategies based on pathophysiologic phases of the injury. J. Matern. Fetal Neonatal Med. 2019, 32, 3685–3692. [Google Scholar] [CrossRef] [PubMed]
- Logan, A.; Frautschy, S.A.; Gonzalez, A.M.; Sporn, M.B.; Baird, A. Enhanced expression of transforming growth factor beta 1 in the rat brain after a localized cerebral injury. Brain Res. 1992, 587, 216–225. [Google Scholar] [CrossRef]
- Clausi, M.G.; Levison, S.W. Delayed ALK5 inhibition improves functional recovery in neonatal brain injury. J. Cereb Blood Flow Metab. 2016. [Google Scholar] [CrossRef]
- Kim, B.H.; Clausi, M.G.; Frondelli, M.; Nnah, I.C.; Saqcena, C.; Dobrowolski, R.; Levison, S.W. Age-dependent effects of ALK5 inhibition and mechanism of neuroprotection in neonatal hypoxic-ischemic brain injury. Dev. Neurosci. 2017, 39, 338–351. [Google Scholar] [CrossRef]
- Vannucci, R.C.; Vannucci, S.J. Perinatal hypoxic-ischemic brain damage: Evolution of an animal model. Dev. Neurosci. 2005, 27, 81–86. [Google Scholar] [CrossRef]
- Bain, J.M.; Ziegler, A.; Yang, Z.; Levison, S.W.; Sen, E. TGFβ1 stimulates the over-production of white matter astrocytes from precursors of the “brain marrow” in a rodent model of neonatal encephalopathy. PLoS ONE 2010, 5, e9567. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, N. Quantification and its applications in fluorescent microscopy imaging. Traffic 2009, 10, 951–961. [Google Scholar] [CrossRef]
- Dunn, K.W.; Kamocka, M.M.; McDonald, J.H. A practical guide to evaluating colocalization in biological microscopy. Am. J. Physiol. Cell Physiol. 2011, 300, C723–C742. [Google Scholar] [CrossRef]
- Fehsel, K.; Kröncke, K.D.; Kolb, H.; Kolb-Bachofen, V. In situ nick-translation detects focal apoptosis in thymuses of glucocorticoid- and lipopolysacharide-treated mice. J. Histochem. Cytochem. 1994, 42, 613–619. [Google Scholar] [CrossRef]
- Tanida, I.; Waguri, S. Measurement of autophagy in cells and tissues. In Protein Misfolding and Cellular Stress in Disease and Aging: Concepts and Protocols; Bross, P., Gregersen, N., Eds.; Humana Press: Totowa, NJ, USA, 2010; pp. 193–214. [Google Scholar]
- Nnah, I.C.; Wang, B.; Saqcena, C.; Weber, G.F.; Bonder, E.M.; Bagley, D.; de Cegli, R.; Napolitano, G.; Medina, D.L.; Ballabio, A.; et al. TFEB-driven endocytosis coordinates MTORC1 signaling and autophagy. Autophagy 2019, 15, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Shoji-Kawata, S.; Sumpter, R.; Leveno, M.; Campbell, G.R.; Zou, Z.; Kinch, L.; Wilkins, A.D.; Sun, Q.; Pallauf, K.; MacDuff, D.; et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature 2013, 494, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Shoji-Kawata, S.; Sumpter, R.M., Jr.; Wei, Y.; Ginet, V.; Zhang, L.; Posner, B.; Tran, K.A.; Green, D.R.; Xavier, R.J.; et al. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc. Natl. Acad. Sci. USA 2013, 110, 20364–20371. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Wang, X.; Huang, Z.; Qiu, L.; Xu, F.; Vahsen, N.; Nilsson, M.; Eriksson, P.S.; Hagberg, H.; Culmsee, C.; et al. Apoptosis-inducing factor is a major contributor to neuronal loss induced by neonatal cerebral hypoxia-ischemia. Cell Death Differ. 2007, 14, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Koike, M.; Shibata, M.; Tadakoshi, M.; Gotoh, K.; Komatsu, M.; Waguri, S.; Kawahara, N.; Kuida, K.; Nagata, S.; Kominami, E.; et al. Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury. Am. J. Pathol. 2008, 172, 454–469. [Google Scholar] [CrossRef]
- Ginet, V.; Spiehlmann, A.; Rummel, C.; Rudinskiy, N.; Grishchuk, Y.; Luthi-Carter, R.; Clarke, P.G.; Truttmann, A.C.; Puyal, J. Involvement of autophagy in hypoxic-excitotoxic neuronal death. Autophagy 2014, 10, 846–860. [Google Scholar] [CrossRef] [PubMed]
- Cui, D.; Sun, D.; Wang, X.; Yi, L.; Kulikowicz, E.; Reyes, M.; Zhu, J.; Yang, Z.J.; Jiang, W.; Koehler, R.C. Impaired autophagosome clearance contributes to neuronal death in a piglet model of neonatal hypoxic-ischemic encephalopathy. Cell Death Dis. 2017, 8, e2919. [Google Scholar] [CrossRef]
- Lechpammer, M.; Tran, Y.P.; Wintermark, P.; Martinez-Cerdeno, V.; Krishnan, V.V.; Ahmed, W.; Berman, R.F.; Jensen, F.E.; Nudler, E.; Zagzag, D. Upregulation of cystathione beta-synthase and p70S6K/S6 in neonatal hypoxic ischemic brain injury. Brain Pathol. 2016. [Google Scholar] [CrossRef]
- Xie, C.; Ginet, V.; Sun, Y.; Koike, M.; Zhou, K.; Li, T.; Li, H.; Li, Q.; Wang, X.; Uchiyama, Y.; et al. Neuroprotection by selective neuronal deletion of Atg7 in neonatal brain injury. Autophagy 2016, 12, 410–423. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.; Cusack, C.L.; Nnah, I.C.; Khayati, K.; Saqcena, C.; Huynh, T.B.; Noggle, S.A.; Ballabio, A.; Dobrowolski, R. Dysregulation of Nutrient Sensing and CLEARance in Presenilin Deficiency. Cell Rep. 2016, 14, 2166–2179. [Google Scholar] [CrossRef] [PubMed]
- Croce, K.R.; Yamamoto, A. A role for autophagy in Huntington’s disease. Neurobiol. Dis. 2019, 122, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Karabiyik, C.; Lee, M.J.; Rubinsztein, D.C. Autophagy impairment in Parkinson’s disease. Essays Biochem. 2017, 61, 711–720. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Ding, Y.; Chu, C.; Tang, J.; Xiao, Q.; Luo, Z.G. Autophagy induction stabilizes microtubules and promotes axon regeneration after spinal cord injury. Proc. Natl. Acad. Sci. USA 2016, 113, 11324–11329. [Google Scholar] [CrossRef]
- Soria, L.R.; Brunetti-Pierri, N. Targeting autophagy for therapy of hyperammonemia. Autophagy 2018, 14, 1273–1275. [Google Scholar] [CrossRef]
- Hylin, M.J.; Zhao, J.; Tangavelou, K.; Rozas, N.S.; Hood, K.N.; MacGowan, J.S.; Moore, A.N.; Dash, P.K. A role for autophagy in long-term spatial memory formation in male rodents. J. Neurosci. Res. 2018, 96, 416–426. [Google Scholar] [CrossRef]
- Hongyun, H.; Tao, G.; Pengyue, Z.; Liqiang, Y.; Yihao, D. Puerarin provides a neuroprotection against transient cerebral ischemia by attenuating autophagy at the ischemic penumbra in neurons but not in astrocytes. Neurosci. Lett. 2017, 643, 45–51. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, B.H.; Jeziorek, M.; Kanal, H.D.; Contu, V.R.; Dobrowolski, R.; Levison, S.W. Moderately Inducing Autophagy Reduces Tertiary Brain Injury after Perinatal Hypoxia-Ischemia. Cells 2021, 10, 898. https://doi.org/10.3390/cells10040898
Kim BH, Jeziorek M, Kanal HD, Contu VR, Dobrowolski R, Levison SW. Moderately Inducing Autophagy Reduces Tertiary Brain Injury after Perinatal Hypoxia-Ischemia. Cells. 2021; 10(4):898. https://doi.org/10.3390/cells10040898
Chicago/Turabian StyleKim, Brian H., Maciej Jeziorek, Hur Dolunay Kanal, Viorica Raluca Contu, Radek Dobrowolski, and Steven W. Levison. 2021. "Moderately Inducing Autophagy Reduces Tertiary Brain Injury after Perinatal Hypoxia-Ischemia" Cells 10, no. 4: 898. https://doi.org/10.3390/cells10040898
APA StyleKim, B. H., Jeziorek, M., Kanal, H. D., Contu, V. R., Dobrowolski, R., & Levison, S. W. (2021). Moderately Inducing Autophagy Reduces Tertiary Brain Injury after Perinatal Hypoxia-Ischemia. Cells, 10(4), 898. https://doi.org/10.3390/cells10040898