
Role of cAMP in Double Switch of Glucagon Secretion





Abstract
1. Introduction
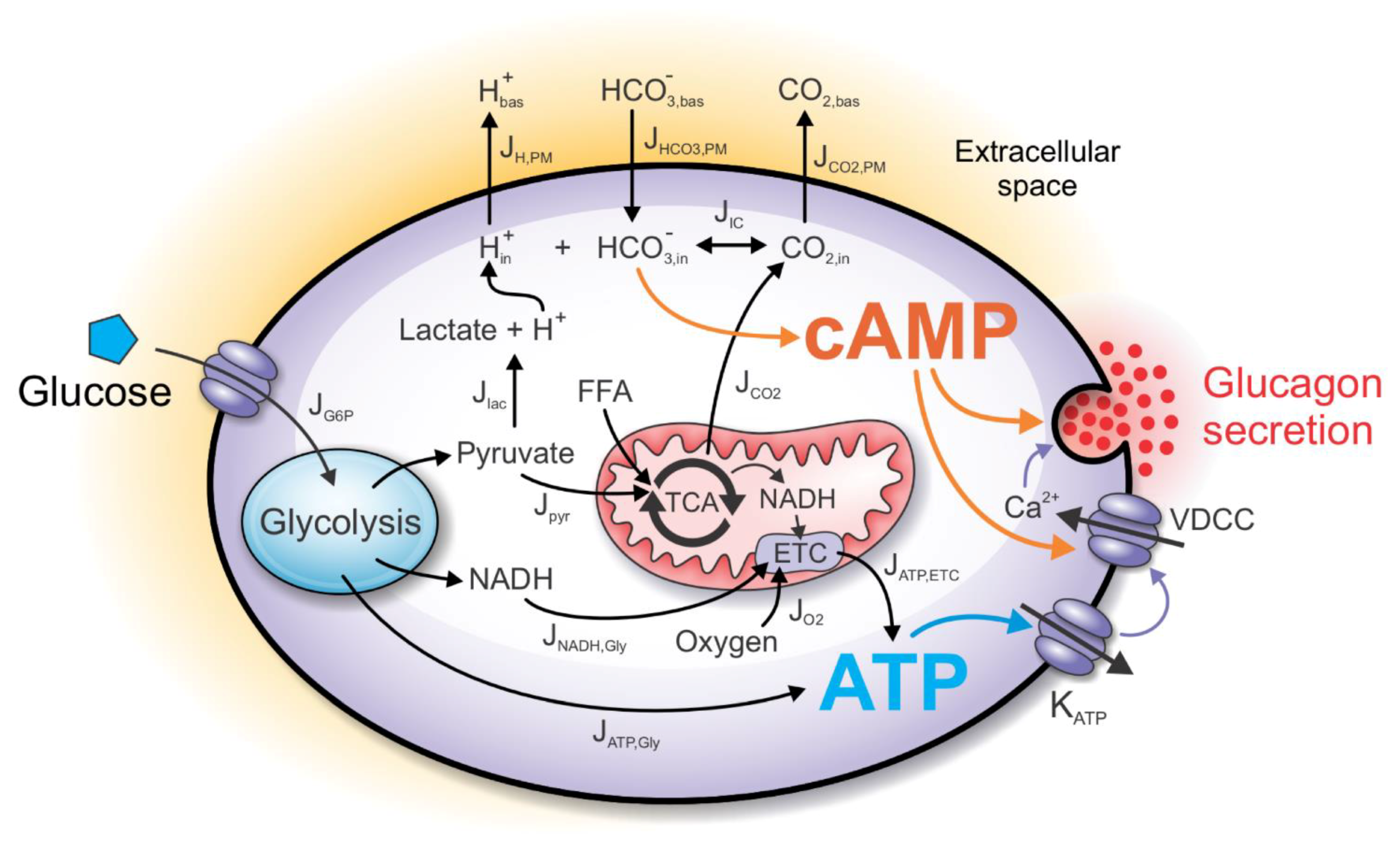
2. Computational Model
2.1. The ATP-Producing Metabolic Component
2.2. The cAMP Signaling Component
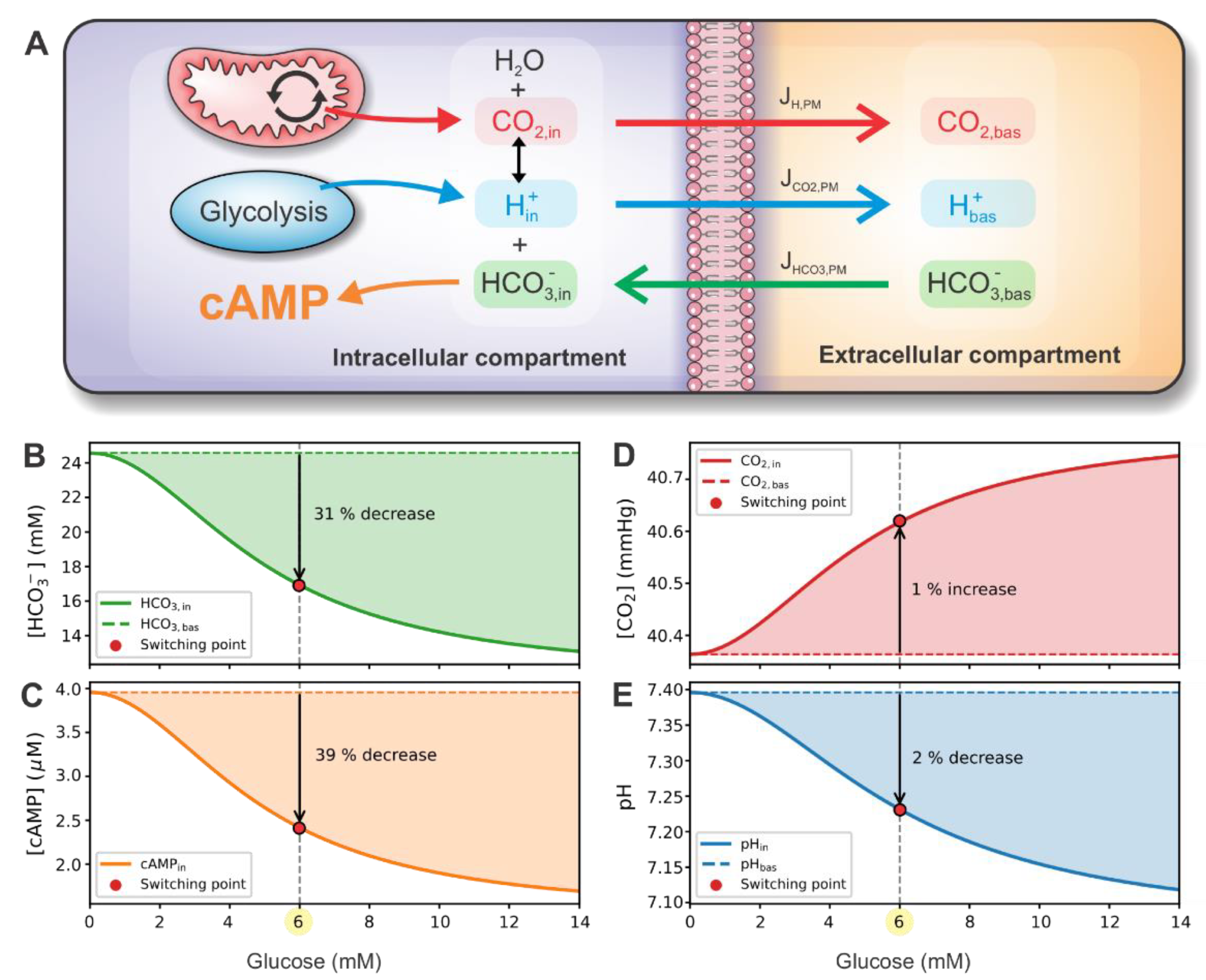
2.3. CO2 and H+ as Signaling Molecules/Ions
2.4. Fluxes through the Plasma Membrane
2.5. The Henderson–Hasselbalch Equilibrium
2.6. Modeling cAMP Levels
2.7. Direct and Indirect Action of cAMP on Exocytosis
2.8. The Glucagon Secretion Component
3. Results
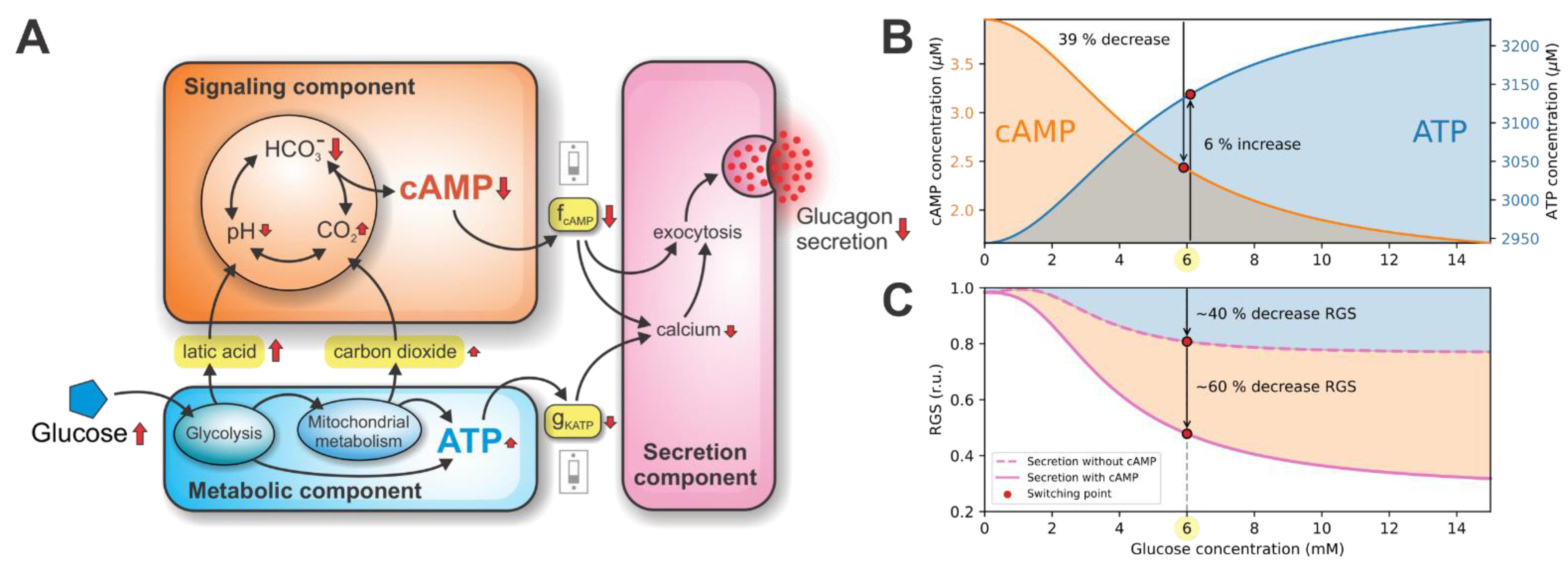
3.1. Double Switch for Glucose-Induced Glucagon Secretion
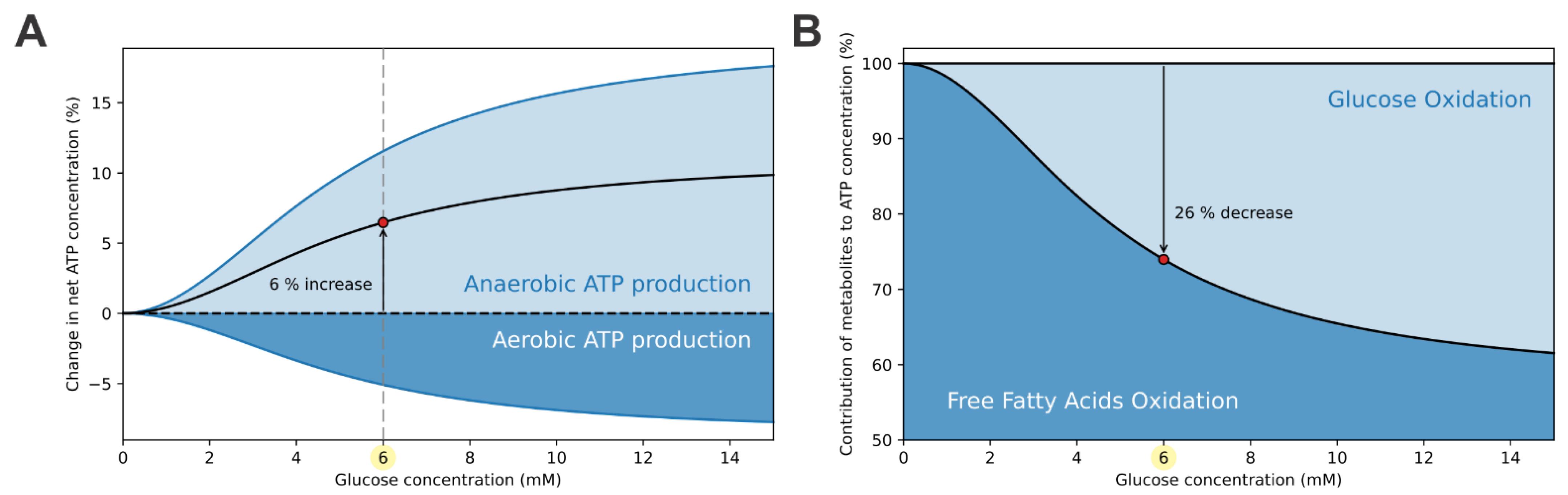
3.2. ATP-Driven Metabolic Switch
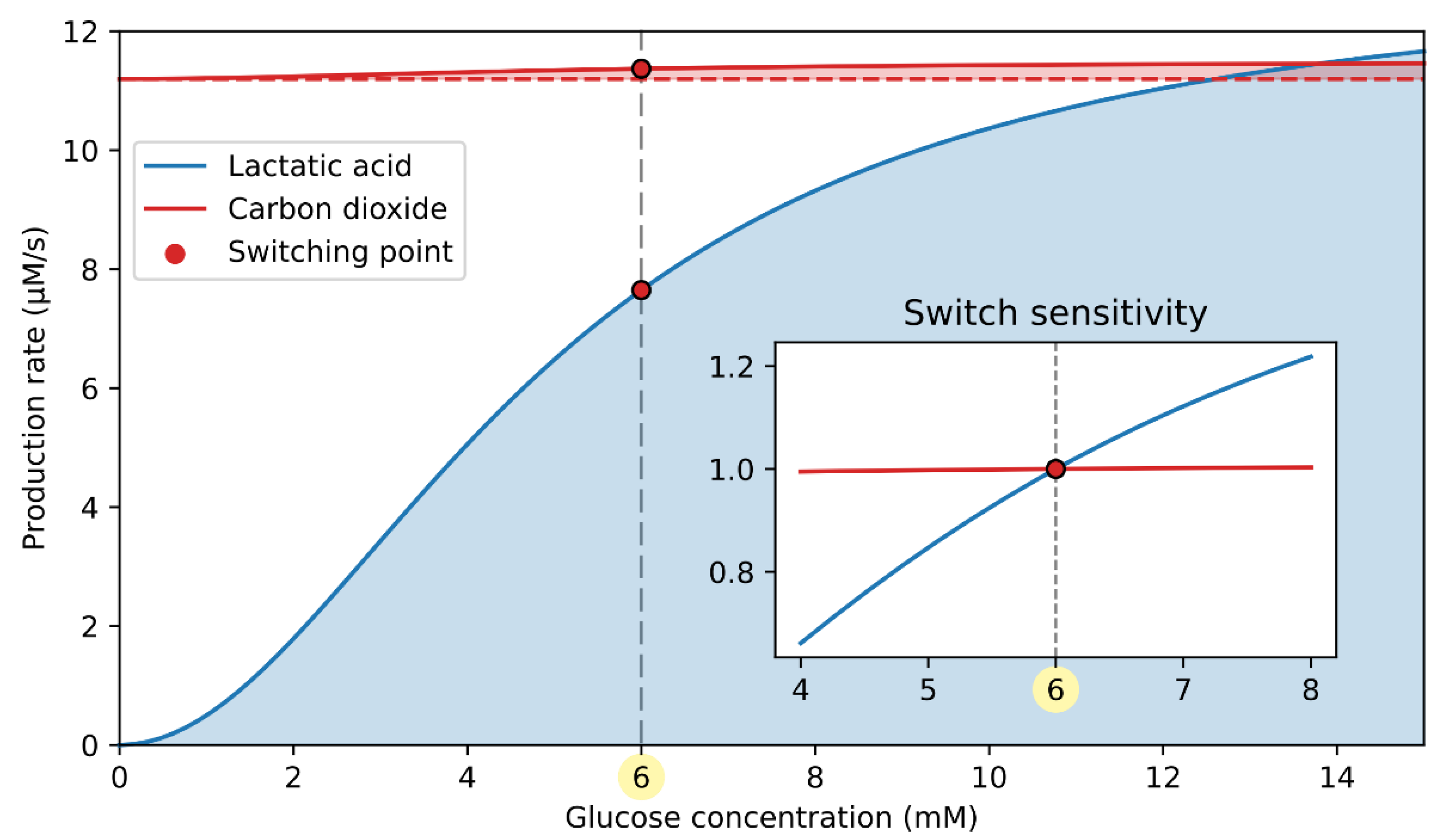
3.3. cAMP-Driven Signaling Switch
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- International Diabetes Federation. IDF Diabetes Atlas Ninth Edition 2019; International Diabetes Federation: Brussels, Belgium, 2019; ISBN 978-2-930229-87-4. [Google Scholar]
- Unger, R.H.; Cherrington, A.D.; Unger, R.H.; Cherrington, A.D. Glucagonocentric restructuring of diabetes: A pathophysiologic and therapeutic makeover Find the latest version: Science in medicine Glucagonocentric restructuring of diabetes: A pathophysiologic and therapeutic makeover. J. Clin. Investig. 2012, 122, 4–12. [Google Scholar] [CrossRef]
- Rorsman, P.; Ashcroft, F.M. Pancreatic β-cell electrical activity and insulin secretion: Of mice and men. Physiol. Rev. 2018, 98, 117–214. [Google Scholar] [CrossRef]
- Quesada, I.; Tudurí, E.; Ripoll, C.; Nadal, Á. Physiology of the pancreatic α-cell and glucagon secretion: Role in glucose homeostasis and diabetes. J. Endocrinol. 2008, 199, 5–19. [Google Scholar] [CrossRef]
- Gao, R.; Yang, T.; Zhang, Q. δ-Cells: The neighborhood watch in the islet community. Biology 2021, 10, 74. [Google Scholar] [CrossRef]
- Vieira, E.; Salehi, A.; Gylfe, E. Glucose inhibits glucagon secretion by a direct effect on mouse pancreatic alpha cells. Diabetologia 2007, 50, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Basco, D.; Zhang, Q.; Salehi, A.; Tarasov, A.; Dolci, W.; Herrera, P.; Spiliotis, I.; Berney, X.; Tarussio, D.; Rorsman, P.; et al. A-cell glucokinase suppresses glucose-regulated glucagon secretion. Nat. Commun. 2018, 9, 546. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yu, Q.; Ahooghalandari, P.; Gribble, F.M.; Reimann, F.; Tengholm, A.; Gylfe, E. Submembrane ATP and Ca2+ kinetics in α-cells: Unexpected signaling for glucagon secretion. FASEB J. 2015, 29, 3379–3388. [Google Scholar] [CrossRef]
- Leclerc, I.; Sun, G.; Morris, C.; Fernandez-Millan, E.; Nyirenda, M.; Rutter, G.A. AMP-activated protein kinase regulates glucagon secretion from mouse pancreatic alpha cells. Diabetologia 2011, 54, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Tengholm, A.; Gylfe, E. cAMP signalling in insulin and glucagon secretion. Diabetes Obes. Metab. 2017, 19, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Onyango, A.N. Mechanisms of the regulation and dysregulation of glucagon secretion. Oxid. Med. Cell. Longev. 2020, 2020, 3089139. [Google Scholar] [CrossRef]
- Gromada, J.; Buschard, K.; Barg, S.; Renström, E.; Bokvist, K.; Rorsman, P.; Ding, W.-G. Adrenaline stimulates glucagon secretion in pancreatic a-cells by increasing the Ca2+ current and the number of granules close to the L-type Ca2+ channels. J. Gen. Physiol. 2002, 110, 217–228. [Google Scholar] [CrossRef]
- Ramracheya, R.; Chapman, C.; Chibalina, M.; Dou, H.; Miranda, C.; González, A.; Moritoh, Y.; Shigeto, M.; Zhang, Q.; Braun, M.; et al. GLP-1 suppresses glucagon secretion in human pancreatic alpha-cells by inhibition of P/Q-type Ca2+ channels. Physiol. Rep. 2018, 6, 1–17. [Google Scholar] [CrossRef]
- Hughes, J.W.; Ustione, A.; Lavagnino, Z.; Piston, D.W. Regulation of islet glucagon secretion: Beyond calcium. Diabetes Obes. Metab. 2018, 20, 127–136. [Google Scholar] [CrossRef]
- Ramos, L.S.; Zippin, J.H.; Kamenetsky, M.; Buck, J.; Levin, L.R. Glucose and GLP-1 stimulate cAMP production via distinct adenylyl cyclases in INS-1E insulinoma cells. J. Gen. Physiol. 2008, 132, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Zippin, J.H.; Chen, Y.; Straub, S.G.; Hess, K.C.; Diaz, A.; Lee, D.; Tso, P.; Holz, G.G.; Sharp, G.W.G.; Levin, L.R.; et al. CO2/HCO3—And calcium-regulated soluble adenylyl cyclase as a physiological ATP sensor. J. Biol. Chem. 2013, 288, 33283–33291. [Google Scholar] [CrossRef] [PubMed]
- Litvin, T.N.; Kamenetsky, M.; Zarifyan, A.; Buck, J.; Levin, L.R. Kinetic properties of “soluble” adenylyl cyclase: Synergism between calcium and bicarbonate. J. Biol. Chem. 2003, 278, 15922–15926. [Google Scholar] [CrossRef]
- Chen, Y.; Cann, M.J.; Litvin, T.N.; Iourgenko, V.; Sinclair, M.L.; Levin, L.R.; Buck, J. Soluble adenylyl cyclase as an evolutionarily conserved bicarbonate sensor. Science 2000, 289, 625–628. [Google Scholar] [CrossRef]
- Zippin, J.H.; Levin, L.R.; Buck, J. CO2/HCO3—Responsive soluble adenylyl cyclase as a putative metabolic sensor. Trends Endocrinol. Metab. 2001, 12, 366–370. [Google Scholar] [CrossRef]
- Chang, J.C.; Oude-Elferink, R.P.J. Role of the bicarbonate-responsive soluble adenylyl cyclase in pH sensing and metabolic regulation. Front. Physiol. 2014, 5, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Tresguerres, M.; Levin, L.R.; Buck, J. Intracellular cAMP signaling by soluble adenylyl cyclase. Kidney Int. 2011, 79, 1277–1288. [Google Scholar] [CrossRef]
- Parkkila, S.; Parkkila, A.K.; Juvonen, T.; Rajaniemi, H. Distribution of the carbonic anhydrase isoenzymes I, II, and VI in the human alimentary tract. Gut 1994, 35, 646–650. [Google Scholar] [CrossRef]
- Yu, Q.; Shuai, H.; Ahooghalandari, P.; Gylfe, E.; Tengholm, A. Glucose controls glucagon secretion by directly modulating cAMP in alpha cells. Diabetologia 2019, 62, 1212–1224. [Google Scholar] [CrossRef]
- Gromada, J.; Ma, X.; Høy, M.; Bokvist, K.; Salehi, A.; Berggren, P.; Rorsman, P. ATP-Sensitive K+ channel—Dependent regulation of glucagon release and electrical activity by glucose in wild-type and SUR1−/− mouse alpha-cells. Diabetes 2004, 53, 181–189. [Google Scholar] [CrossRef]
- Rorsman, P.; Ramracheya, R.; Rorsman, N.J.G.; Zhang, Q. ATP-regulated potassium channels and voltage-gated calcium channels in pancreatic alpha and beta cells: Similar functions but reciprocal effects on secretion. Diabetologia 2014, 57, 1749–1761. [Google Scholar] [CrossRef]
- MacDonald, P.E.; De Marinis, Y.Z.; Ramracheya, R.; Salehi, A.; Ma, X.; Johnson, P.R.V.; Cox, R.; Eliasson, L.; Rorsman, P. A KATPchannel-dependent pathway within α cells regulates glucagon release from both rodent and human islets of langerhans. PLoS Biol. 2007, 5, 1236–1247. [Google Scholar] [CrossRef]
- Ramracheya, R.; Ward, C.; Shigeto, M.; Walker, J.N.; Amisten, S.; Zhang, Q.; Johnson, P.R.; Rorsman, P.; Braun, M. Membrane potential-dependent inactivation of voltage-gated ion channels in α-cells inhibits glucagon secretion from human islets. Diabetes 2010, 59, 2198–2208. [Google Scholar] [CrossRef] [PubMed]
- Le Marchand, S.J.; Piston, D.W. Glucose decouples intracellular Ca2+ activity from glucagon secretion in mouse pancreatic islet alpha-cells. PLoS ONE 2012, 7, e047084. [Google Scholar] [CrossRef] [PubMed]
- Heimberg, H.; De Vos, A.; Moens, K.; Quartier, E.; Bouwens, L.; Pipeleers, D.; Van Schaftingen, E.; Madsen, O.; Schuit, F. The glucose sensor protein glucokinase is expressed in glucagon-producing a-cells. Proc. Natl. Acad. Sci. USA 1996, 93, 7036–7041. [Google Scholar] [CrossRef] [PubMed]
- Detimary, P.; Dejonghe, S.; Ling, Z.; Pipeleers, D.; Schuit, F.; Henquin, J.C. The changes in adenine nucleotides measured in glucose-stimulated rodent islets occur in beta cells but not in alpha cells and are also observed in human islets. J. Biol. Chem. 1998, 273, 33905–33908. [Google Scholar] [CrossRef]
- Schuit, F.; De Vos, A.; Farfari, S.; Moens, K.; Pipeleers, D.; Brun, T.; Prentki, M. Metabolic fate of glucose in purified islet cells. Glucose-regulated anaplerosis in β cells. J. Biol. Chem. 1997, 272, 18572–18579. [Google Scholar] [CrossRef] [PubMed]
- Kristinsson, H.; Sargsyan, E.; Manell, H.; Smith, D.M.; Göpel, S.O.; Bergsten, P. Basal hypersecretion of glucagon and insulin from palmitate-exposed human islets depends on FFAR1 but not decreased somatostatin secretion. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Olofsson, C.S.; Salehi, A.; Göpel, S.O.; Holm, C.; Rorsman, P. Palmitate stimulation of glucagon secretion in mouse pancreatic α-cells results from activation of L-type calcium channels and elevation of cytoplasmic calcium. Diabetes 2004, 53, 2836–2843. [Google Scholar] [CrossRef] [PubMed]
- Briant, L.J.B.; Dodd, M.S.; Chibalina, M.V.; Rorsman, N.J.G.; Johnson, P.R.V.; Carmeliet, P.; Rorsman, P.; Knudsen, J.G. CPT1a-dependent long-chain fatty acid oxidation contributes to maintaining glucagon secretion from pancreatic islets. Cell Rep. 2018, 23, 3300–3311. [Google Scholar] [CrossRef] [PubMed]
- Watts, M.; Sherman, A. Modeling the pancreatic α-cell: Dual mechanisms of glucose suppression of glucagon secretion. Biophys. J. 2014, 106, 741–751. [Google Scholar] [CrossRef]
- Montefusco, F.; Pedersen, M.G. Mathematical modelling of local calcium and regulated exocytosis during inhibition and stimulation of glucagon secretion from pancreatic alpha-cells. J. Physiol. 2015, 593, 4519–4530. [Google Scholar] [CrossRef] [PubMed]
- Diderichsen, P.M.; Göpel, S.O. Modelling the electrical activity of pancreatic α-cells based on experimental data from intact mouse islets. J. Biol. Phys. 2006, 32, 209–229. [Google Scholar] [CrossRef] [PubMed]
- Grubelnik, V.; Zmazek, J.; Markovič, R.; Gosak, M.; Marhl, M. Modelling of energy-driven switch for glucagon and insulin secretion. J. Theor. Biol. 2020, 493, 110213. [Google Scholar] [CrossRef]
- Elliott, A.D.; Ustione, A.; Piston, D.W. Somatostatin and insulin mediate glucose-inhibited glucagon secretion in the pancreatic α-cell by lowering cAMP. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E130–E143. [Google Scholar] [CrossRef]
- Gilon, P. The role of α-cells in islet function and glucose homeostasis in health and type 2 diabetes. J. Mol. Biol. 2020, 432, 1367–1394. [Google Scholar] [CrossRef]
- Wilson, D.F.; Cember, A.T.J.; Matschinsky, F.M. The thermodynamic basis of glucose-stimulated insulin release: A model of the core mechanism. Physiol. Rep. 2017, 5, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Diao, J.; Allister, E.M.; Koshkin, V.; Lee, S.C.; Bhattacharjee, A.; Tang, C.; Giacca, A.; Chan, C.B.; Wheeler, M.B. UCP2 is highly expressed in pancreatic α-cells and influences secretion and survival. Proc. Natl. Acad. Sci. USA 2008, 105, 12057–12062. [Google Scholar] [CrossRef]
- Jaswal, J.S.; Ussher, J.R.; Lopaschuk, G.D. Myocardial fatty acid utilization as a determinant of cardiac efficiency and function. Future Lipidol. 2009, 4, 379–389. [Google Scholar] [CrossRef]
- Grubelnik, V.; Markovič, R.; Lipovšek, S.; Leitinger, G.; Gosak, M.; Dolenšek, J.; Valladolid-Acebes, I.; Berggren, P.O.; Stožer, A.; Perc, M.; et al. Modelling of dysregulated glucagon secretion in type 2 diabetes by considering mitochondrial alterations in pancreatic α-cells. R. Soc. Open Sci. 2020, 7, 191171. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.E.; Chun, J.N.; Kwon, M.J.; Ji, Y.S.; Jeong, M.H.; Kim, H.H.; Park, S.H.; Rah, J.C.; Kang, J.S.; Lee, S.H.; et al. Endocytosis of KATP channels drives glucose-stimulated excitation of pancreatic β cells. Cell Rep. 2018, 22, 471–481. [Google Scholar] [CrossRef]
- Zhang, Q.; Ramracheya, R.; Lahmann, C.; Tarasov, A.; Bengtsson, M.; Braha, O.; Braun, M.; Brereton, M.; Collins, S.; Galvanovskis, J.; et al. Role of KATPchannels in glucose-regulated glucagon secretion and impaired counterregulation in type 2 diabetes. Cell Metab. 2013, 18, 871–882. [Google Scholar] [CrossRef]
- Grubelnik, V.; Zmazek, J.; Markovič, R.; Gosak, M.; Marhl, M. Mitochondrial dysfunction in pancreatic alpha and beta cells associated with type 2 diabetes mellitus. Life 2020, 10, 348. [Google Scholar] [CrossRef]
- Karwi, Q.G.; Uddin, G.M.; Ho, K.L.; Lopaschuk, G.D. Loss of metabolic flexibility in the failing heart. Front. Cardiovasc. Med. 2018, 5, 68. [Google Scholar] [CrossRef]
- Yarwood, S.J. Special issue on “new advances in cyclic amp signalling”—An editorial overview. Cells 2020, 9, 2274. [Google Scholar] [CrossRef] [PubMed]
- Endeward, V.; Al-Samir, S.; Itel, F.; Gros, G. How does carbon dioxide permeate cell membranes? A discussion of concepts, results and methods. Front. Physiol. 2014, 4, 382. [Google Scholar] [CrossRef]
- McMurtrie, H.L.; Cleary, H.J.; Alvarez, B.V.; Loiselle, F.B.; Sterling, D.; Morgan, P.E.; Johnson, D.E.; Casey, J.R. The bicarbonate transport metabolon. J. Enzyme Inhib. Med. Chem. 2004, 19, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Romero, M.F.; Chen, A.P.; Parker, M.D.; Boron, W.F. The SLC4 family of bicarbonate (HCO3−) transporters. Mol. Asp. Med. 2013, 34, 159–182. [Google Scholar] [CrossRef]
- Cao, L.H.; Xia, C.C.; Shi, Z.C.; Wang, N.; Gu, Z.H.; Yu, L.Z.; Wan, Q.; De, W. Na+/HCO3− cotransporter is expressed on β and α cells during rat pancreatic development. World J. Gastroenterol. 2016, 22, 9525–9533. [Google Scholar] [CrossRef]
- Soyfoo, M.S.; Bulur, N.; Virreira, M.; Louchami, K.; Lybaert, P.; Crutzen, R.; Perret, J.; Delporte, C.; Roussa, E.; Thevenod, F.; et al. Expression of the electrogenic Na+-HCO3−—Cotransporters NBCe1-A and NBCe1-B in rat pancreatic islet cells. Endocrine 2009, 35, 449–458. [Google Scholar] [CrossRef][Green Version]
- Boyd, C.E. Water Quality; Springer International Publishing: Cham, Switzerland, 2020; ISBN 978-3-030-23334-1. [Google Scholar]
- Ohadi, D.; Schmitt, D.L.; Calabrese, B.; Halpain, S.; Zhang, J.; Rangamani, P. Computational modeling reveals frequency modulation of calcium-cAMP/PKA pathway in dendritic spines. Biophys. J. 2019, 117, 1963–1980. [Google Scholar] [CrossRef]
- Degerman, E.; Belfrage, P.; Manganiello, V.C. Structure, localization, and regulation of cGMP-inhibited phosphodiesterase (PDE3). J. Biol. Chem. 1997, 272, 6823–6826. [Google Scholar] [CrossRef]
- Leroy, M.J.; Lugnier, C.; Merezak, J.; Tanguy, G.; Olivier, S.; Le Bec, A.; Ferré, F. Isolation and characterization of the rolipram-sensitive cyclic AMP-specific phosphodiesterase (type IV PDE) in human term myometrium. Cell. Signal. 1994, 6, 405–412. [Google Scholar] [CrossRef]
- Fridlyand, L.E.; Philipson, L.H. Pancreatic beta cell G-protein coupled receptors and second messenger interactions: A systems biology computational analysis. PLoS ONE 2016, 11, e0152869. [Google Scholar] [CrossRef]
- Koeppen, B.M.; Stanton, B.A. (Eds.) Berne & Levy Physiology, 7th ed.; Elsevier Inc.: Philadelphia, PA, USA, 2018; ISBN 9780323443388. [Google Scholar]
- Ress, K.L.; Koerbin, G.; Li, L.; Chesher, D.; Bwititi, P.; Horvath, A.R. Reference intervals for venous blood gas measurement in adults. Clin. Chem. Lab. Med. 2020, 59, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Kraynik, S.M.; Miyaoka, R.S.; Beavo, J.A. PDE3 and PDE4 isozyme-selective inhibitors are both required for synergistic activation of brown adipose tissues. Mol. Pharmacol. 2013, 83, 1155–1165. [Google Scholar] [CrossRef]
- Sekine, N.; Cirullis, V.; Regazzi, R.; Brown, L.J.; Ginen, E.; Tamarit-rodrigued, J.; Girotti, M.; Marie, S.; Macdonaldg, M.J.; Wollheim, C.B.; et al. Low lactate dehydrogenase and high mitochondrial glycerol phosphate dehydrogenase in pancreatic p-cells. J. Biol. Chem. 1994, 269, 4895–4902. [Google Scholar] [CrossRef]
- Thorrez, L.; Laudadio, I.; Van Deun, K.; Quintens, R.; Hendrickx, N.; Granvik, M.; Lemaire, K.; Schraenen, A.; Van Lommel, L.; Lehnert, S.; et al. Tissue-specific disallowance of housekeeping genes: The other face of cell differentiation. Genome Res. 2011, 21, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Holst, J.J.; Albrechtsen, N.J.W.; Pedersen, J.; Knop, F.K. Glucagon and amino acids are linked in a mutual feedback cycle: The liver-α-cell axis. Diabetes 2017, 66, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Janah, L.; Kjeldsen, S.; Galsgaard, K.D.; Winther-Sørensen, M.; Stojanovska, E.; Pedersen, J.; Knop, F.K.; Holst, J.J.; Wewer Albrechtsen, N.J. Glucagon receptor signaling and glucagon resistance. Int. J. Mol. Sci. 2019, 20, 3314. [Google Scholar] [CrossRef]
- Unger, R.H.; Ohneda, A.; Aguilar-Parada, E.; Eisentraut, A.M. The role of aminogenic glucagon secretion in blood glucose homeostasis. J. Clin. Investig. 1969, 48, 810–822. [Google Scholar] [CrossRef]
- Danielle Dean, E. A primary role for α-cells as amino acid sensors. Diabetes 2020, 69, 542–549. [Google Scholar] [CrossRef]
- Dean, E.D.; Li, M.; Prasad, N.; Wisniewski, S.N.; Von Deylen, A.; Spaeth, J.; Maddison, L.; Botros, A.; Sedgeman, L.R.; Bozadjieva, N.; et al. Interrupted glucagon signaling reveals hepatic α cell axis and role for l-glutamine in α cell proliferation. Cell Metab. 2017, 25, 1362.e5–1373.e5. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, J.G.; Hamilton, A.; Ramracheya, R.; Tarasov, A.I.; Brereton, M.; Haythorne, E.; Chibalina, M.V.; Spégel, P.; Mulder, H.; Zhang, Q.; et al. Dysregulation of glucagon secretion by hyperglycemia-induced sodium-dependent reduction of ATP production. Cell Metab. 2019, 29, 430.e4–442.e4. [Google Scholar] [CrossRef]
- Rebolledo, O.R.; Gagliardino, J.J. Glucagon and insulin secretion during acid-base alterations. Acta Diabetol. Lat. 1983, 20, 211–220. [Google Scholar] [CrossRef]
- Mokuda, O.; Sakamoto, Y. Acidotic pH augments glucagon secretion and gluconeogenesis in the isolated perfused rat pancreas and liver. Exp. Clin. Endocrinol. Diabetes 2000, 108, 353–357. [Google Scholar] [CrossRef]
- Krapf, R.; Berry, C.A.; Alpern, R.J.; Rector, F.C. Regulation of cell pH by ambient bicarbonate, carbon dioxide tension, and pH in the rabbit proximal convoluted tubule. J. Clin. Investig. 1988, 81, 381–389. [Google Scholar] [CrossRef]
- Tian, G.; Sandler, S.; Gylfe, E.; Tengholm, A. Glucose- and hormone-induced cAMP oscillations in α- and β-cells within intact pancreatic islets. Diabetes 2011, 60, 1535–1543. [Google Scholar] [CrossRef] [PubMed]
- Tengholm, A. Cyclic AMP dynamics in the pancreatic β-cell. Ups. J. Med. Sci. 2012, 117, 355–369. [Google Scholar] [CrossRef]
- Briant, L.; Salehi, A.; Vergari, E.; Zhang, Q.; Rorsman, P. Glucagon secretion from pancreatic α-cells. Ups. J. Med. Sci. 2016, 121, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Briant, L.J.B.; Reinbothe, T.M.; Spiliotis, I.; Miranda, C.; Rodriguez, B.; Rorsman, P. Δ-cells and β-cells are electrically coupled and regulate α-cell activity via somatostatin. J. Physiol. 2018, 596, 197–215. [Google Scholar] [CrossRef] [PubMed]
- Gylfe, E. Glucose control of glucagon secretion—‘There’s a brand-new gimmick every year’. Ups. J. Med. Sci. 2016, 121, 120–132. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Value | References |
|---|---|---|
| Jmax | 7.2 μM s−1 | [38,47], fitted according to [31] |
| Km | 5 mM | |
| pTCA | 1 | [38,47], estimated according to [31] |
| pL | 0.9 | [38,47], estimated according to [31,41] |
| RP/O,G | 2.5 | [48] |
| RP/O,FFA | 2.3 | |
| −0.2 | [38,47], fitted to match relative ATP elevation [8] | |
| 16 μM s−1 | ||
| bATPase | 0.025 s−1 | Estimated to match ATP range [30] |
| Atot | 4000 μM | [30,38] |
| AKATP | 0.65 nS | KATP conductance constants are obtained by fitting values at 1 mM glucose (0.27 nS) and 6 mM glucose (0.20 nS) as measured experimentally [46], cf. [44] |
| BKATP | 0.21 |
| Parameter | Value | References |
|---|---|---|
| Ka | 10−6.1 M | Known dissociation constant [60] |
| [CO2]bas | 40 mmHg = 1.232 mM | Typical values for venous blood [60,61] |
| [H+]bas | 10−7.4 M | |
| 24.6 mM | ||
| 1 s−1 | Fitted to match the minimal drop in pH [23] and physiological intracellular ranges in CO2 and HCO3− concentrations [60] | |
| 106 s−1 | ||
| 103 s−1 | ||
| Km,sAC | 11 mM | [17] |
| Km,PDE3B | 0.4 µM | [57] |
| Km,PDE4 | 4.4 µM | [58] |
| rsAC | 1.0 | Fitted to match the representative absolute cAMP values in islet cells [59] |
| rPDE3B | 0.5 | Assumed 50% of overall PDE activity [39,62] |
| rPDE4 | 0.5 |
| Parameter | Value | References |
|---|---|---|
| kindirect | 0.05 | Sensitivity analysis (see Supplementary Materials, Figure S2) |
| kdirect | 0.6 | |
| gL,min | 0.20 nS | [36] |
| gL,1/2 | 0.25 nS | |
| nh | 9 | [38] |
| ΔgL | 0.04 nS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zmazek, J.; Grubelnik, V.; Markovič, R.; Marhl, M. Role of cAMP in Double Switch of Glucagon Secretion. Cells 2021, 10, 896. https://doi.org/10.3390/cells10040896
Zmazek J, Grubelnik V, Markovič R, Marhl M. Role of cAMP in Double Switch of Glucagon Secretion. Cells. 2021; 10(4):896. https://doi.org/10.3390/cells10040896
Chicago/Turabian StyleZmazek, Jan, Vladimir Grubelnik, Rene Markovič, and Marko Marhl. 2021. "Role of cAMP in Double Switch of Glucagon Secretion" Cells 10, no. 4: 896. https://doi.org/10.3390/cells10040896
APA StyleZmazek, J., Grubelnik, V., Markovič, R., & Marhl, M. (2021). Role of cAMP in Double Switch of Glucagon Secretion. Cells, 10(4), 896. https://doi.org/10.3390/cells10040896