
Zebrafish Models of Autosomal Recessive Ataxias

 , , ,
, , , 
Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. Autosomal Recessive Cerebellar Ataxias
- Ataxia-telangiectasia (AT).
- Boucher–Neuhäuser syndrome and spastic paraplegia type 39 (SPG39).
- Marinesco–Sjögren syndrome.
- Spinocerebellar ataxia autosomal recessive type 20 (SCAR20).
- Ataxia with isolated vitamin E deficiency (AVED).
- Wolfram syndrome (WFS).
3.2. Less Frequent Autosomal Recessive Ataxias
- Polyneuropathy, hearing loss, ataxia, retinitis pigmentosa and cataract (PHARC).
- Cayman ataxia.
- Spinocerebellar ataxia autosomal recessive type 20 (SCAR25).
- Spastic paraplegia type 76 (SPG76).
- Spinocerebellar ataxia autosomal recessive type 17 (SCAR17).
- EAST (epilepsy, ataxia, sensorineural deafness and tubulopathy) syndrome.
- Poretti–Boltshauser syndrome.
- Mitochondrial recessive ataxia syndrome (MIRAS).
- Pancreatic and cerebellar agenesis (PACA).
- Gordon Holmes syndrome.
- Childhood-onset neurodegeneration with ataxia, dystonia, and gaze palsy (NADGP).
- Spinocerebellar ataxia autosomal recessive type 7 (SCAR7) and ceroid lipofuscinosis neuronal 2 (CLN2).
- Spinocerebellar ataxia autosomal recessive type 24 (SCAR24).
- Galloway-Mowat syndrome.
- Spinocerebellar ataxia autosomal recessive type 12 (SCAR12).
3.3. Recessive Inherited Disorders Related to Ataxia
- Niemann–Pick disease type C (NPC).
- Cerebellar ataxia and mental retardation with or without quadrupedal locomotion 3 (CAMRQ3).
- Joubert syndrome.
- Pontocerebellar hypoplasia.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Evidente, V.G.; Gwinn-Hardy, K.A.; Caviness, J.N.; Gilman, S. Hereditary Ataxias. Mayo Clin. Proc. 2000, 75, 475–490. [Google Scholar] [CrossRef]
- Beaudin, M.; Klein, C.J.; Rouleau, G.A.; Dupré, N. Systematic Review of Autosomal Recessive Ataxias and Proposal for a Classification. Cerebellum Ataxias 2017, 4, 3. [Google Scholar] [CrossRef]
- Beaudin, M.; Matilla-Dueñas, A.; Soong, B.-W.; Pedroso, J.L.; Barsottini, O.G.; Mitoma, H.; Tsuji, S.; Schmahmann, J.D.; Manto, M.; Rouleau, G.A.; et al. The Classification of Autosomal Recessive Cerebellar Ataxias: A Consensus Statement from the Society for Research on the Cerebellum and Ataxias Task Force. Cerebellum 2019, 18, 1098–1125. [Google Scholar] [CrossRef]
- Quelle-Regaldie, A.; Sobrido-Cameán, D.; Barreiro-Iglesias, A.; Sobrido, M.J.; Sánchez, L. Zebrafish Models of Autosomal Dominant Ataxias. Cells 2021, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Synofzik, M.; Németh, A.H. Recessive Ataxias. Handb. Clin. Neurol. 2018, 155, 73–89. [Google Scholar] [CrossRef]
- Manto, M.; Marmolino, D. Cerebellar Ataxias. Curr. Opin. Neurol. 2009, 22, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Di Donato, S.; Gellera, C.; Mariotti, C. The Complex Clinical and Genetic Classification of Inherited Ataxias. II. Autosomal Recessive Ataxias. Neurol. Sci. 2001, 22, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Fogel, B.L.; Perlman, S. Clinical Features and Molecular Genetics of Autosomal Recessive Cerebellar Ataxias. Lancet Neurol. 2007, 6, 245–257. [Google Scholar] [CrossRef]
- Dawson, T.M.; Golde, T.E.; Lagier-Tourenne, C. Animal Models of Neurodegenerative Diseases. Nat. Neurosci. 2018, 21, 1370–1379. [Google Scholar] [CrossRef] [PubMed]
- Hoxha, E.; Balbo, I.; Miniaci, M.C.; Tempia, F. Purkinje Cell Signaling Deficits in Animal Models of Ataxia. Front. Synaptic Neurosci. 2018, 10, 6. [Google Scholar] [CrossRef] [PubMed]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The Zebrafish Reference Genome Sequence and Its Relationship to the Human Genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef]
- White, R.; Rose, K.; Zon, L. Zebrafish Cancer: The State of the Art and the Path Forward. Nat. Rev. Cancer 2013, 13, 624–636. [Google Scholar] [CrossRef]
- Kabashi, E.; Brustein, E.; Champagne, N.; Drapeau, P. Zebrafish Models for the Functional Genomics of Neurogenetic Disorders. Biochim. Biophys. Acta 2011, 1812, 335–345. [Google Scholar] [CrossRef]
- Volgin, A.D.; Yakovlev, O.A.; Demin, K.A.; de Abreu, M.S.; Alekseeva, P.A.; Friend, A.J.; Lakstygal, A.M.; Amstislavskaya, T.G.; Bao, W.; Song, C.; et al. Zebrafish Models for Personalized Psychiatry: Insights from Individual, Strain and Sex Differences, and Modeling Gene x Environment Interactions. J. Neurosci. Res. 2019, 97, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Bakkers, J. Zebrafish as a Model to Study Cardiac Development and Human Cardiac Disease. Cardiovasc. Res. 2011, 91, 279–288. [Google Scholar] [CrossRef]
- Barreiro-Iglesias, A.; Mysiak, K.S.; Scott, A.L.; Reimer, M.M.; Yang, Y.; Becker, C.G.; Becker, T. Serotonin Promotes Development and Regeneration of Spinal Motor Neurons in Zebrafish. Cell Rep. 2015, 13, 924–932. [Google Scholar] [CrossRef] [PubMed]
- Da Silva-Álvarez, S.; Guerra-Varela, J.; Sobrido-Cameán, D.; Quelle, A.; Barreiro-Iglesias, A.; Sánchez, L.; Collado, M. Cell Senescence Contributes to Tissue Regeneration in Zebrafish. Aging Cell 2020, 19, e13052. [Google Scholar] [CrossRef]
- Lieschke, G.J.; Currie, P.D. Animal Models of Human Disease: Zebrafish Swim into View. Nat. Rev. Genet. 2007, 8, 353–367. [Google Scholar] [CrossRef]
- Kozol, R.A.; Abrams, A.J.; James, D.M.; Buglo, E.; Yan, Q.; Dallman, J.E. Function Over Form: Modeling Groups of Inherited Neurological Conditions in Zebrafish. Front. Mol. Neurosci. 2016, 9, 55. [Google Scholar] [CrossRef] [PubMed]
- Nasevicius, A.; Ekker, S.C. Effective Targeted Gene “knockdown” in Zebrafish. Nat. Genet. 2000, 26, 216–220. [Google Scholar] [CrossRef]
- de Bruijn, E.; Cuppen, E.; Feitsma, H. Highly Efficient ENU Mutagenesis in Zebrafish. Methods Mol. Biol. 2009, 546, 3–12. [Google Scholar] [CrossRef]
- Hruscha, A.; Krawitz, P.; Rechenberg, A.; Heinrich, V.; Hecht, J.; Haass, C.; Schmid, B. Efficient CRISPR/Cas9 Genome Editing with Low off-Target Effects in Zebrafish. Development 2013, 140, 4982–4987. [Google Scholar] [CrossRef]
- Bird, T.D. Hereditary Ataxia Overview. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Lavin, M.F.; Shiloh, Y. The Genetic Defect in Ataxia-Telangiectasia. Annu. Rev. Immunol. 1997, 15, 177–202. [Google Scholar] [CrossRef]
- Synofzik, M.; Gonzalez, M.A.; Lourenco, C.M.; Coutelier, M.; Haack, T.B.; Rebelo, A.; Hannequin, D.; Strom, T.M.; Prokisch, H.; Kernstock, C.; et al. PNPLA6 Mutations Cause Boucher-Neuhauser and Gordon Holmes Syndromes as Part of a Broad Neurodegenerative Spectrum. Brain 2014, 137, 69–77. [Google Scholar] [CrossRef]
- Byrne, S.; Dlamini, N.; Lumsden, D.; Pitt, M.; Zaharieva, I.; Muntoni, F.; King, A.; Robert, L.; Jungbluth, H. SIL1-Related Marinesco-Sjoegren Syndrome (MSS) with Associated Motor Neuronopathy and Bradykinetic Movement Disorder. Neuromuscul. Disord. 2015, 25, 585–588. [Google Scholar] [CrossRef]
- Akizu, N.; Cantagrel, V.; Zaki, M.S.; Al-Gazali, L.; Wang, X.; Rosti, R.O.; Dikoglu, E.; Gelot, A.B.; Rosti, B.; Vaux, K.K.; et al. Biallelic Mutations in SNX14 Cause a Syndromic Form of Cerebellar Atrophy and Lysosome-Autophagosome Dysfunction. Nat. Genet. 2015, 47, 528–534. [Google Scholar] [CrossRef] [PubMed]
- Mariotti, C.; Gellera, C.; Rimoldi, M.; Mineri, R.; Uziel, G.; Zorzi, G.; Pareyson, D.; Piccolo, G.; Gambi, D.; Piacentini, S.; et al. Ataxia with Isolated Vitamin E Deficiency: Neurological Phenotype, Clinical Follow-up and Novel Mutations in TTPA Gene in Italian Families. Neurol. Sci. 2004, 25, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Becker, L.; Deck, J. Evidence of Widespread Axonal Pathology in Wolfram Syndrome. Acta Neuropathol. 1999, 98, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Rigoli, L.; Lombardo, F.; Di Bella, C. Wolfram Syndrome and WFS1 Gene. Clin. Genet. 2011, 79, 103–117. [Google Scholar] [CrossRef]
- Fiskerstrand, T.; H’mida-Ben Brahim, D.; Johansson, S.; M’zahem, A.; Haukanes, B.I.; Drouot, N.; Zimmermann, J.; Cole, A.J.; Vedeler, C.; Bredrup, C.; et al. Mutations in ABHD12 Cause the Neurodegenerative Disease PHARC: An Inborn Error of Endocannabinoid Metabolism. Am. J. Hum. Genet. 2010, 87, 410–417. [Google Scholar] [CrossRef]
- Bomar, J.M.; Benke, P.J.; Slattery, E.L.; Puttagunta, R.; Taylor, L.P.; Seong, E.; Nystuen, A.; Chen, W.; Albin, R.L.; Patel, P.D.; et al. Mutations in a Novel Gene Encoding a CRAL-TRIO Domain Cause Human Cayman Ataxia and Ataxia/Dystonia in the Jittery Mouse. Nat. Genet. 2003, 35, 264–269. [Google Scholar] [CrossRef]
- Kim, M.; Sandford, E.; Gatica, D.; Qiu, Y.; Liu, X.; Zheng, Y.; Schulman, B.A.; Xu, J.; Semple, I.; Ro, S.-H.; et al. Mutation in ATG5 Reduces Autophagy and Leads to Ataxia with Developmental Delay. Elife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Shetty, A.; Gan-Or, Z.; Ashtiani, S.; Ruskey, J.A.; van de Warrenburg, B.; Wassenberg, T.; Kamsteeg, E.-J.; Rouleau, G.A.; Suchowersky, O. CAPN1 Mutations: Expanding the CAPN1-Related Phenotype: From Hereditary Spastic Paraparesis to Spastic Ataxia. Eur. J. Med. Genet. 2019, 62, 103605. [Google Scholar] [CrossRef]
- Burns, R.; Majczenko, K.; Xu, J.; Peng, W.; Yapici, Z.; Dowling, J.J.; Li, J.Z.; Burmeister, M. Homozygous Splice Mutation in CWF19L1 in a Turkish Family with Recessive Ataxia Syndrome. Neurology 2014, 83, 2175–2182. [Google Scholar] [CrossRef] [PubMed]
- Scholl, U.I.; Choi, M.; Liu, T.; Ramaekers, V.T.; Häusler, M.G.; Grimmer, J.; Tobe, S.W.; Farhi, A.; Nelson-Williams, C.; Lifton, R.P. Seizures, Sensorineural Deafness, Ataxia, Mental Retardation, and Electrolyte Imbalance (SeSAME Syndrome) Caused by Mutations in KCNJ10. Proc. Natl. Acad. Sci. USA 2009, 106, 5842–5847. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, F.; Mozere, M.; Zdebik, A.A.; Stanescu, H.C.; Tobin, J.; Beales, P.L.; Kleta, R.; Bockenhauer, D.; Russell, C. Generation and Validation of a Zebrafish Model of EAST (Epilepsy, Ataxia, Sensorineural Deafness and Tubulopathy) Syndrome. Dis. Model. Mech. 2013, 6, 652–660. [Google Scholar] [CrossRef]
- Aldinger, K.A.; Mosca, S.J.; Tétreault, M.; Dempsey, J.C.; Ishak, G.E.; Hartley, T.; Phelps, I.G.; Lamont, R.E.; O’Day, D.R.; Basel, D.; et al. Mutations in LAMA1 Cause Cerebellar Dysplasia and Cysts with and without Retinal Dystrophy. Am. J. Hum. Genet. 2014, 95, 227–234. [Google Scholar] [CrossRef]
- Mancuso, M.; Filosto, M.; Bellan, M.; Liguori, R.; Montagna, P.; Baruzzi, A.; DiMauro, S.; Carelli, V. POLG Mutations Causing Ophthalmoplegia, Sensorimotor Polyneuropathy, Ataxia, and Deafness. Neurology 2004, 62, 316–318. [Google Scholar] [CrossRef]
- Horvath, R.; Hudson, G.; Ferrari, G.; Fütterer, N.; Ahola, S.; Lamantea, E.; Prokisch, H.; Lochmüller, H.; McFarland, R.; Ramesh, V.; et al. Phenotypic Spectrum Associated with Mutations of the Mitochondrial Polymerase Gamma Gene. Brain 2006, 129, 1674–1684. [Google Scholar] [CrossRef]
- Sellick, G.S.; Barker, K.T.; Stolte-Dijkstra, I.; Fleischmann, C.; Coleman, R.J.; Garrett, C.; Gloyn, A.L.; Edghill, E.L.; Hattersley, A.T.; Wellauer, P.K.; et al. Mutations in PTF1A Cause Pancreatic and Cerebellar Agenesis. Nat. Genet. 2004, 36, 1301–1305. [Google Scholar] [CrossRef]
- Margolin, D.H.; Kousi, M.; Chan, Y.-M.; Lim, E.T.; Schmahmann, J.D.; Hadjivassiliou, M.; Hall, J.E.; Adam, I.; Dwyer, A.; Plummer, L.; et al. Ataxia, Dementia, and Hypogonadotropism Caused by Disordered Ubiquitination. N. Engl. J. Med. 2013, 368, 1992–2003. [Google Scholar] [CrossRef]
- Muto, V.; Flex, E.; Kupchinsky, Z.; Primiano, G.; Galehdari, H.; Dehghani, M.; Cecchetti, S.; Carpentieri, G.; Rizza, T.; Mazaheri, N.; et al. Biallelic SQSTM1 Mutations in Early-Onset, Variably Progressive Neurodegeneration. Neurology 2018, 91, e319–e330. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Almomani, R.; Breedveld, G.J.; Santen, G.W.E.; Aten, E.; Lefeber, D.J.; Hoff, J.I.; Brusse, E.; Verheijen, F.W.; Verdijk, R.M.; et al. Autosomal Recessive Spinocerebellar Ataxia 7 (SCAR7) Is Caused by Variants in TPP1, the Gene Involved in Classic Late-Infantile Neuronal Ceroid Lipofuscinosis 2 Disease (CLN2 Disease). Hum. Mutat. 2013, 34, 706–713. [Google Scholar] [CrossRef] [PubMed]
- Duan, R.; Shi, Y.; Yu, L.; Zhang, G.; Li, J.; Lin, Y.; Guo, J.; Wang, J.; Shen, L.; Jiang, H.; et al. UBA5 Mutations Cause a New Form of Autosomal Recessive Cerebellar Ataxia. PLoS ONE 2016, 11, e0149039. [Google Scholar] [CrossRef]
- Ben-Omran, T.; Fahiminiya, S.; Sorfazlian, N.; Almuriekhi, M.; Nawaz, Z.; Nadaf, J.; Khadija, K.A.; Zaineddin, S.; Kamel, H.; Majewski, J.; et al. Nonsense Mutation in the WDR73 Gene Is Associated with Galloway-Mowat Syndrome. J. Med. Genet. 2015, 52, 381–390. [Google Scholar] [CrossRef]
- Mallaret, M.; Synofzik, M.; Lee, J.; Sagum, C.A.; Mahajnah, M.; Sharkia, R.; Drouot, N.; Renaud, M.; Klein, F.A.C.; Anheim, M.; et al. The Tumour Suppressor Gene WWOX Is Mutated in Autosomal Recessive Cerebellar Ataxia with Epilepsy and Mental Retardation. Brain 2014, 137, 411–419. [Google Scholar] [CrossRef]
- Schrock, M.S.; Batar, B.; Lee, J.; Druck, T.; Ferguson, B.; Cho, J.H.; Akakpo, K.; Hagrass, H.; Heerema, N.A.; Xia, F.; et al. Wwox-Brca1 Interaction: Role in DNA Repair Pathway Choice. Oncogene 2017, 36, 2215–2227. [Google Scholar] [CrossRef]
- Johannsen, J.; Kortüm, F.; Rosenberger, G.; Bokelmann, K.; Schirmer, M.A.; Denecke, J.; Santer, R. A Novel Missense Variant in the SDR Domain of the WWOX Gene Leads to Complete Loss of WWOX Protein with Early-Onset Epileptic Encephalopathy and Severe Developmental Delay. Neurogenetics 2018, 19, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Vanier, M.T. Niemann-Pick Disease Type C. Orphanet J. Rare. Dis. 2010, 5, 16. [Google Scholar] [CrossRef]
- Türkmen, S.; Guo, G.; Garshasbi, M.; Hoffmann, K.; Alshalah, A.J.; Mischung, C.; Kuss, A.; Humphrey, N.; Mundlos, S.; Robinson, P.N. CA8 Mutations Cause a Novel Syndrome Characterized by Ataxia and Mild Mental Retardation with Predisposition to Quadrupedal Gait. PLoS Genet. 2009, 5, e1000487. [Google Scholar] [CrossRef]
- Aspatwar, A.; Tolvanen, M.E.E.; Jokitalo, E.; Parikka, M.; Ortutay, C.; Harjula, S.-K.E.; Rämet, M.; Vihinen, M.; Parkkila, S. Abnormal Cerebellar Development and Ataxia in CARP VIII Morphant Zebrafish. Hum. Mol. Genet. 2013, 22, 417–432. [Google Scholar] [CrossRef] [PubMed]
- Romani, M.; Micalizzi, A.; Valente, E.M. Joubert Syndrome: Congenital Cerebellar Ataxia with the Molar Tooth. Lancet Neurol. 2013, 12, 894–905. [Google Scholar] [CrossRef]
- Qian, Y.; Wang, H.; Jin, T.; Wang, Y.; Fang, L.; Chen, Y.; Chen, L. A Familial Late-onset Hereditary Ataxia Mimicking Pontocerebellar Hypoplasia Caused by a Novel TSEN54 Mutation. Mol. Med. Rep. 2014, 10, 1423–1425. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Imamura, S.; Kishi, S. Molecular Cloning and Functional Characterization of Zebrafish ATM. Int. J. Biochem. Cell Biol. 2005, 37, 1105–1116. [Google Scholar] [CrossRef] [PubMed]
- Vierstraete, J.; Fieuws, C.; Creytens, D.; Van Dorpe, J.; Willaert, A.; Vral, A.; Claes, K. Atm Deficient Zebrafish Model Reveals Conservation of the Tumour Suppressor Function. In Proceedings of the 20th BESHG Meeting: Genome for All, Brussels, Belgium, 6 March 2020. [Google Scholar]
- Hufnagel, R.B.; Arno, G.; Hein, N.D.; Hersheson, J.; Prasad, M.; Anderson, Y.; Krueger, L.A.; Gregory, L.C.; Stoetzel, C.; Jaworek, T.J.; et al. Neuropathy Target Esterase Impairments Cause Oliver-McFarlane and Laurence-Moon Syndromes. J. Med. Genet. 2015, 52, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Wang, M.; Mao, F.; Shao, M.; Zhao, B.; Song, Z.; Shao, C.; Gong, Y. Knockdown of Pnpla6 Protein Results in Motor Neuron Defects in Zebrafish. Dis. Model. Mech. 2013, 6, 404–413. [Google Scholar] [CrossRef]
- Kawahara, G.; Hayashi, Y.K. Characterization of Zebrafish Models of Marinesco-Sjögren Syndrome. PLoS ONE 2016, 11, e0165563. [Google Scholar] [CrossRef]
- Phan, V.; Cox, D.; Cipriani, S.; Spendiff, S.; Buchkremer, S.; O’Connor, E.; Horvath, R.; Goebel, H.H.; Hathazi, D.; Lochmüller, H.; et al. SIL1 Deficiency Causes Degenerative Changes of Peripheral Nerves and Neuromuscular Junctions in Fish, Mice and Human. Neurobiol. Dis. 2019, 124, 218–229. [Google Scholar] [CrossRef]
- Bryant, D.; Seda, M.; Peskett, E.; Maurer, C.; Pomeranz, G.; Ghosh, M.; Hawkins, T.A.; Cleak, J.; Datta, S.; Hariri, H.; et al. Diverse Species-Specific Phenotypic Consequences of Loss of Function Sorting Nexin 14 Mutations. Sci. Rep. 2020, 10, 13763. [Google Scholar] [CrossRef]
- Stainier, D.Y.R.; Raz, E.; Lawson, N.D.; Ekker, S.C.; Burdine, R.D.; Eisen, J.S.; Ingham, P.W.; Schulte-Merker, S.; Yelon, D.; Weinstein, B.M.; et al. Guidelines for Morpholino Use in Zebrafish. PLoS Genet. 2017, 13, e1007000. [Google Scholar] [CrossRef] [PubMed]
- Miller, G.W.; Ulatowski, L.; Labut, E.M.; Lebold, K.M.; Manor, D.; Atkinson, J.; Barton, C.L.; Tanguay, R.L.; Traber, M.G. The α-Tocopherol Transfer Protein Is Essential for Vertebrate Embryogenesis. PLoS ONE 2012, 7, e47402. [Google Scholar] [CrossRef]
- Miller, G.W.; Labut, E.M.; Lebold, K.M.; Floeter, A.; Tanguay, R.L.; Traber, M.G. Zebrafish (Danio Rerio) Fed Vitamin E-Deficient Diets Produce Embryos with Increased Morphologic Abnormalities and Mortality. J. Nutr. Biochem. 2012, 23, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Head, B.; La Du, J.; Tanguay, R.L.; Kioussi, C.; Traber, M.G. Vitamin E Is Necessary for Zebrafish Nervous System Development. Sci. Rep. 2020, 10, 15028. [Google Scholar] [CrossRef]
- O’Hare, E.A.; Yerges-Armstrong, L.M.; Perry, J.A.; Shuldiner, A.R.; Zaghloul, N.A. Assignment of Functional Relevance to Genes at Type 2 Diabetes-Associated Loci Through Investigation of β-Cell Mass Deficits. Mol. Endocrinol. 2016, 30, 429–445. [Google Scholar] [CrossRef]
- Cairns, G.E. Characterisation of a Zebrafish Model of Wolfram Syndrome. Ph.D. Thesis, Newcastle University, Newcastle upon Tyne, UK, 2019. [Google Scholar]
- Gonzaga-Jauregui, C.; Harel, T.; Gambin, T.; Kousi, M.; Griffin, L.B.; Francescatto, L.; Ozes, B.; Karaca, E.; Jhangiani, S.N.; Bainbridge, M.N.; et al. Exome Sequence Analysis Suggests That Genetic Burden Contributes to Phenotypic Variability and Complex Neuropathy. Cell Rep. 2015, 12, 1169–1183. [Google Scholar] [CrossRef] [PubMed]
- Tingaud-Sequeira, A.; Raldúa, D.; Lavie, J.; Mathieu, G.; Bordier, M.; Knoll-Gellida, A.; Rambeau, P.; Coupry, I.; André, M.; Malm, E.; et al. Functional Validation of ABHD12 Mutations in the Neurodegenerative Disease PHARC. Neurobiol. Dis. 2017, 98, 36–51. [Google Scholar] [CrossRef]
- Sun, J.; Pan, C.Q.; Chew, T.W.; Liang, F.; Burmeister, M.; Low, B.C. BNIP-H Recruits the Cholinergic Machinery to Neurite Terminals to Promote Acetylcholine Signaling and Neuritogenesis. Dev. Cell 2015, 34, 555–568. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Zhang, J.; Zhang, Q. Expression Pattern and Functions of Autophagy-Related Gene Atg5 in Zebrafish Organogenesis. Autophagy 2011, 7, 1514–1527. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Koo, Y.; Ng, A.; Wei, Y.; Luby-Phelps, K.; Juraszek, A.; Xavier, R.J.; Cleaver, O.; Levine, B.; Amatruda, J.F. Autophagy Is Essential for Cardiac Morphogenesis during Vertebrate Development. Autophagy 2014, 10, 572–587. [Google Scholar] [CrossRef]
- Varga, M.; Sass, M.; Papp, D.; Takács-Vellai, K.; Kobolak, J.; Dinnyés, A.; Klionsky, D.J.; Vellai, T. Autophagy Is Required for Zebrafish Caudal Fin Regeneration. Cell Death Differ. 2014, 21, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Saera-Vila, A.; Kish, P.E.; Louie, K.W.; Grzegorski, S.J.; Klionsky, D.J.; Kahana, A. Autophagy Regulates Cytoplasmic Remodeling during Cell Reprogramming in a Zebrafish Model of Muscle Regeneration. Autophagy 2016, 12, 1864–1875. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.-Y.; Ma, T.-L.; Hung, C.-C.; Tian, Y.-C.; Chen, Y.-C.; Yang, C.-W.; Cheng, Y.-C. Metformin Inhibits Cyst Formation in a Zebrafish Model of Polycystin-2 Deficiency. Sci. Rep. 2017, 7, 7161. [Google Scholar] [CrossRef]
- Zhu, P.; Sieben, C.J.; Xu, X.; Harris, P.C.; Lin, X. Autophagy Activators Suppress Cystogenesis in an Autosomal Dominant Polycystic Kidney Disease Model. Hum. Mol. Genet. 2017, 26, 158–172. [Google Scholar] [CrossRef]
- Hu, Z.-Y.; Chen, B.; Zhang, J.-P.; Ma, Y.-Y. Up-Regulation of Autophagy-Related Gene 5 (ATG5) Protects Dopaminergic Neurons in a Zebrafish Model of Parkinson’s Disease. J. Biol. Chem. 2017, 292, 18062–18074. [Google Scholar] [CrossRef]
- Gan-Or, Z.; Bouslam, N.; Birouk, N.; Lissouba, A.; Chambers, D.B.; Vérièpe, J.; Androschuk, A.; Laurent, S.B.; Rochefort, D.; Spiegelman, D.; et al. Mutations in CAPN1 Cause Autosomal-Recessive Hereditary Spastic Paraplegia. Am. J. Hum. Genet. 2016, 98, 1038–1046. [Google Scholar] [CrossRef]
- Zdebik, A.A.; Mahmood, F.; Stanescu, H.C.; Kleta, R.; Bockenhauer, D.; Russell, C. Epilepsy in Kcnj10 Morphant Zebrafish Assessed with a Novel Method for Long-Term EEG Recordings. PLoS ONE 2013, 8, e79765. [Google Scholar] [CrossRef] [PubMed]
- Sicca, F.; Ambrosini, E.; Marchese, M.; Sforna, L.; Servettini, I.; Valvo, G.; Brignone, M.S.; Lanciotti, A.; Moro, F.; Grottesi, A.; et al. Gain-of-Function Defects of Astrocytic Kir4.1 Channels in Children with Autism Spectrum Disorders and Epilepsy. Sci. Rep. 2016, 6, 34325. [Google Scholar] [CrossRef] [PubMed]
- Zinkevich, N.S.; Bosenko, D.V.; Link, B.A.; Semina, E.V. Laminin Alpha 1 Gene Is Essential for Normal Lens Development in Zebrafish. BMC Dev. Biol. 2006, 6, 13. [Google Scholar] [CrossRef]
- Karlstrom, R.O.; Trowe, T.; Klostermann, S.; Baier, H.; Brand, M.; Crawford, A.D.; Grunewald, B.; Haffter, P.; Hoffmann, H.; Meyer, S.U.; et al. Zebrafish Mutations Affecting Retinotectal Axon Pathfinding. Development 1996, 123, 427–438. [Google Scholar]
- Paulus, J.D.; Halloran, M.C. Zebrafish Bashful/Laminin-Alpha 1 Mutants Exhibit Multiple Axon Guidance Defects. Dev. Dyn. 2006, 235, 213–224. [Google Scholar] [CrossRef]
- Pollard, S.M.; Parsons, M.J.; Kamei, M.; Kettleborough, R.N.W.; Thomas, K.A.; Pham, V.N.; Bae, M.-K.; Scott, A.; Weinstein, B.M.; Stemple, D.L. Essential and Overlapping Roles for Laminin Alpha Chains in Notochord and Blood Vessel Formation. Dev. Biol. 2006, 289, 64–76. [Google Scholar] [CrossRef]
- Semina, E.V.; Bosenko, D.V.; Zinkevich, N.C.; Soules, K.A.; Hyde, D.R.; Vihtelic, T.S.; Willer, G.B.; Gregg, R.G.; Link, B.A. Mutations in Laminin Alpha 1 Result in Complex, Lens-Independent Ocular Phenotypes in Zebrafish. Dev. Biol. 2006, 299, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Biehlmaier, O.; Makhankov, Y.; Neuhauss, S.C.F. Impaired Retinal Differentiation and Maintenance in Zebrafish Laminin Mutants. Investig. Ophthalmol. Vis. Sci. 2007, 48, 2887–2894. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sztal, T.E.; Sonntag, C.; Hall, T.E.; Currie, P.D. Epistatic Dissection of Laminin-Receptor Interactions in Dystrophic Zebrafish Muscle. Hum. Mol. Genet. 2012, 21, 4718–4731. [Google Scholar] [CrossRef] [PubMed]
- Bryan, C.D.; Chien, C.-B.; Kwan, K.M. Loss of Laminin Alpha 1 Results in Multiple Structural Defects and Divergent Effects on Adhesion during Vertebrate Optic Cup Morphogenesis. Dev. Biol. 2016, 416, 324–337. [Google Scholar] [CrossRef] [PubMed]
- Rahn, J.J.; Bestman, J.E.; Stackley, K.D.; Chan, S.S.L. Zebrafish Lacking Functional DNA Polymerase Gamma Survive to Juvenile Stage, despite Rapid and Sustained Mitochondrial DNA Depletion, Altered Energetics and Growth. Nucleic Acids Res. 2015, 43, 10338–10352. [Google Scholar] [CrossRef] [PubMed]
- Facchinello, N.; Laquatra, C.; Locatello, L.; Beffagna, G.; Brañas Casas, R.; Fornetto, C.; Dinarello, A.; Martorano, L.; Vettori, A.; Risato, G.; et al. Efficient Clofilium Tosylate-Mediated Rescue of POLG-Related Disease Phenotypes in Zebrafish. Cell Death Dis. 2021, 12, 100. [Google Scholar] [CrossRef]
- Zecchin, E.; Mavropoulos, A.; Devos, N.; Filippi, A.; Tiso, N.; Meyer, D.; Peers, B.; Bortolussi, M.; Argenton, F. Evolutionary Conserved Role of Ptf1a in the Specification of Exocrine Pancreatic Fates. Dev. Biol. 2004, 268, 174–184. [Google Scholar] [CrossRef]
- Lin, J.W.; Biankin, A.V.; Horb, M.E.; Ghosh, B.; Prasad, N.B.; Yee, N.S.; Pack, M.A.; Leach, S.D. Differential Requirement for Ptf1a in Endocrine and Exocrine Lineages of Developing Zebrafish Pancreas. Dev. Biol. 2004, 274, 491–503. [Google Scholar] [CrossRef]
- Dong, P.D.S.; Provost, E.; Leach, S.D.; Stainier, D.Y.R. Graded Levels of Ptf1a Differentially Regulate Endocrine and Exocrine Fates in the Developing Pancreas. Genes Dev. 2008, 22, 1445–1450. [Google Scholar] [CrossRef]
- Jusuf, P.R.; Almeida, A.D.; Randlett, O.; Joubin, K.; Poggi, L.; Harris, W.A. Origin and Determination of Inhibitory Cell Lineages in the Vertebrate Retina. J. Neurosci. 2011, 31, 2549–2562. [Google Scholar] [CrossRef] [PubMed]
- Randlett, O.; MacDonald, R.B.; Yoshimatsu, T.; Almeida, A.D.; Suzuki, S.C.; Wong, R.O.; Harris, W.A. Cellular Requirements for Building a Retinal Neuropil. Cell Rep. 2013, 3, 282–290. [Google Scholar] [CrossRef]
- Pashos, E.; Park, J.T.; Leach, S.; Fisher, S. Distinct Enhancers of Ptf1a Mediate Specification and Expansion of Ventral Pancreas in Zebrafish. Dev. Biol. 2013, 381, 471–481. [Google Scholar] [CrossRef]
- Lattante, S.; de Calbiac, H.; Le Ber, I.; Brice, A.; Ciura, S.; Kabashi, E. Sqstm1 Knock-down Causes a Locomotor Phenotype Ameliorated by Rapamycin in a Zebrafish Model of ALS/FTLD. Hum. Mol. Genet. 2015, 24, 1682–1690. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Yang, D.; He, Q.; Songyang, Z. Zebrafish as a Model System to Study the Physiological Function of Telomeric Protein TPP1. PLoS ONE 2011, 6, e16440. [Google Scholar] [CrossRef]
- Mahmood, F.; Fu, S.; Cooke, J.; Wilson, S.W.; Cooper, J.D.; Russell, C. A Zebrafish Model of CLN2 Disease Is Deficient in Tripeptidyl Peptidase 1 and Displays Progressive Neurodegeneration Accompanied by a Reduction in Proliferation. Brain 2013, 136, 1488–1507. [Google Scholar] [CrossRef]
- Mahmood, F.; Zdebik, A.; Au, A.; Cooke, J.; Russell, C. Valproic ACID Extends Lifespan of the Zebrafish Model of CLN2 Disease (Late Infantile Neuronal Ceroid Lipofuscinosis). J. Neurol. Neurosurg. Psychiatry 2015, 86, 86:e4. [Google Scholar] [CrossRef]
- Colin, E.; Daniel, J.; Ziegler, A.; Wakim, J.; Scrivo, A.; Haack, T.B.; Khiati, S.; Denommé, A.-S.; Amati-Bonneau, P.; Charif, M.; et al. Biallelic Variants in UBA5 Reveal That Disruption of the UFM1 Cascade Can Result in Early-Onset Encephalopathy. Am. J. Hum. Genet. 2016, 99, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Tsuruwaka, Y.; Konishi, M.; Shimada, E. Loss of Wwox Expression in Zebrafish Embryos Causes Edema and Alters Ca(2+) Dynamics. PeerJ 2015, 3, e727. [Google Scholar] [CrossRef] [PubMed]
- Schwend, T.; Loucks, E.J.; Snyder, D.; Ahlgren, S.C. Requirement of Npc1 and Availability of Cholesterol for Early Embryonic Cell Movements in Zebrafish. J. Lipid Res. 2011, 52, 1328–1344. [Google Scholar] [CrossRef]
- Louwette, S.; Régal, L.; Wittevrongel, C.; Thys, C.; Vandeweeghde, G.; Decuyper, E.; Leemans, P.; De Vos, R.; Van Geet, C.; Jaeken, J.; et al. NPC1 Defect Results in Abnormal Platelet Formation and Function: Studies in Niemann-Pick Disease Type C1 Patients and Zebrafish. Hum. Mol. Genet. 2013, 22, 61–73. [Google Scholar] [CrossRef]
- Lin, Y.; Cai, X.; Wang, G.; Ouyang, G.; Cao, H. Model Construction of Niemann-Pick Type C Disease in Zebrafish. Biol. Chem. 2018, 399, 903–910. [Google Scholar] [CrossRef]
- Tseng, W.-C.; Loeb, H.E.; Pei, W.; Tsai-Morris, C.-H.; Xu, L.; Cluzeau, C.V.; Wassif, C.A.; Feldman, B.; Burgess, S.M.; Pavan, W.J.; et al. Modeling Niemann-Pick Disease Type C1 in Zebrafish: A Robust Platform for in Vivo Screening of Candidate Therapeutic Compounds. Dis. Model. Mech. 2018, 11. [Google Scholar] [CrossRef]
- Huang, M.-S.; Wang, T.-K.; Liu, Y.-W.; Li, Y.-T.; Chi, T.-H.; Chou, C.-W.; Hsieh, M. Roles of Carbonic Anhydrase 8 in Neuronal Cells and Zebrafish. Biochim. Biophys. Acta 2014, 1840, 2829–2842. [Google Scholar] [CrossRef]
- Khanna, H.; Davis, E.E.; Murga-Zamalloa, C.A.; Estrada-Cuzcano, A.; Lopez, I.; den Hollander, A.I.; Zonneveld, M.N.; Othman, M.I.; Waseem, N.; Chakarova, C.F.; et al. A Common Allele in RPGRIP1L Is a Modifier of Retinal Degeneration in Ciliopathies. Nat. Genet. 2009, 41, 739–745. [Google Scholar] [CrossRef]
- Bachmann-Gagescu, R.; Phelps, I.G.; Stearns, G.; Link, B.A.; Brockerhoff, S.E.; Moens, C.B.; Doherty, D. The Ciliopathy Gene Cc2d2a Controls Zebrafish Photoreceptor Outer Segment Development through a Role in Rab8-Dependent Vesicle Trafficking. Hum. Mol. Genet. 2011, 20, 4041–4055. [Google Scholar] [CrossRef] [PubMed]
- Murga-Zamalloa, C.A.; Ghosh, A.K.; Patil, S.B.; Reed, N.A.; Chan, L.S.; Davuluri, S.; Peränen, J.; Hurd, T.W.; Rachel, R.A.; Khanna, H. Accumulation of the Raf-1 Kinase Inhibitory Protein (Rkip) Is Associated with Cep290-Mediated Photoreceptor Degeneration in Ciliopathies. J. Biol. Chem. 2011, 286, 28276–28286. [Google Scholar] [CrossRef]
- Luo, N.; Lu, J.; Sun, Y. Evidence of a Role of Inositol Polyphosphate 5-Phosphatase INPP5E in Cilia Formation in Zebrafish. Vision Res. 2012, 75, 98–107. [Google Scholar] [CrossRef]
- Song, P.; Dudinsky, L.; Fogerty, J.; Gaivin, R.; Perkins, B.D. Arl13b Interacts With Vangl2 to Regulate Cilia and Photoreceptor Outer Segment Length in Zebrafish. Investig. Ophthalmol. Vis. Sci. 2016, 57, 4517–4526. [Google Scholar] [CrossRef] [PubMed]
- Ojeda Naharros, I.; Cristian, F.B.; Zang, J.; Gesemann, M.; Ingham, P.W.; Neuhauss, S.C.F.; Bachmann-Gagescu, R. The Ciliopathy Protein TALPID3/KIAA0586 Acts Upstream of Rab8 Activation in Zebrafish Photoreceptor Outer Segment Formation and Maintenance. Sci. Rep. 2018, 8, 2211. [Google Scholar] [CrossRef] [PubMed]
- Lessieur, E.M.; Song, P.; Nivar, G.C.; Piccillo, E.M.; Fogerty, J.; Rozic, R.; Perkins, B.D. Ciliary Genes Arl13b, Ahi1 and Cc2d2a Differentially Modify Expression of Visual Acuity Phenotypes but Do Not Enhance Retinal Degeneration Due to Mutation of Cep290 in Zebrafish. PLoS ONE 2019, 14, e0213960. [Google Scholar] [CrossRef] [PubMed]
- Simms, R.J.; Hynes, A.M.; Eley, L.; Inglis, D.; Chaudhry, B.; Dawe, H.R.; Sayer, J.A. Modelling a Ciliopathy: Ahi1 Knockdown in Model Systems Reveals an Essential Role in Brain, Retinal, and Renal Development. Cell Mol. Life Sci. 2012, 69, 993–1009. [Google Scholar] [CrossRef]
- Lessieur, E.M.; Fogerty, J.; Gaivin, R.J.; Song, P.; Perkins, B.D. The Ciliopathy Gene Ahi1 Is Required for Zebrafish Cone Photoreceptor Outer Segment Morphogenesis and Survival. Investig. Ophthalmol. Vis. Sci. 2017, 58, 448–460. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhu, L.; Chen, L.; Yan, L.; Perkins, B.D.; Li, S.; Li, B.; Xu, H.A.; Li, X.-J. Mutant Ahi1 Affects Retinal Axon Projection in Zebrafish via Toxic Gain of Function. Front. Cell. Neurosci. 2019, 13, 81. [Google Scholar] [CrossRef]
- Sayer, J.A.; Otto, E.A.; O’Toole, J.F.; Nurnberg, G.; Kennedy, M.A.; Becker, C.; Hennies, H.C.; Helou, J.; Attanasio, M.; Fausett, B.V.; et al. The Centrosomal Protein Nephrocystin-6 Is Mutated in Joubert Syndrome and Activates Transcription Factor ATF4. Nat. Genet. 2006, 38, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, T.; Pütz, M.; Lienkamp, S.; Ganner, A.; Bergbreiter, A.; Ramachandran, H.; Gieloff, V.; Gerner, M.; Mattonet, C.; Czarnecki, P.G.; et al. Genetic and Physical Interaction between the NPHP5 and NPHP6 Gene Products. Hum. Mol. Genet. 2008, 17, 3655–3662. [Google Scholar] [CrossRef] [PubMed]
- Tobin, J.L.; Beales, P.L. Restoration of Renal Function in Zebrafish Models of Ciliopathies. Pediatric Nephrol. 2008, 23, 2095–2099. [Google Scholar] [CrossRef]
- Pearson, C.G.; Osborn, D.P.S.; Giddings, T.H.; Beales, P.L.; Winey, M. Basal Body Stability and Ciliogenesis Requires the Conserved Component Poc1. J. Cell Biol. 2009, 187, 905–920. [Google Scholar] [CrossRef]
- Beck, B.B.; Phillips, J.B.; Bartram, M.P.; Wegner, J.; Thoenes, M.; Pannes, A.; Sampson, J.; Heller, R.; Göbel, H.; Koerber, F.; et al. Mutation of POC1B in a Severe Syndromic Retinal Ciliopathy. Hum. Mutat. 2014, 35, 1153–1162. [Google Scholar] [CrossRef] [PubMed]
- Tuz, K.; Bachmann-Gagescu, R.; O’Day, D.R.; Hua, K.; Isabella, C.R.; Phelps, I.G.; Stolarski, A.E.; O’Roak, B.J.; Dempsey, J.C.; Lourenco, C.; et al. Mutations in CSPP1 Cause Primary Cilia Abnormalities and Joubert Syndrome with or without Jeune Asphyxiating Thoracic Dystrophy. Am. J. Hum. Genet. 2014, 94, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kecskés, A.; Copmans, D.; Langlois, M.; Crawford, A.D.; Ceulemans, B.; Lagae, L.; de Witte, P.A.M.; Esguerra, C.V. Pharmacological Characterization of an Antisense Knockdown Zebrafish Model of Dravet Syndrome: Inhibition of Epileptic Seizures by the Serotonin Agonist Fenfluramine. PLoS ONE 2015, 10, e0125898. [Google Scholar] [CrossRef] [PubMed]
- Roosing, S.; Rosti, R.O.; Rosti, B.; de Vrieze, E.; Silhavy, J.L.; van Wijk, E.; Wakeling, E.; Gleeson, J.G. Identification of a Homozygous Nonsense Mutation in KIAA0556 in a Consanguineous Family Displaying Joubert Syndrome. Hum. Genet. 2016, 135, 919–921. [Google Scholar] [CrossRef]
- Frikstad, K.-A.M.; Molinari, E.; Thoresen, M.; Ramsbottom, S.A.; Hughes, F.; Letteboer, S.J.F.; Gilani, S.; Schink, K.O.; Stokke, T.; Geimer, S.; et al. A CEP104-CSPP1 Complex Is Required for Formation of Primary Cilia Competent in Hedgehog Signaling. Cell. Rep. 2019, 28, 1907–1922. [Google Scholar] [CrossRef] [PubMed]
- Kasher, P.R.; Namavar, Y.; van Tijn, P.; Fluiter, K.; Sizarov, A.; Kamermans, M.; Grierson, A.J.; Zivkovic, D.; Baas, F. Impairment of the TRNA-Splicing Endonuclease Subunit 54 (Tsen54) Gene Causes Neurological Abnormalities and Larval Death in Zebrafish Models of Pontocerebellar Hypoplasia. Hum. Mol. Genet. 2011, 20, 1574–1584. [Google Scholar] [CrossRef] [PubMed]
- Charlesworth, G.; Balint, B.; Mencacci, N.E.; Carr, L.; Wood, N.W.; Bhatia, K.P. SLC25A46 Mutations Underlie Progressive Myoclonic Ataxia with Optic Atrophy and Neuropathy. Mov. Disord. 2016, 31, 1249–1251. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Steffen, J.; Yourshaw, M.; Mamsa, H.; Andersen, E.; Rudnik-Schöneborn, S.; Pope, K.; Howell, K.B.; McLean, C.A.; Kornberg, A.J.; et al. Loss of Function of SLC25A46 Causes Lethal Congenital Pontocerebellar Hypoplasia. Brain 2016, 139, 2877–2890. [Google Scholar] [CrossRef] [PubMed]
- Abrams, A.J.; Hufnagel, R.B.; Rebelo, A.; Zanna, C.; Patel, N.; Gonzalez, M.A.; Campeanu, I.J.; Griffin, L.B.; Groenewald, S.; Strickland, A.V.; et al. Mutations in SLC25A46, Encoding a UGO1-like Protein, Cause an Optic Atrophy Spectrum Disorder. Nat. Genet. 2015, 47, 926–932. [Google Scholar] [CrossRef]
- Buglo, E.; Sarmiento, E.; Martuscelli, N.B.; Sant, D.W.; Danzi, M.C.; Abrams, A.J.; Dallman, J.E.; Züchner, S. Genetic Compensation in a Stable Slc25a46 Mutant Zebrafish: A Case for Using F0 CRISPR Mutagenesis to Study Phenotypes Caused by Inherited Disease. PLoS ONE 2020, 15, e0230566. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome Engineering Using the CRISPR-Cas9 System. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]



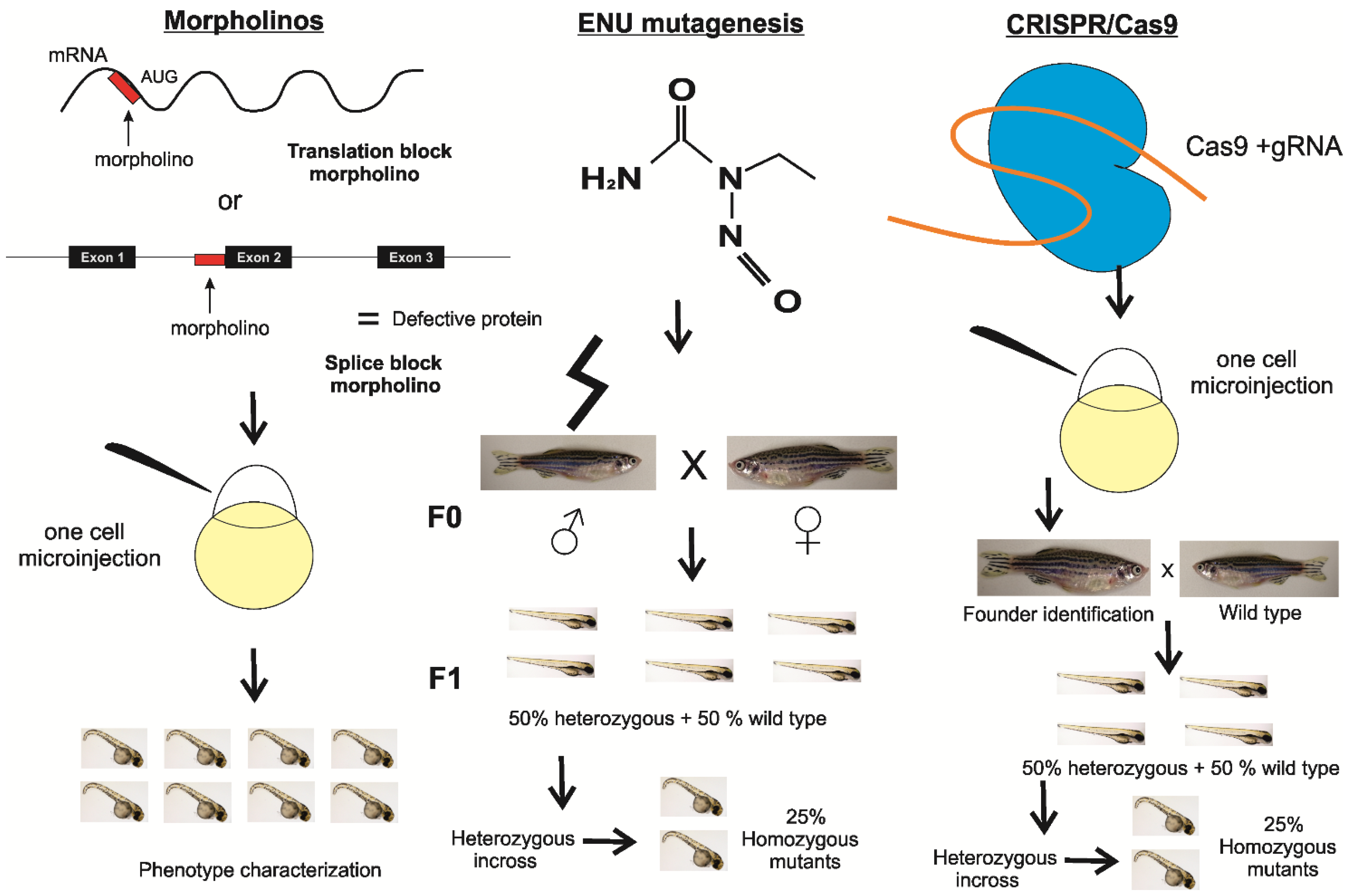{kind=link}
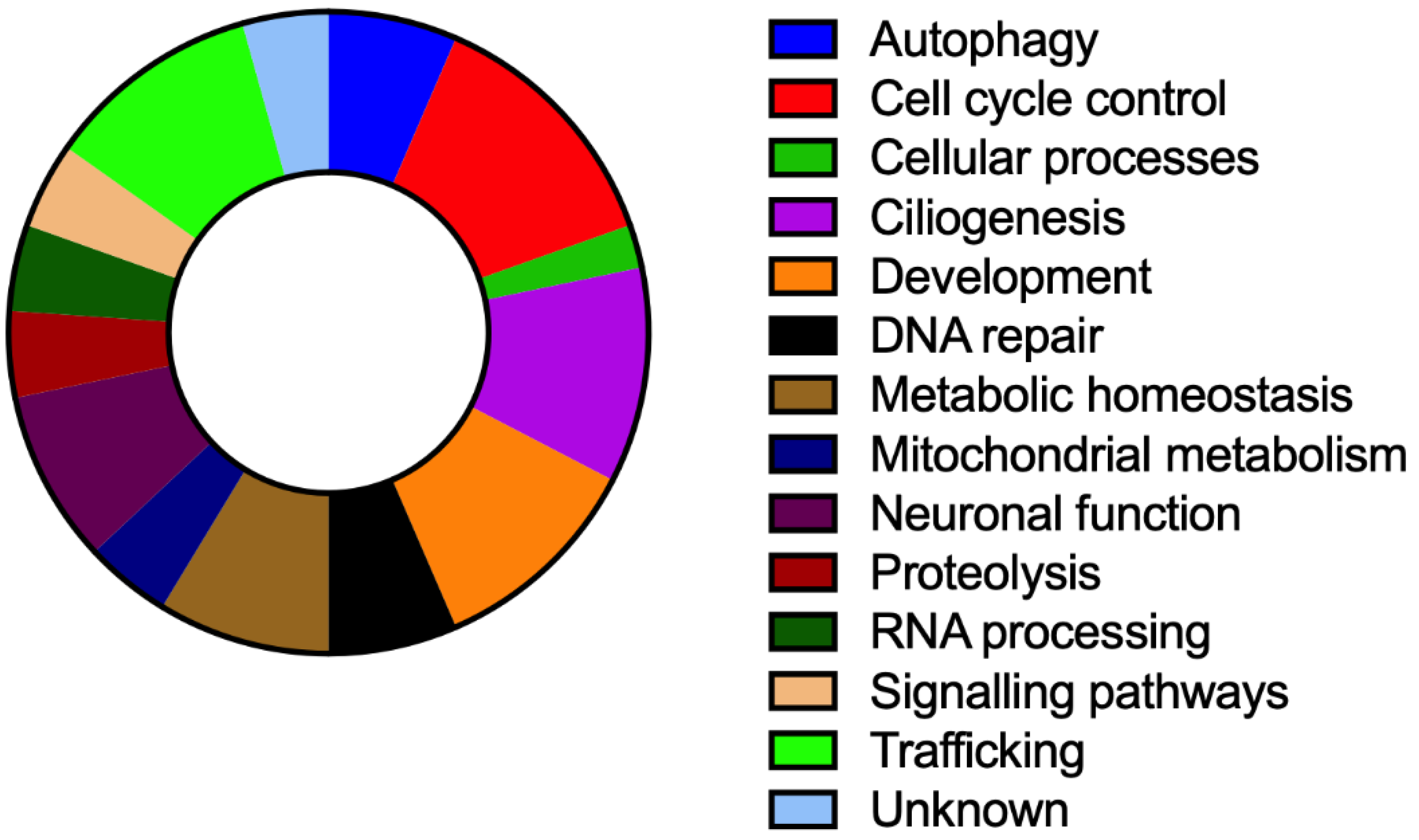{kind=link}
| Disease | Gene | Protein Function | Clinical Features Other Than Ataxia * | References |
|---|---|---|---|---|
| Ataxia-telangiectasia (AT) | ATM | Cellular responses to DNA damage and cell cycle control | Oculocutaneous telangiectasia, radiosensitivity, predisposition to lymphoid malignancies, cellular and humoral immunodeficiency | [24] |
| Boucher–Neuhäuser syndrome; Spastic paraplegia type 39 (SPG39) | PNPLA6 | Phospholipase | Hypogonadotropic hypogonadism, chorioretinal dystrophy | [25] |
| Marinesco-Sjögren syndrome | SIL1 | Nuclear exchange factor for the endoplasmic reticulum resident chaperone BiP | Cataracts, skeletal muscle myopathy, developmental delay | [26] |
| Spinocerebellar ataxia autosomal recessive type 20 (SCAR20) | SNX14 | Autophagosome function | Intellectual disability, coarse facial features, hearing loss, epilepsy, macrocephaly | [27] |
| Ataxia with isolated vitamin E deficiency (AVED) | TTPA | Distribution of vitamin E | Hyporeflexia, decreased vibration sense, cardiomyopath, retinitis pigmentosa | [28] |
| Wolfram syndrome (WFS) | WFS1 | Membrane trafficking, endoplasmic reticulum stress and calcium homeostasis | Mental retardation, seizures, psychiatric symptoms, pigmentary retinopathy, Diabetes mellitus, optic atrophy, sensorineural hearing loss, urinary tract abnormalities | [29,30] |
| Polyneuropathy, hearing loss, ataxia, retinitis pigmentosa and cataract (PHARC) | ABHD12 | Endocannabinoid and phospholipid metabolism | Cataracts, hearing loss, retinitis pigmentosa, demyelinating sensorimotor polyneuropathy | [31] |
| Cayman ataxia | ATCAY | Neural development | Hypotonia, psychomotor retardation | [32] |
| Spinocerebellar ataxia autosomal recessive type 25 (SCAR25) | ATG5 | Autophagy | Mental retardation, developmental delay | [33] |
| Spastic paraplegia type 76 (SPG76) | CAPN1 | Neuronal plasticity, migration, microtubular regulation | Skeletal abnormalities, peripheral neuropathy, amyotrophy, spastic paraplegia | [34] |
| Spinocerebellar ataxia autosomal recessive (SCAR17) | CWF19L1 | Cell Cycle Control | Developmental delay, mental retardation | [35] |
| EAST syndrome | KCNJ10 | Potassium channel | Short stature, epilepsy, sensorineural deafness, mental retardation, renal tubulopathy | [36,37] |
| Poretti-Boltshauser syndrome | LAMA1 | Cell attachment, migration | Retinal dystrophy | [38] |
| Mitochondrial recessive ataxia syndrome (MIRAS) | POLG | Catalytic subunit of mitochondrial DNA polymerase | Ophthalmoplegia, polyneuropathy, myopathy, encephalopathy, liver failure, muscle pain, sensorineural hearing loss, epilepsy | [39,40] |
| Pancreatic and cerebellar agenesis (PACA) | PTF1A | Pancreatic development | Neonatal diabetes mellitus, pancreatic and cerebellar agenesis | [41] |
| Gordon Holmes syndrome | RNF216 | Ubiquitination | Chorea, dementia, hypogonadotropic hypogonadism | [42] |
| Childhood-onset neurodegeneration with ataxia, dystonia, and gaze palsy (NADGP) | SQSTM1 | Intracellular signalling, oxidative stress response, apoptosis and autophagy | Dystonia, gaze palsy, dyskinesia, cognitive decline | [43] |
| Spinocerebellar ataxia autosomal recessive type 7 (SCAR7), Ceroid lipofuscinosis neuronal 2 (CLN2) | TPP1 | Serine peptidase | SCAR7: Slowly progressive spasticity CLN2: Rapidly progressive myoclonus, developmental regression, early death, epilepsy, loss of vision | [44] |
| Spinocerebellar ataxia autosomal recessive type 24 (SCAR24) | UBA5 | Ubiquitin-like | Cataracts, demyelinating sensorimotor neuropathy | [45] |
| Galloway-Mowat syndrome | WDR73 | Unknown | Intellectual disability, short stature, microcephaly, facial dysmorphism, hypotonia, nephrotic synrome, cataracts, skin hypopigmentation | [46] |
| Spinocerebellar ataxia autosomal recessive type 12 (SCAR12) | WWOX | DNA repair | Epilepsy, mental retardation, developmental delay. microcephaly | [47,48,49] |
| Niemann–Pick disease type C (NPC) | NPC1 | Cholesterol trafficking | Learning difficulties, neuropsychiatric symptoms, dementia, seizures, dystonia, supranuclear gaze palsy, neonatal cholestasis, hepatosplenomegaly | [50] |
| Cerebellar ataxia and mental retardation with or without quadrupedal locomotion 3 (CAMRQ3) | CA8 | Unknown | Mental retardation quadrupedal gait | [51,52] |
| Joubert syndrome | Several genes | Diverse functions | Hypotonia, Developmental delay, oculomotor apraxia, breathing dysregulation, retinal abnormalities, renal cysts, polydactily, hepatic fibrosis | [53] |
| Pontocerebellar hypoplasia | Several genes | Diverse functions | Hypoplasia of the cerebellum or the ventral pons, progressive microcephaly, motorneuron disease, resiratory insuficiency, mental retardation | [54] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quelle-Regaldie, A.; Sobrido-Cameán, D.; Barreiro-Iglesias, A.; Sobrido, M.J.; Sánchez, L. Zebrafish Models of Autosomal Recessive Ataxias. Cells 2021, 10, 836. https://doi.org/10.3390/cells10040836
Quelle-Regaldie A, Sobrido-Cameán D, Barreiro-Iglesias A, Sobrido MJ, Sánchez L. Zebrafish Models of Autosomal Recessive Ataxias. Cells. 2021; 10(4):836. https://doi.org/10.3390/cells10040836
Chicago/Turabian StyleQuelle-Regaldie, Ana, Daniel Sobrido-Cameán, Antón Barreiro-Iglesias, María Jesús Sobrido, and Laura Sánchez. 2021. "Zebrafish Models of Autosomal Recessive Ataxias" Cells 10, no. 4: 836. https://doi.org/10.3390/cells10040836
APA StyleQuelle-Regaldie, A., Sobrido-Cameán, D., Barreiro-Iglesias, A., Sobrido, M. J., & Sánchez, L. (2021). Zebrafish Models of Autosomal Recessive Ataxias. Cells, 10(4), 836. https://doi.org/10.3390/cells10040836