
Concentrated Secretome of Adipose Stromal Cells Limits Influenza A Virus-Induced Lung Injury in Mice


Abstract
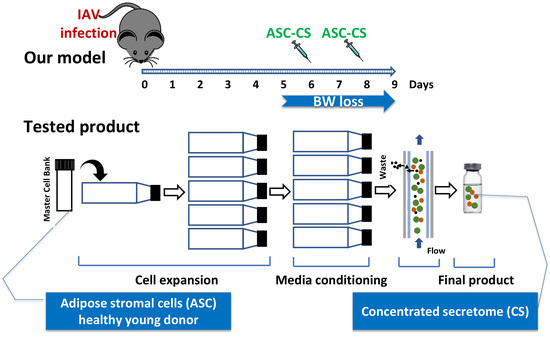
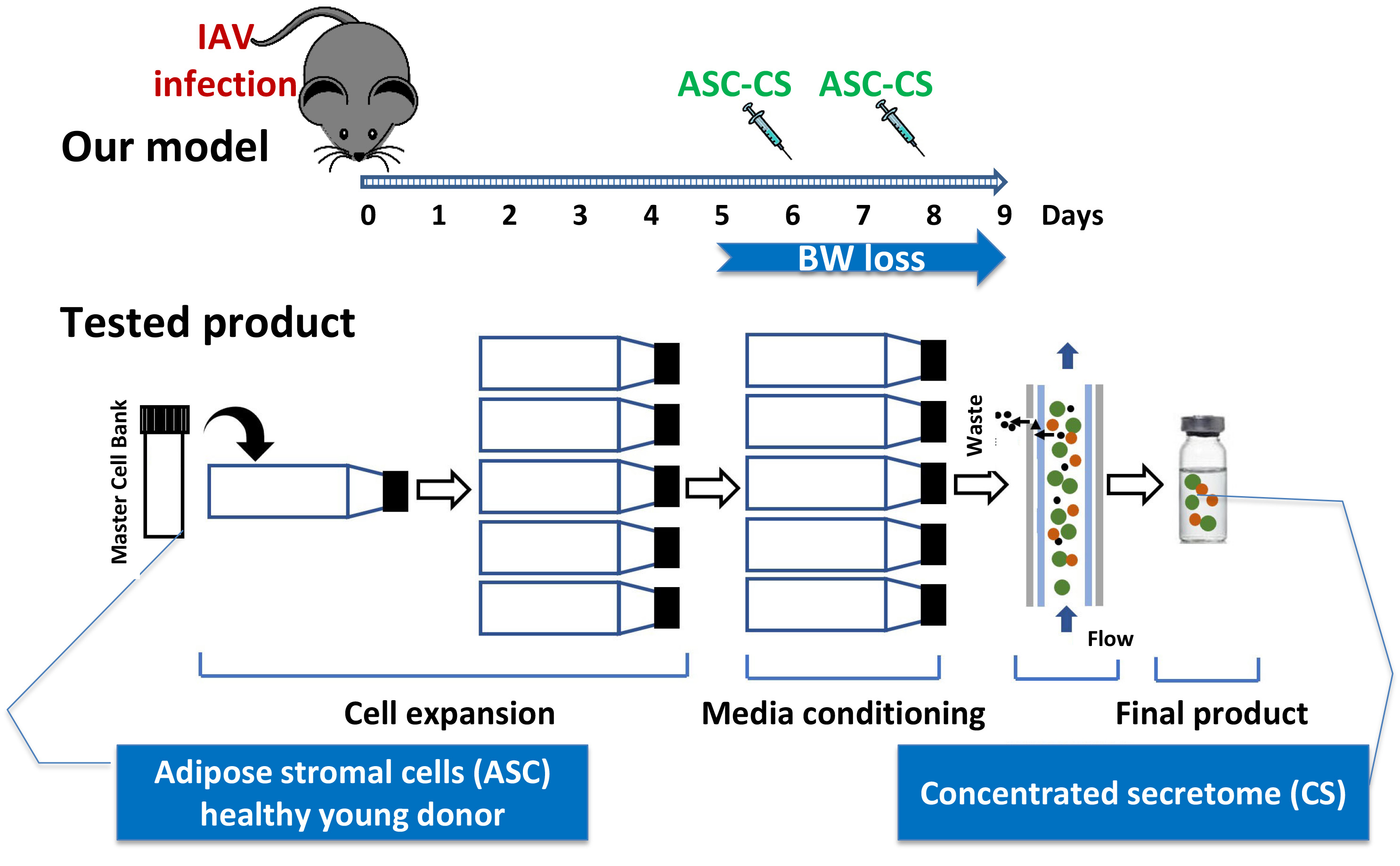{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Preparation of Secretome
2.3. Animals
2.4. Viral Infection, Administration of Therapeutic Material, and Assessment of Viral Antigen
2.5. Blood Oxygenation Monitoring
2.6. Analysis of Lung Injury Indices
2.7. Analysis of Angiopoietin 2 (Angpt2) and PDL1 Pathway
2.8. Statistical Analyses
3. Results
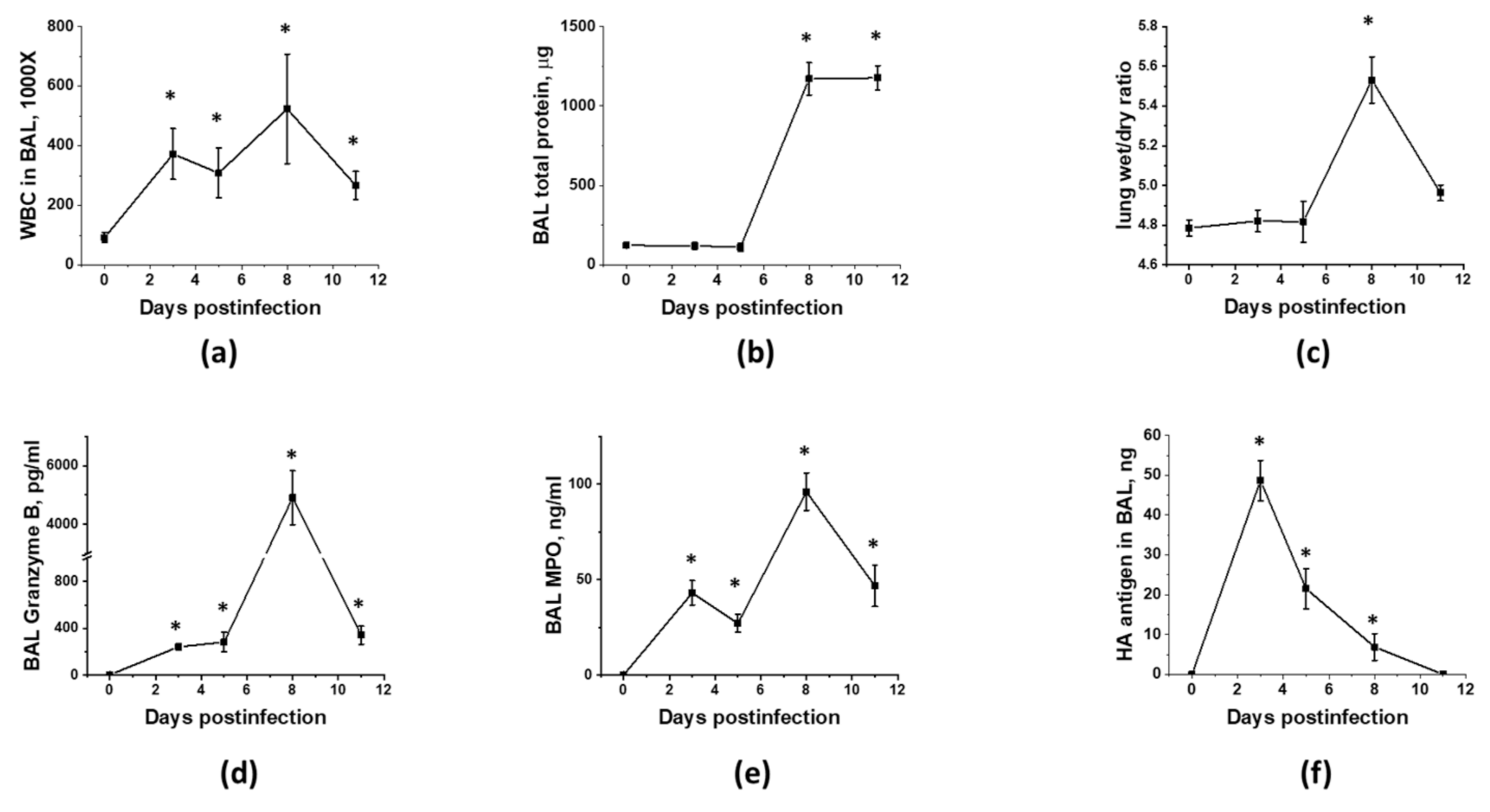
3.1. Optimization of the Timeline for Therapy Administration
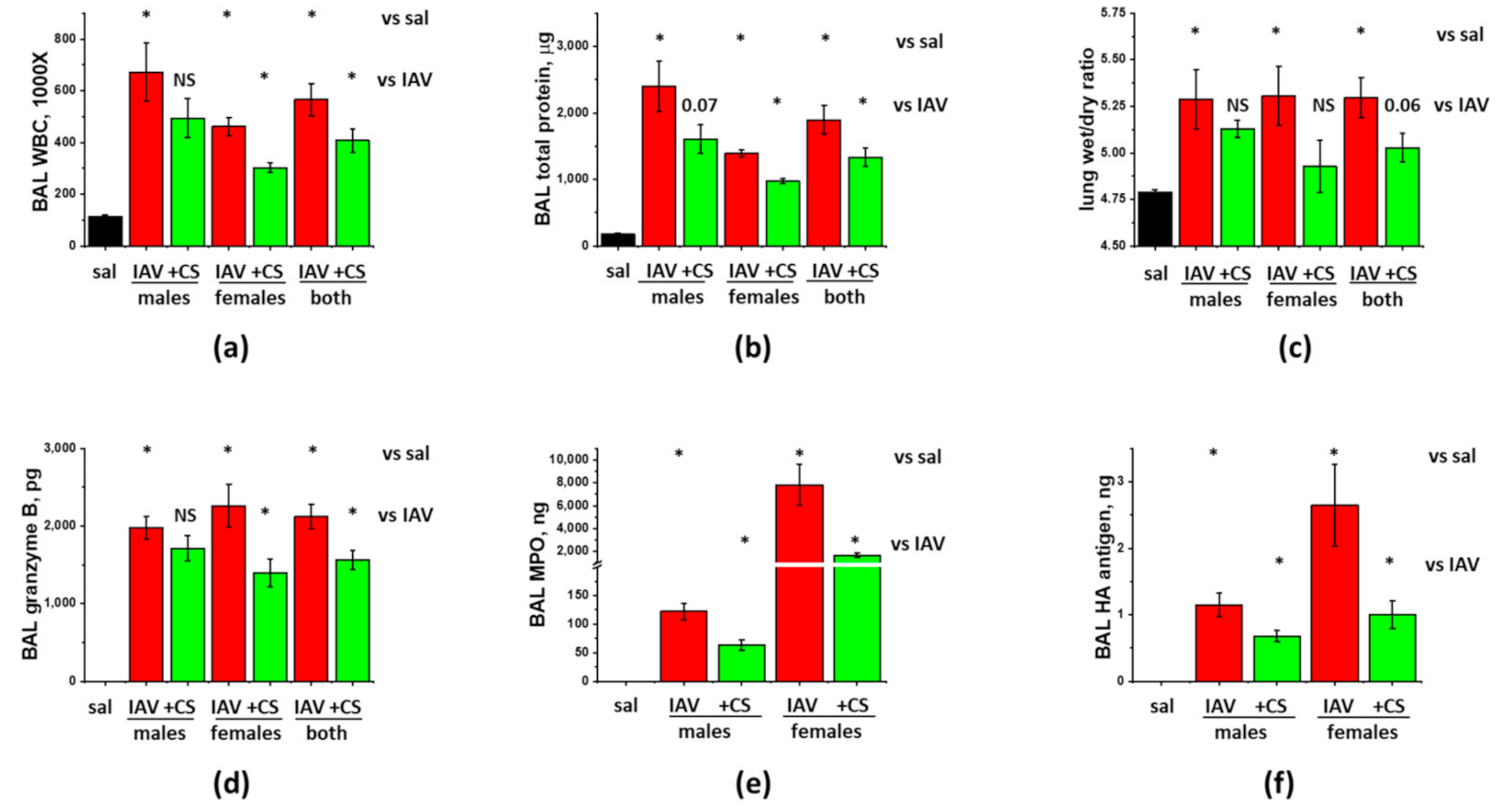
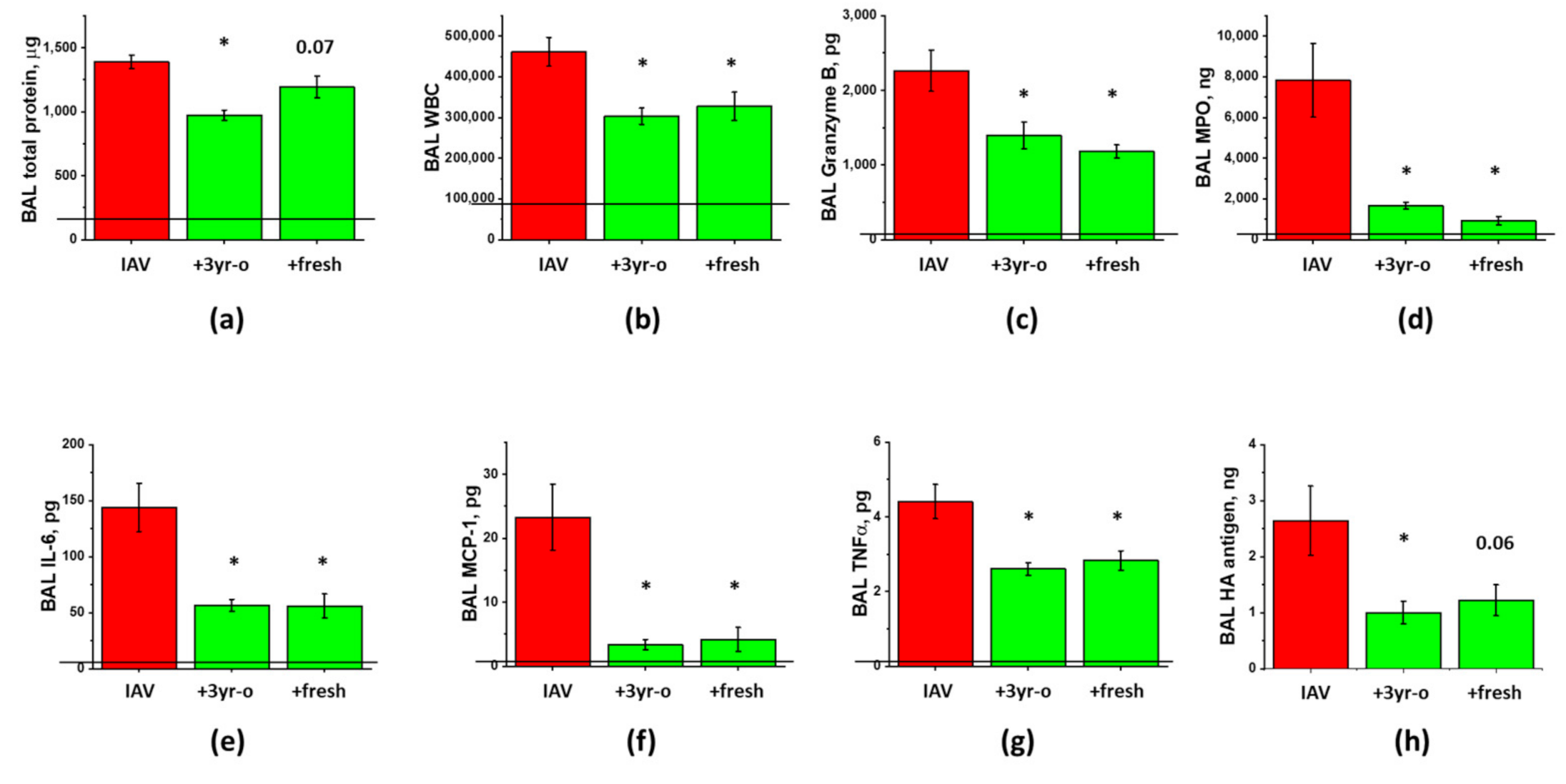
3.2. Sex-Dependent Responses to ASC-CS in the Model of IAV-Induced Lung Injury
3.3. The Effect of the Long-Term Storage at −80 °C on ARDS-Mitigating Potency of ASC Secretome Preparations
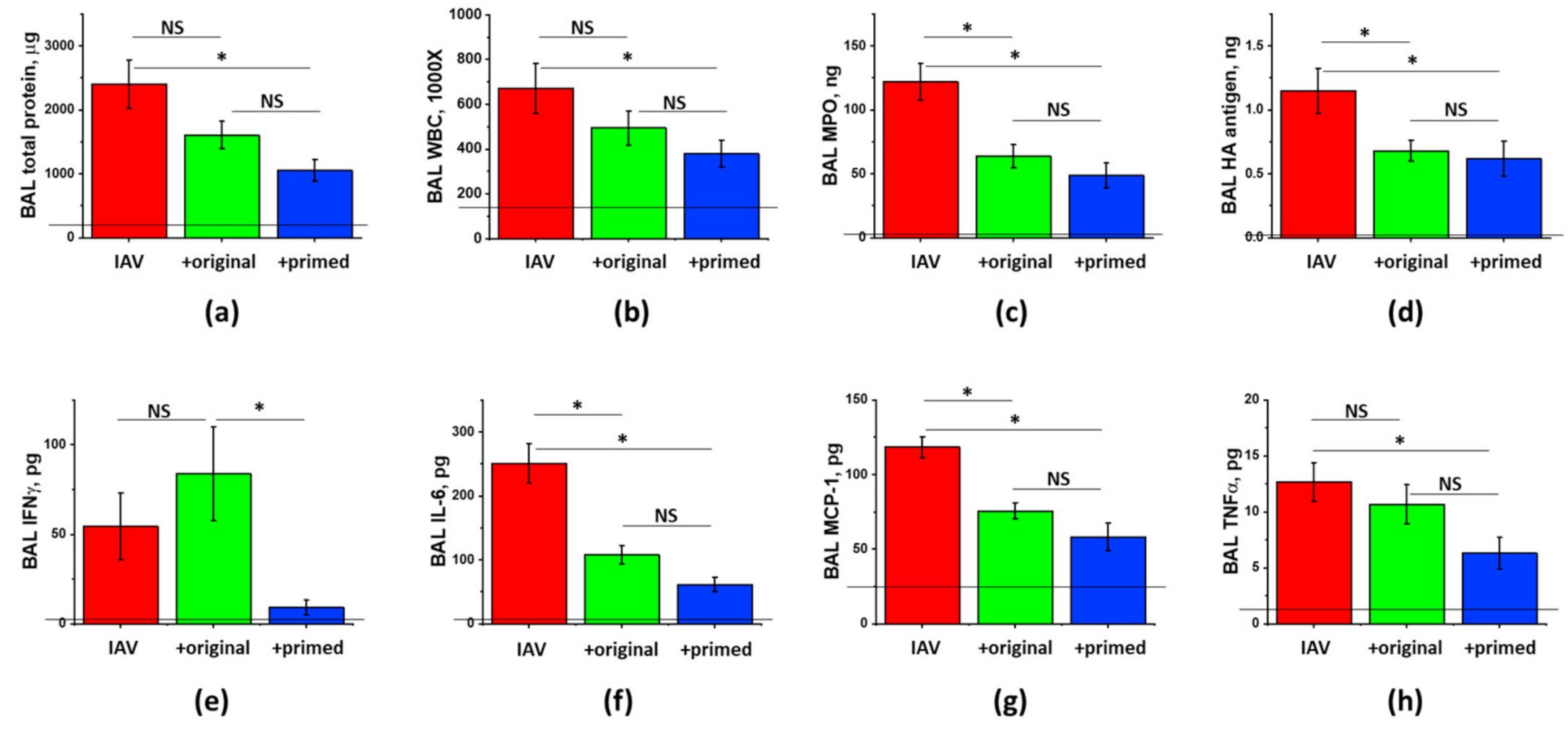
3.4. The Effect of ASC Inflammatory Priming on the ARDS-Mitigating Potency of ASC Secretome Preparations
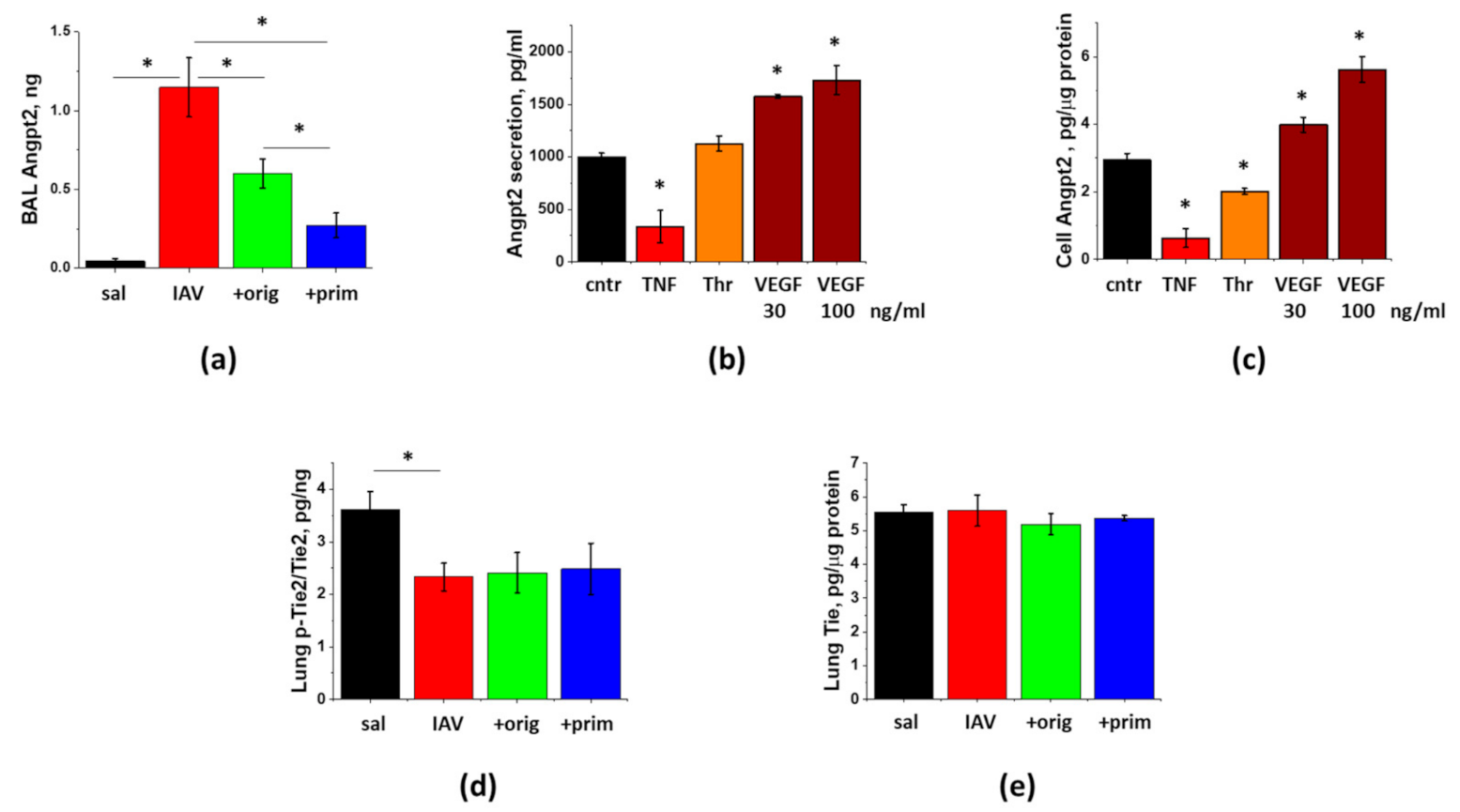
3.5. IAV-Induced Lung Injury Is Associated with Angpt2 Release, Which Can Be Suppressed by ASC Secretome
3.6. PDL1 Ablation during Lung Injury Phase Does Not Attenuate Angpt2 Release, Protein Leak, or Inflammation in IAV-Infected Mice
4. Discussion
5. Conclusions
6. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Angpt2 | Angiopoietin 2 |
| ARDS | acute respiratory distress syndrome |
| ASC | adipose stromal cells |
| BAL | bronchoalveolar lavage |
| BM | bone marrow |
| cGMP | current Good Manufacturing Practices |
| CS | concentrated secretome |
| HPAEC | human pulmonary artery endothelial cells |
| IAV | influenza A virus |
| ICU | intensive care unit |
| MPO | myeloperoxidase |
| MSC | mesenchymal stromal cells |
| PDL1 | programmed cell death ligand |
| SQ | subcutaneous |
| WBC | white blood cells |
References
- Cassini, A.; Colzani, E.; Pini, A.; Mangen, M.-J.J.; Plass, D.; McDonald, S.A.; Maringhini, G.; Van Lier, A.; Haagsma, J.A.; Havelaar, A.H.; et al. Impact of infectious diseases on population health using incidence-based disability-adjusted life years (DALYs): Results from the Burden of Communicable Diseases in Europe study, European Union and European Economic Area countries, 2009 to 2013. Eurosurveillance 2018, 23, 17–00454. [Google Scholar] [CrossRef]
- Troeger, C.E.; Blacker, B.F.; Khalil, I.A.; Zimsen, S.R.M.; Albertson, S.B.; Abate, D.; Abdela, J.; Adhikari, T.B.; Aghayan, S.A.; Agrawal, S.; et al. Mortality, morbidity, and hospitalisations due to influenza lower respiratory tract infections, 2017: An analysis for the Global Burden of Disease Study 2017. Lancet Respir. Med. 2019, 7, 69–89. [Google Scholar] [CrossRef]
- Beumer, M.; Koch, R.; Van Beuningen, D.; Oudelashof, A.; Van De Veerdonk, F.; Kolwijck, E.; Van Der Hoeven, J.; Bergmans, D.; Hoedemaekers, C. Influenza virus and factors that are associated with ICU admission, pulmonary co-infections and ICU mortality. J. Crit. Care 2019, 50, 59–65. [Google Scholar] [CrossRef]
- Ebell, M.H. Baloxavir Reduces Symptom Duration Similar to Oseltamivir, Primarily Within 24 Hours of Symptom Onset. Am. Fam. Physician 2019, 100. [Google Scholar]
- Muthuri, S.G.; Venkatesan, S.; Myles, P.R.; Leonardi-Bee, J.; Lim, W.S.; Al Mamun, A.; Anovadiya, A.P.; Araújo, W.N.; Azziz-Baumgartner, E.; Báez, C.; et al. Impact of neuraminidase inhibitors on influenza A(H1N1)pdm09-related pneumonia: An individual participant data meta-analysis. Influ. Other Respir. Viruses 2016, 10, 192–204. [Google Scholar] [CrossRef]
- Viasus, D.; Paño-Pardo, J.R.; Cordero, E.; Campins, A.; López-Medrano, F.; Villoslada, A.; Fariñas, M.C.; Moreno, A.; Rodríguez-Baño, J.; Oteo, J.A. Effect of immunomodulatory therapies in patients with pandemic influenza A (H1N1) 2009 complicated by pneumonia. J. Infect. 2011, 62, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, H.; Horie, S.; Laffey, J.G. Emerging cellular and pharmacologic therapies for acute respiratory distress syndrome. Curr. Opin. Crit. Care 2021, 27, 20–28. [Google Scholar] [CrossRef]
- Huppert, L.A.; Matthay, M.A.; Ware, L.B. Pathogenesis of Acute Respiratory Distress Syndrome. Semin. Respir. Crit. Care Med. 2019, 40, 031–039. [Google Scholar] [CrossRef] [PubMed]
- Matthay, M.A.; Pati, S.; Lee, J.-W. Concise Review: Mesenchymal Stem (Stromal) Cells: Biology and Preclinical Evidence for Therapeutic Potential for Organ Dysfunction Following Trauma or Sepsis. Stem Cells 2017, 35, 316–324. [Google Scholar] [CrossRef]
- Matthay, M.A.; Calfee, C.S.; Zhuo, H.; Thompson, B.T.; Wilson, J.G.; Levitt, J.E.; Rogers, A.J.; Gotts, J.E.; Wiener-Kronish, J.P.; Bajwa, E.K.; et al. Treatment with allogeneic mesenchymal stromal cells for moderate to severe acute respiratory distress syndrome (START study): A randomised phase 2a safety trial. Lancet Respir. Med. 2019, 7, 154–162. [Google Scholar] [CrossRef]
- Zheng, G.; Huang, L.; Tong, H.; Shu, Q.; Hu, Y.; Ge, M.; Deng, K.; Zhang, L.; Zou, B.; Cheng, B.; et al. Treatment of acute respiratory distress syndrome with allogeneic adipose-derived mesenchymal stem cells: A randomized, placebo-controlled pilot study. Respir. Res. 2014, 15, 39. [Google Scholar] [CrossRef]
- Chen, J.; Hu, C.; Chen, L.; Tang, L.; Zhu, Y.; Xu, X.; Chen, L.; Gao, H.; Lu, X.; Yu, L.; et al. Clinical study of mesenchymal stem cell treatment for acute respiratory distress syndrome induced by epidemic influenza A (H7N9) infection: A hint for COVID-19 treatment. Engineering 2020, 6, 1153–1161. [Google Scholar] [CrossRef] [PubMed]
- Hashemian, S.-M.R.; Aliannejad, R.; Zarrabi, M.; Soleimani, M.; Vosough, M.; Hosseini, S.-E.; Hossieni, H.; Keshel, S.H.; Naderpour, Z.; Hajizadeh-Saffar, E.; et al. Mesenchymal stem cells derived from perinatal tissues for treatment of critically ill COVID-19-induced ARDS patients: A case series. Stem Cell Res. Ther. 2021, 12, 1–12. [Google Scholar] [CrossRef]
- Sengupta, V.; Sengupta, S.; Lazo, A.; Woods, P.; Nolan, A.; Bremer, N. Exosomes derived from bone marrow mesenchymal stem cells as treatment for severe COVID-19. Stem Cells Dev. 2020, 29, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Jiang, Y.; Zhu, M.; Chen, L.; Zhou, X.; Zhou, C.; Ye, P.; Chen, X.; Wang, B.; Xu, Z.; et al. Clinical study using mesenchymal stem cells for the treatment of patients with severe COVID-19. Front. Med. 2020, 14, 664–673. [Google Scholar] [CrossRef] [PubMed]
- Chan, M.C.; Kuok, D.I.; Leung, C.Y.; Hui, K.P.; Valkenburg, S.A.; Lau, E.H.; Nicholls, J.M.; Fang, X.; Guan, Y.; Lee, J.W.; et al. Human mesenchymal stromal cells reduce influenza A H5N1-associated acute lung injury In vitro and In vivo. Proc. Natl. Acad. Sci. USA 2016, 113, 3621–3626. [Google Scholar] [CrossRef] [PubMed]
- Loy, H.; Kuok, D.I.T.; Hui, K.P.Y.; Choi, M.H.L.; Yuen, W.; Nicholls, J.M.; Peiris, J.S.M.; Chan, M.C.W. Therapeutic implications of human umbilical cord mesenchymal stromal cells in attenuating influenza A(H5N1) virus-associated acute lung injury. J. Infect. Dis. 2019, 219, 186–196. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, J.; Shi, W.; Chen, C.; Shao, Y.; Zhu, L.; Lu, W.; Han, X. Mesenchymal stromal cell treatment prevents H9N2 avian influenza virus-induced acute lung injury in mice. Stem Cell Res. Ther. 2016, 7, 1–11. [Google Scholar] [CrossRef]
- Darwish, I.; Banner, D.; Mubareka, S.; Kim, H.; Besla, R.; Kelvin, D.J.; Kain, K.C.; Liles, W.C. Mesenchymal stromal (stem) cell therapy fails to improve outcomes in experimental severe influenza. PLoS ONE 2013, 8, e71761. [Google Scholar] [CrossRef]
- Gotts, J.E.; Abbott, J.; Matthay, M.A. Influenza causes prolonged disruption of the alveolar-capillary barrier in mice unresponsive to mesenchymal stem cell therapy. Am. J. Physiol. Lung. Cell Mol. Physiol. 2014, 307, L395–L406. [Google Scholar] [CrossRef]
- Lv, H.; Chen, W.; Xiang, A.P.; Zhang, Q.; Yang, Y.; Yi, H. Mesenchymal stromal cells as a salvage treatment for confirmed acute respiratory distress syndrome: Preliminary data from a single-arm study. Intensiv. Care Med. 2020, 46, 1944–1947. [Google Scholar] [CrossRef]
- Bahsoun, S.; Coopman, K.; Akam, E.C. The impact of cryopreservation on bone marrow-derived mesenchymal stem cells: A systematic review. J. Transl. Med. 2019, 17, 397. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Chelvanambi, S.; Poirier, C.; Saliba, J.; March, K.L.; Clauss, M.; Bogatcheva, N.V. EMAPII monoclonal antibody ameliorates influenza a virus-induced lung injury. Mol. Ther. 2018, 26, 2060–2069. [Google Scholar] [CrossRef] [PubMed]
- Ortega, H.; Yancey, S.; Cozens, S. Pharmacokinetics and absolute bioavailability of mepolizumab following administration at subcutaneous and intramuscular sites. Clin. Pharmacol. Drug Dev. 2013, 3, 57–62. [Google Scholar] [CrossRef]
- Morgan, R.; Klein, S.L. The intersection of sex and gender in the treatment of influenza. Curr. Opin. Virol. 2019, 35, 35–41. [Google Scholar] [CrossRef] [PubMed]
- François, M.; Romieu-Mourez, R.; Li, M.; Galipeau, J. Human MSC suppression correlates with cytokine induction of indoleamine 2,3-dioxygenase and bystander M2 macrophage differentiation. Mol. Ther. 2012, 20, 187–195. [Google Scholar] [CrossRef]
- Redondo-Castro, E.; Cunningham, C.; Miller, J.; Martuscelli, L.; Aoulad-Ali, S.; Rothwell, N.J.; Kielty, C.M.; Allan, S.M.; Pinteaux, E. Interleukin-1 primes human mesenchymal stem cells towards an anti-inflammatory and pro-trophic phenotype in vitro. Stem Cell Res. Ther. 2017, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hegen, A.; Koidl, S.; Weindel, K.; Marmé, D.; Augustin, H.G.; Fiedler, U. Expression of angiopoietin-2 in endothelial cells is controlled by positive and negative regulatory promoter elements. Arter. Thromb. Vasc. Biol. 2004, 24, 1803–1809. [Google Scholar] [CrossRef] [PubMed]
- Lomas-Neira, J.; Monaghan, S.F.; Huang, X.; Fallon, E.A.; Chung, C.S.; Ayala, A. novel role for PD-1:PD-L1 as dediator of pulmonary vascular endothelial cell functions in pathogenesis of indirect ARDS in mice. Front. Immunol. 2018, 9, 3030. [Google Scholar] [CrossRef]
- Mandriota, S.J.; Pepper, M.S. Regulation of angiopoietin-2 mRNA levels in bovine microvascular endothelial cells by cytokines and hypoxia. Circ. Res. 1998, 83, 852–859. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Poirier, C.; Cook, T.; O Traktuev, D.; Merfeld-Clauss, S.; Lease, B.; Petrache, I.; March, K.L.; Bogatcheva, N.V. Conditioned media from adipose stromal cells limit lipopolysaccharide-induced lung injury, endothelial hyperpermeability and apoptosis. J. Transl. Med. 2015, 13, 1–15. [Google Scholar] [CrossRef]
- Griffin, D.O.; Brennan-Rieder, D.; Ngo, B.; Kory, P.; Confalonieri, M.; Shapiro, L.; Iglesias, J.; Dube, M.; Nanda, N.; In, G.K.; et al. The Importance of Understanding the Stages of COVID-19 in Treatment and Trials. Aids Rev. 2021, 10. [Google Scholar] [CrossRef]
- Nedel, W.L.; Nora, D.G.; Salluh, J.I.F.; Lisboa, T.; Póvoa, P. Corticosteroids for severe influenza pneumonia: A critical appraisal. World J. Crit. Care Med. 2016, 5, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sun, W.; Svendsen, E.R.; Tang, S.; MacIntyre, R.C.; Yang, P.; Zhang, D.; Wang, Q. Do corticosteroids reduce the mortality of influenza A (H1N1) infection? A meta-analysis. Crit. Care 2015, 19, 46. [Google Scholar] [CrossRef] [PubMed]
- Blanchon, T.; Mentré, F.; Charlois-Ou, C.; Domic, Q.; Mosnier, A.; Bouscambert, M.; Carrat, F.; Duval, X.; Enouf, V.; Leport, C.; et al. Factors associated with clinical and virological response in patients treated with oseltamivir or zanamivir for influenza A during the 2008–2009 winter. Clin. Microbiol. Infect. 2013, 19, 196–203. [Google Scholar] [CrossRef]
- Jensen-Fangel, S.; Mohey, R.; Johnsen, S.P.; Andersen, P.L.; Sørensen, H.T.; Østergaard, L. Gender differences in hospitalization rates for respiratory tract infections in Danish youth. Scand. J. Infect. Dis. 2004, 36, 31–36. [Google Scholar] [CrossRef]
- Heffernan, D.S.; Dossett, L.A.; Lightfoot, M.A.; Fremont, R.D.; Ware, L.B.; Sawyer, R.G.; May, A.K. Gender and acute respiratory distress syndrome in critically injured adults: A prospective study. J. Trauma Inj. Infect. Crit. Care 2011, 71, 878–885. [Google Scholar] [CrossRef]
- McNicholas, B.A.; Madotto, F.; Pham, T.; Rezoagli, E.; Masterson, C.H.; Horie, S.; Bellani, G.; Brochard, L.; Laffey, J.G.; LUNG SAFE Investigators and the ESICM Trials Group. Demographics, management and outcome of females and males with acute respiratory distress syndrome in the LUNG SAFE prospective cohort study. Eur. Respir. J. 2019, 54. [Google Scholar] [CrossRef]
- Robinson, D.P.; Lorenzo, M.E.; Jian, W.; Klein, S.L. Elevated 17β-estradiol protects females from influenza A virus pathogenesis by suppressing inflammatory responses. PLoS Pathog. 2011, 7, e1002149. [Google Scholar] [CrossRef]
- Celestino, I.; Checconi, P.; Amatore, D.; De Angelis, M.; Coluccio, P.; Dattilo, R.; Alunni Fegatelli, D.; Clemente, A.M.; Matarrese, P.; Torcia, M.G.; et al. Differential redox state contributes to sex disparities in the response to influenza virus infection in male and female mice. Front. Immun. 2018, 9, 1747. [Google Scholar] [CrossRef]
- Zeller, C.N.; Wang, Y.; Markel, T.A.; Weil, B.; Abarbanell, A.; Herrmann, J.L.; Kelly, M.L.; Coffey, A.; Meldrum, D.R. Role of tumor necrosis factor receptor 1 in sex differences of stem cell mediated cardioprotection. Ann. Thorac. Surg. 2009, 87, 812–819. [Google Scholar] [CrossRef] [PubMed]
- Crisostomo, P.R.; Markel, T.A.; Wang, M.; Lahm, T.; Lillemoe, K.D.; Meldrum, D.R. In the adult mesenchymal stem cell population, source gender is a biologically relevant aspect of protective power. Surgery 2007, 142, 215–221. [Google Scholar] [CrossRef]
- Ock, S.A.; Lee, Y.M.; Park, J.S.; Shivakumar, S.B.; Moon, S.W.; Sung, N.J.; Lee, W.J.; Jang, S.J.; Park, J.M.; Lee, S.C.; et al. Evaluation of phenotypic, functional and molecular characteristics of porcine mesenchymal stromal/stem cells depending on donor age, gender and tissue source. J. Vet. Med Sci. 2016, 78, 987–995. [Google Scholar] [CrossRef]
- Bianconi, E.; Casadei, R.; Frabetti, F.; Ventura, C.; Facchin, F.; Canaider, S. Sex-specific transcriptome differences in human adipose mesenchymal stem cells. Genes 2020, 11, 909. [Google Scholar] [CrossRef] [PubMed]
- Stolzing, A.; Jones, E.; McGonagle, D.; Scutt, A. Age-related changes in human bone marrow-derived mesenchymal stem cells: Consequences for cell therapies. Mech. Ageing Dev. 2008, 129, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Badimon, L.; Cubedo, J. Adipose tissue depots and inflammation: Effects on plasticity and resident mesenchymal stem cell function. Cardiovasc. Res. 2017, 113, 1064–1073. [Google Scholar] [CrossRef]
- Cramer, C.; Freisinger, E.; Jones, R.K.; Slakey, D.P.; Dupin, C.L.; Newsome, E.R.; Alt, E.U.; Izadpanah, R. Persistent high glucose concentrations alter the regenerative potential of mesenchymal stem cells. Stem Cells Dev. 2010, 19, 1875–1884. [Google Scholar] [CrossRef] [PubMed]
- Stern-Straeter, J.; Bonaterra, G.A.; Juritz, S.; Birk, R.; Goessler, U.R.; Bieback, K.; Bugert, P.; Schultz, J.; Hörmann, K.; Kinscherf, R.; et al. Evaluation of the effects of different culture media on the myogenic differentiation potential of adipose tissue- or bone marrow-derived human mesenchymal stem cells. Int. J. Mol. Med. 2014, 33, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Noronha, N.C.; Mizukami, A.; Caliári-Oliveira, C.; Cominal, J.G.; Rocha, J.L.M.; Covas, D.T.; Swiech, K.; Malmegrim, K.C.R. Priming approaches to improve the efficacy of mesenchymal stromal cell-based therapies. Stem Cell Res. Ther. 2019, 10, 131. [Google Scholar] [CrossRef]
- Ong, T.; McClintock, D.E.; Kallet, R.H.; Ware, L.B.; Matthay, M.A.; Liu, K.D. Ratio of angiopoietin-2 to angiopoietin-1 as a predictor of mortality in acute lung injury patients. Crit. Care Med. 2010, 38, 1845–1851. [Google Scholar] [CrossRef]
- Orfanos, S.E.; Kotanidou, A.; Glynos, C.; Athanasiou, C.; Tsigkos, S.; Dimopoulou, I.; Sotiropoulou, C.; Zakynthinos, S.; Armaganidis, A.; Papapetropoulos, A.; et al. Angiopoietin-2 is increased in severe sepsis: Correlation with inflammatory mediators. Crit. Care Med. 2007, 35, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Parikh, S.M.; Mammoto, T.; Schultz, A.; Yuan, H.T.; Christiani, D.; Karumanchi, S.A.; Sukhatme, V.P. Excess circulating angiopoietin-2 may contribute to pulmonary vascular leak in sepsis in humans. PLoS Med. 2006, 3, e46. [Google Scholar] [CrossRef] [PubMed]
- van der Heijden, M.; van Nieuw Amerongen, G.P.; Koolwijk, P.; van Hinsbergh, V.W.; Groeneveld, A.B. Angiopoietin-2, permeability oedema, occurrence and severity of ALI/ARDS in septic and non-septic critically ill patients. Thorax 2008, 63, 903–909. [Google Scholar] [CrossRef]
- Stiehl, T.; Thamm, K.; Kaufmann, J.; Schaeper, U.; Kirsch, T.; Haller, H.; Santel, A.; Ghosh, C.C.; Parikh, S.M.; David, S. Lung-targeted RNA interference against angiopoietin-2 ameliorates multiple organ dysfunction and death in sepsis. Crit. Care Med. 2014, 42, e654–e662. [Google Scholar] [CrossRef]
- Souma, T.; Thomson, B.R.; Heinen, S.; Carota, I.A.; Yamaguchi, S.; Onay, T.; Liu, P.; Ghosh, A.K.; Li, C.; Eremina, V.; et al. Context-dependent functions of angiopoietin 2 are determined by the endothelial phosphatase VEPTP. Proc. Natl. Acad. Sci. USA 2018, 115, 1298–1303. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, C.C.; David, S.; Zhang, R.; Berghelli, A.; Milam, K.; Higgins, S.J.; Hunter, J.; Mukherjee, A.; Wei, Y.; Tran, M.; et al. Gene control of tyrosine kinase TIE2 and vascular manifestations of infections. Proc. Natl. Acad. Sci. USA 2016, 113, 2472–2477. [Google Scholar] [CrossRef]
- Sugiyama, M.G.; Armstrong, S.M.; Wang, C.; Hwang, D.; Leong-Poi, H.; Advani, A.; Advani, S.; Zhang, H.; Szaszi, K.; Tabuchi, A.; et al. The TIE2-agonist vasculotide rescues mice from influenza virus infection. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef]
- Xu, S.; Yang, Q.; Bai, J.; Tao, T.; Tang, L.; Chen, Y.; Chung, C.S.; Fallon, E.A.; Ayala, A. Blockade of endothelial, but not epithelial, cell expression of PD-L1 following severe shock attenuates the development of indirect acute lung injury in mice. Am. J. Physiol. Cell. Mol. Physiol. 2020, 318, L801–L812. [Google Scholar] [CrossRef]
- FDA. Remestemcel-L for Treatment of Steroid Refractory Acute Graft Versus Host Disease in Pediatric Patients. Available online: https://www.fda.gov/media/140996/download (accessed on 19 March 2021).
- Bogatcheva, N.V.; Coleman, M.E. Conditioned medium of mesenchymal stromal cells: A new class of therapeutics. Biochemistry 2019, 84, 1375–1389. [Google Scholar] [CrossRef]
- Lu, H.; Merfeld-Clauss, S.; Jawed, Y.; March, K.L.; Coleman, M.E.; Bogatcheva, N.V. Distinct factors secreted by adipose stromal cells protect the endothelium from barrier dysfunction and apoptosis. Front. Cell Dev. Biol. 2020, 8, 4653. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bogatcheva, N.V.; Coleman, M.E. Concentrated Secretome of Adipose Stromal Cells Limits Influenza A Virus-Induced Lung Injury in Mice. Cells 2021, 10, 720. https://doi.org/10.3390/cells10040720
Bogatcheva NV, Coleman ME. Concentrated Secretome of Adipose Stromal Cells Limits Influenza A Virus-Induced Lung Injury in Mice. Cells. 2021; 10(4):720. https://doi.org/10.3390/cells10040720
Chicago/Turabian StyleBogatcheva, Natalia V., and Michael E. Coleman. 2021. "Concentrated Secretome of Adipose Stromal Cells Limits Influenza A Virus-Induced Lung Injury in Mice" Cells 10, no. 4: 720. https://doi.org/10.3390/cells10040720
APA StyleBogatcheva, N. V., & Coleman, M. E. (2021). Concentrated Secretome of Adipose Stromal Cells Limits Influenza A Virus-Induced Lung Injury in Mice. Cells, 10(4), 720. https://doi.org/10.3390/cells10040720