
Assessment of the Neuroprotective and Stemness Properties of Human Wharton’s Jelly-Derived Mesenchymal Stem Cells under Variable (5% vs. 21%) Aerobic Conditions

 , , ,
, , ,  and
and
Abstract
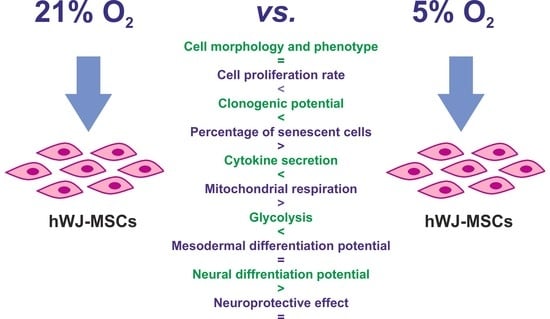
:
{kind=link}
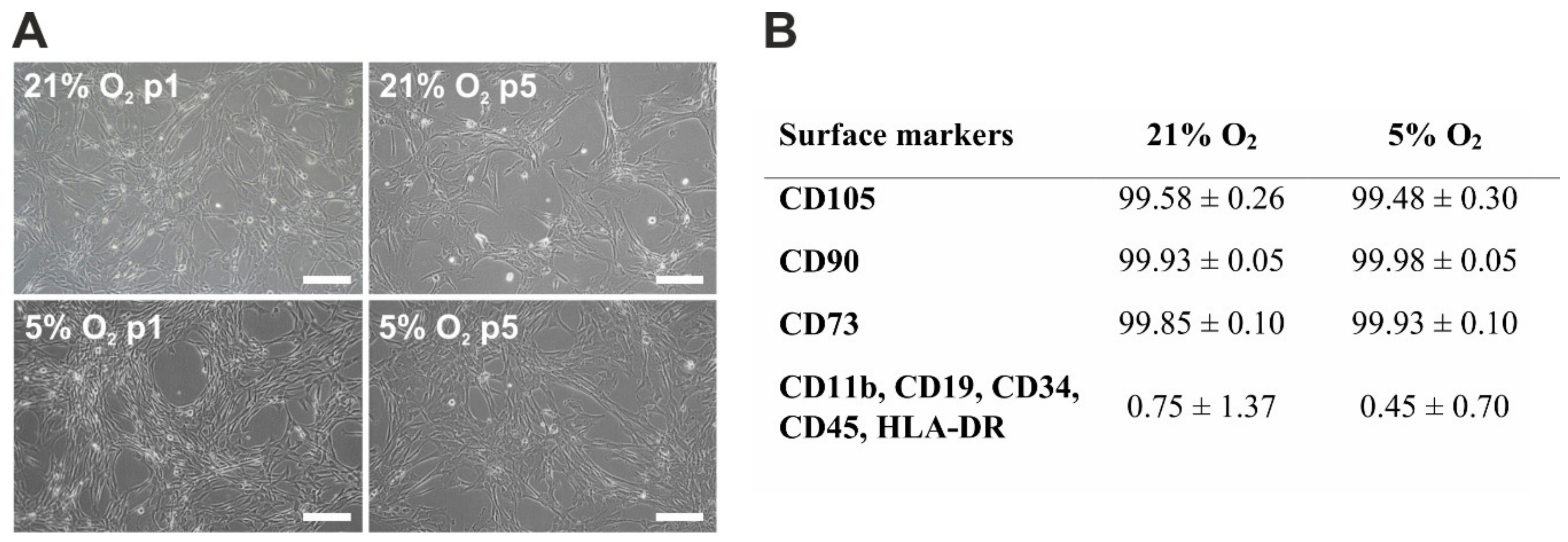{kind=link}
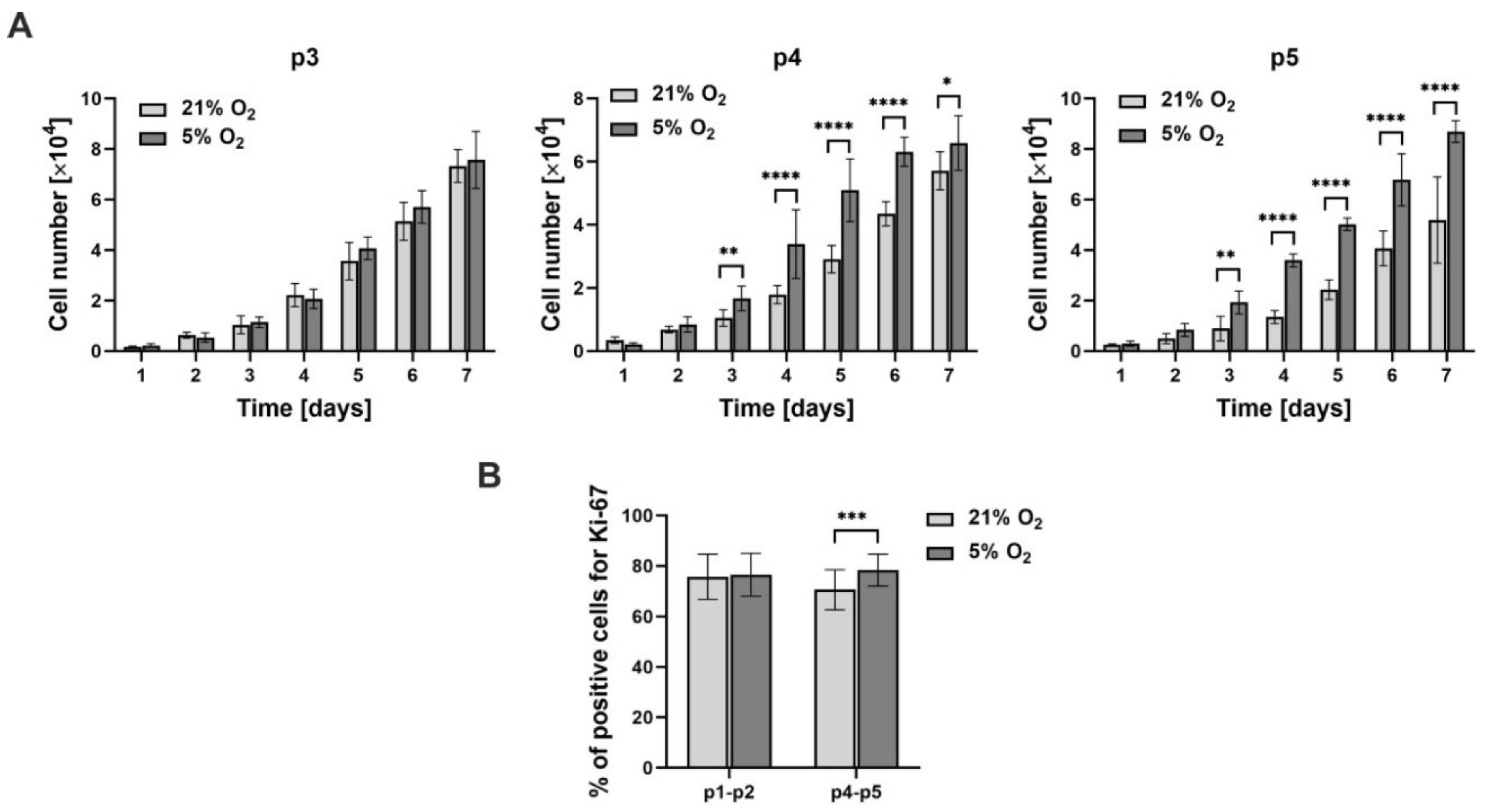{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Isolation and Culture of Human WJ-MSCs
2.2. Flow Cytometry Analysis
2.3. Cell Proliferation Assays
2.3.1. WST-1 Assay
2.3.2. Analysis of Ki67 Marker Expression
2.4. Colony Forming Unit (CFU) Assay
2.5. Senescence-Associated β-Galactosidase Assay
2.6. Mesodermal Differentiation Ability of hWJ-MSCs
2.7. Analysis of hWJ-MSCs Secretome
2.8. Determination of the Oxygen Consumption Rate (OCR) and Extracellular Acidification Rate (ECAR)
2.9. Neural Differentiation Ability of hWJ-MSCs
2.10. Neuroprotective Properties of hWJ-MSCs
2.11. Microscopic Observations
2.12. Statistical Analysis
3. Results
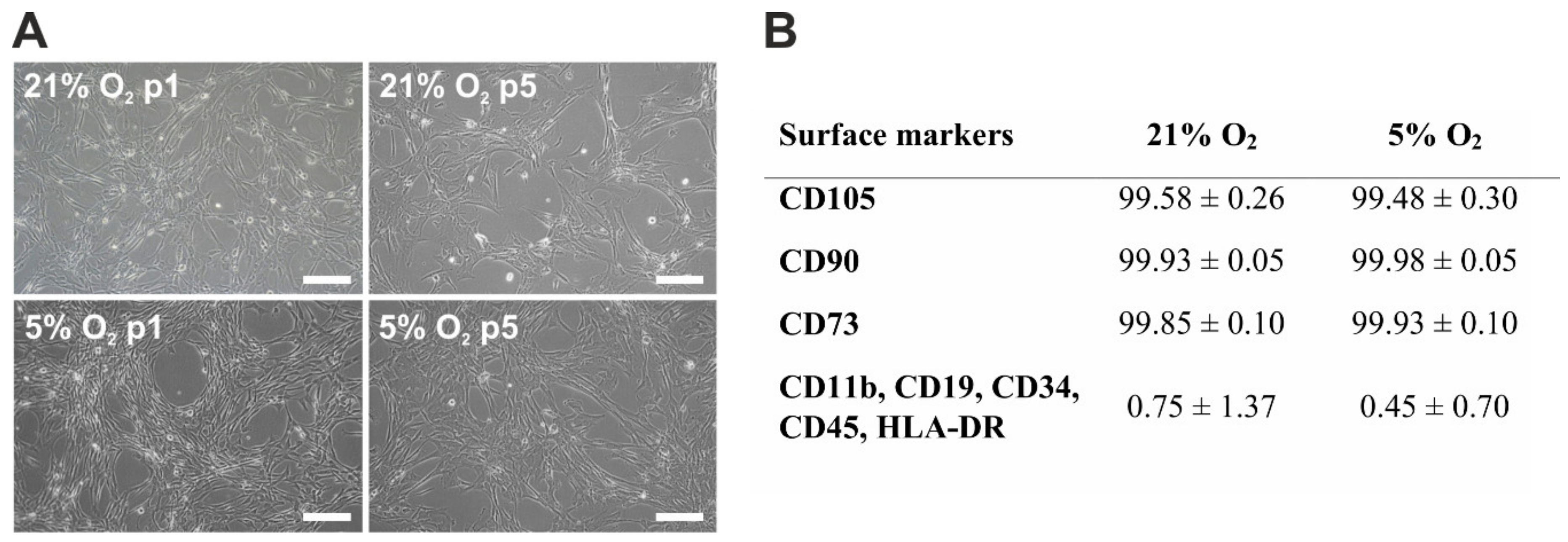
3.1. Morphology and Immunophenotype of hWJ-MSCs
3.2. Cell Proliferation
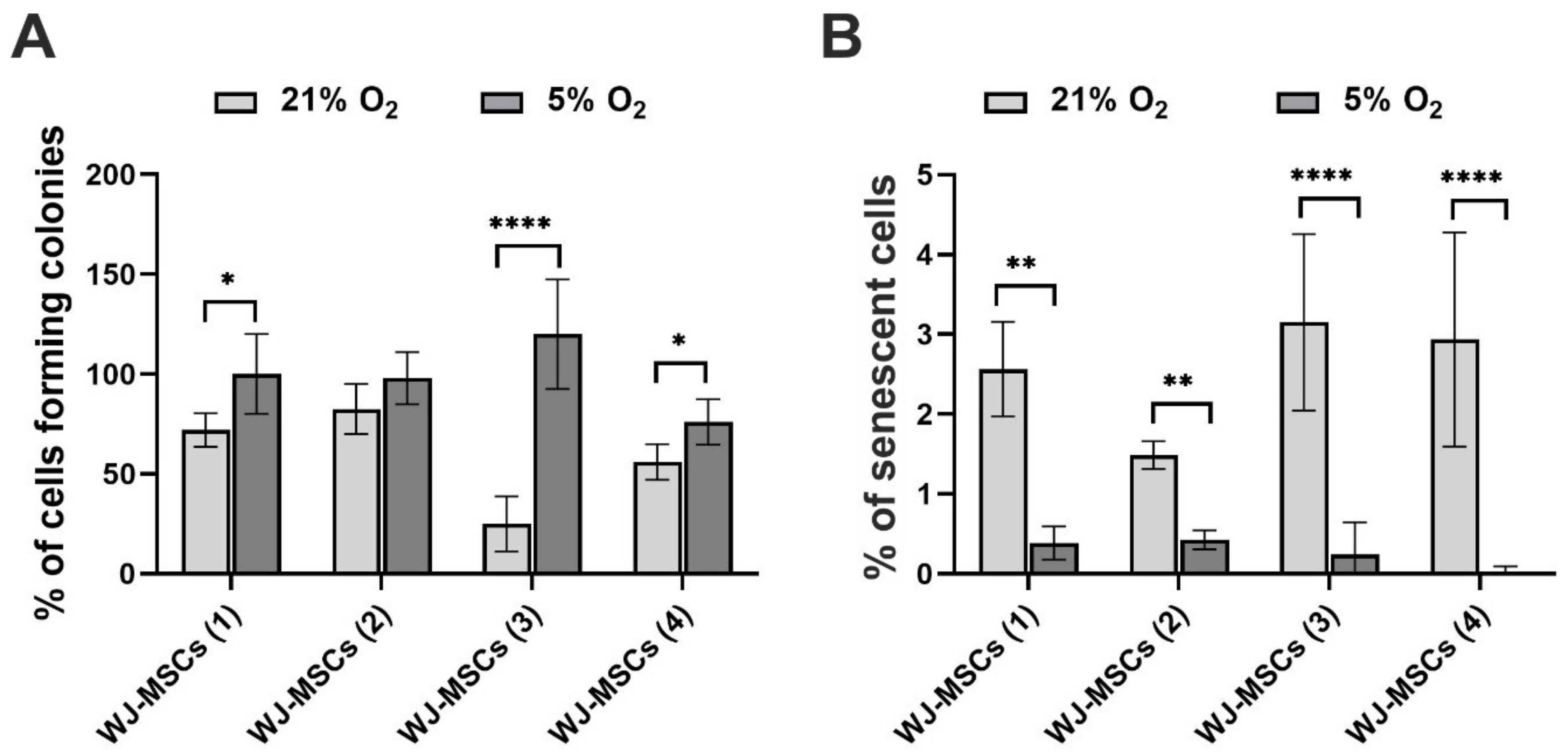
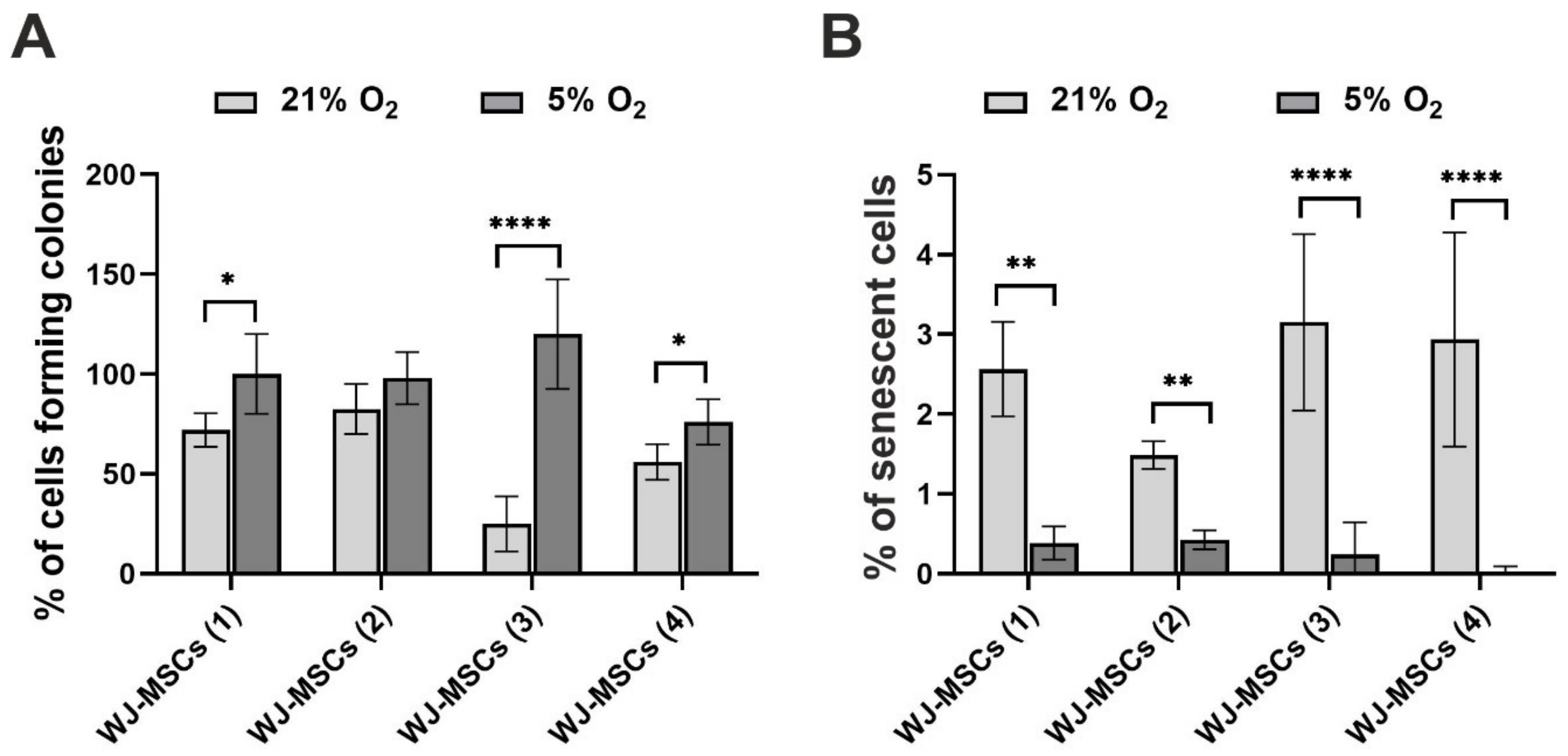
3.3. Clonogenicity and Cellular Senescence of hWJ-MSCs
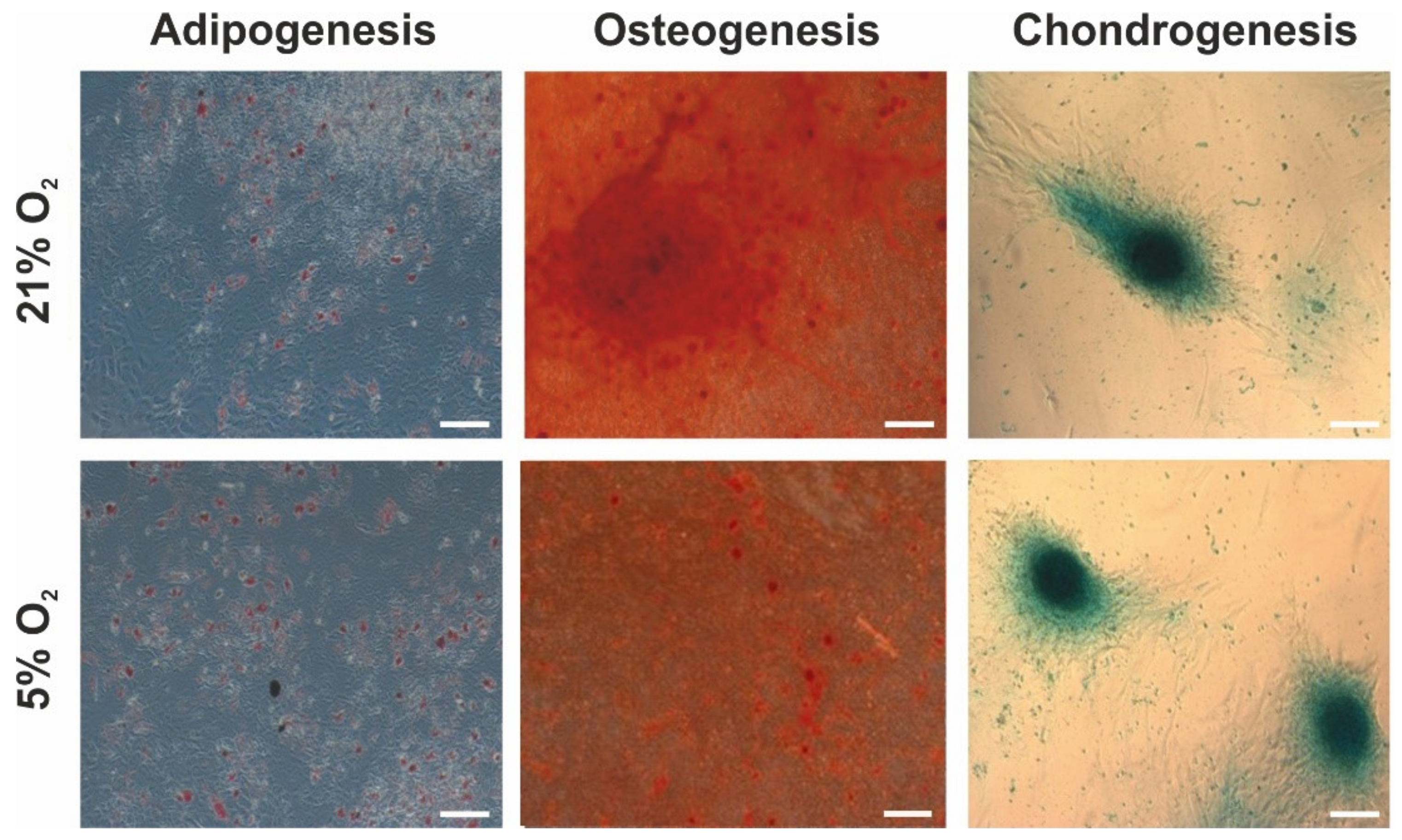
3.4. Adipogenic, Osteogenic, and Chondrogenic Differentiation Potential of hWJ-MSCs
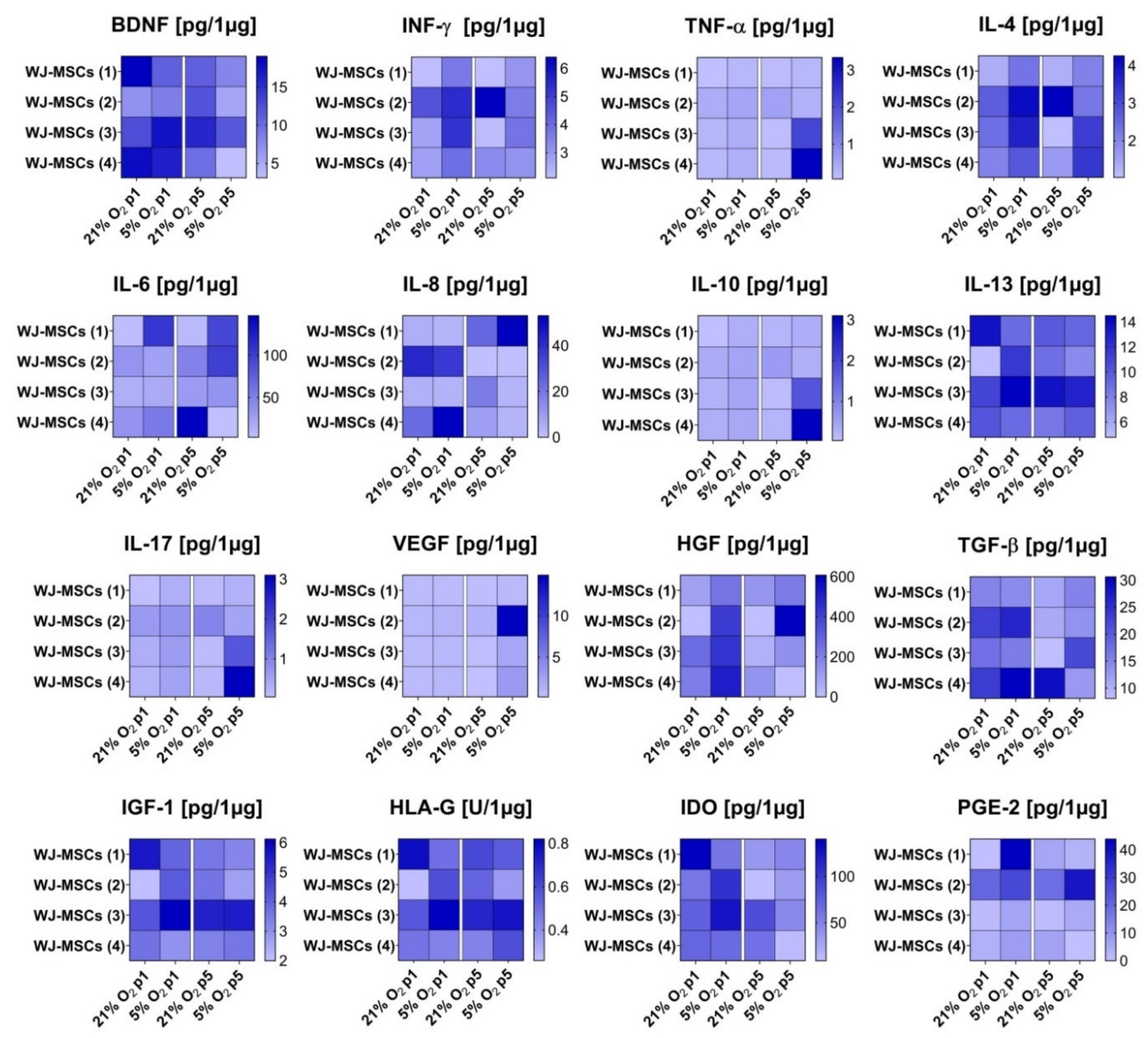
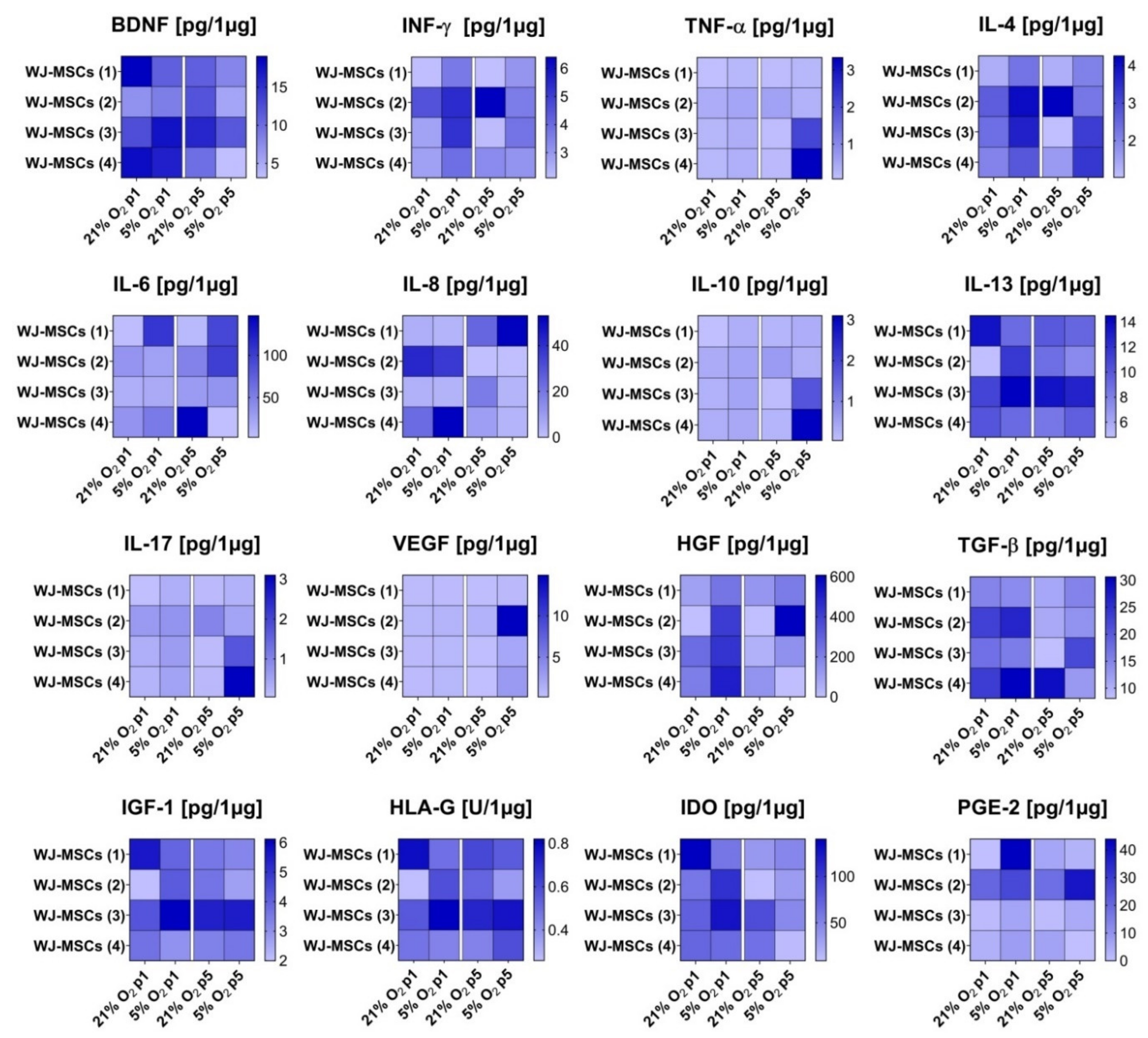
3.5. Secretory Profile of hWJ-MSCs
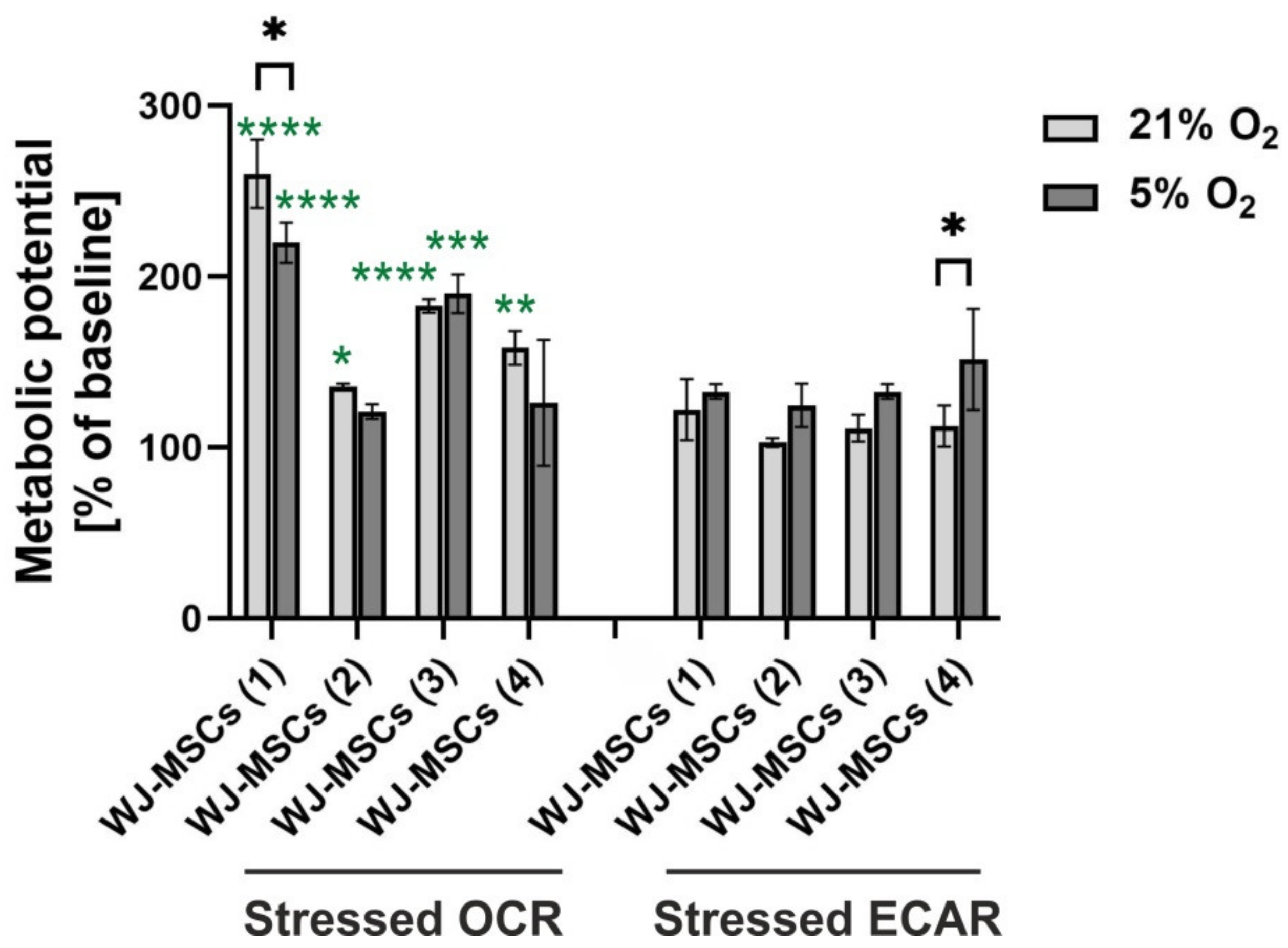
3.6. Metabolic Potential of hWJ-MSCs
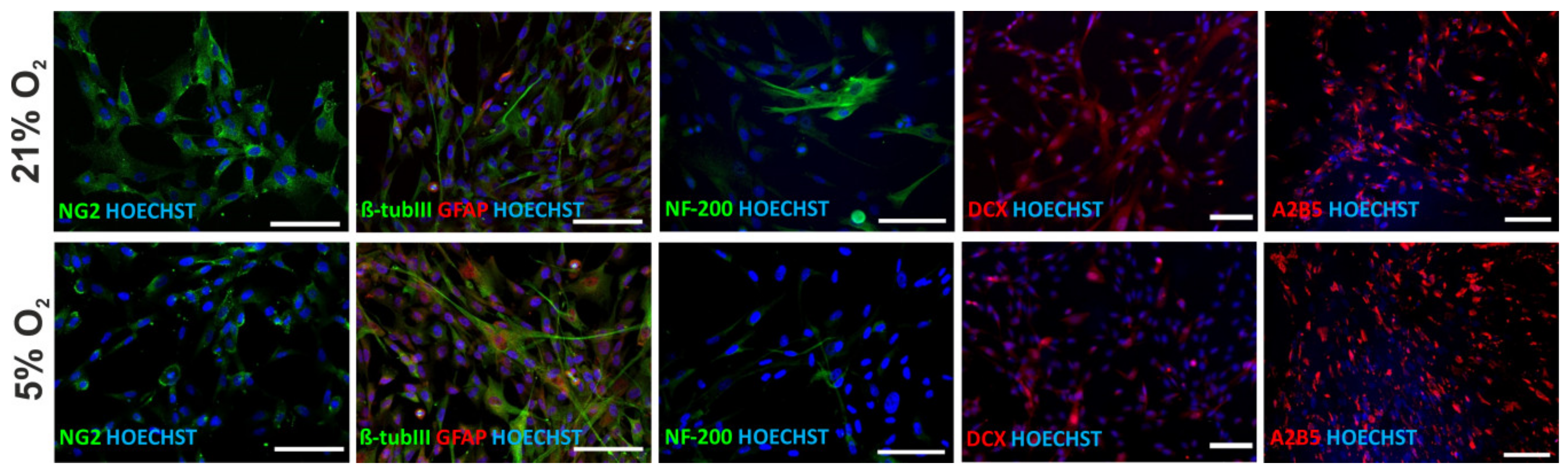
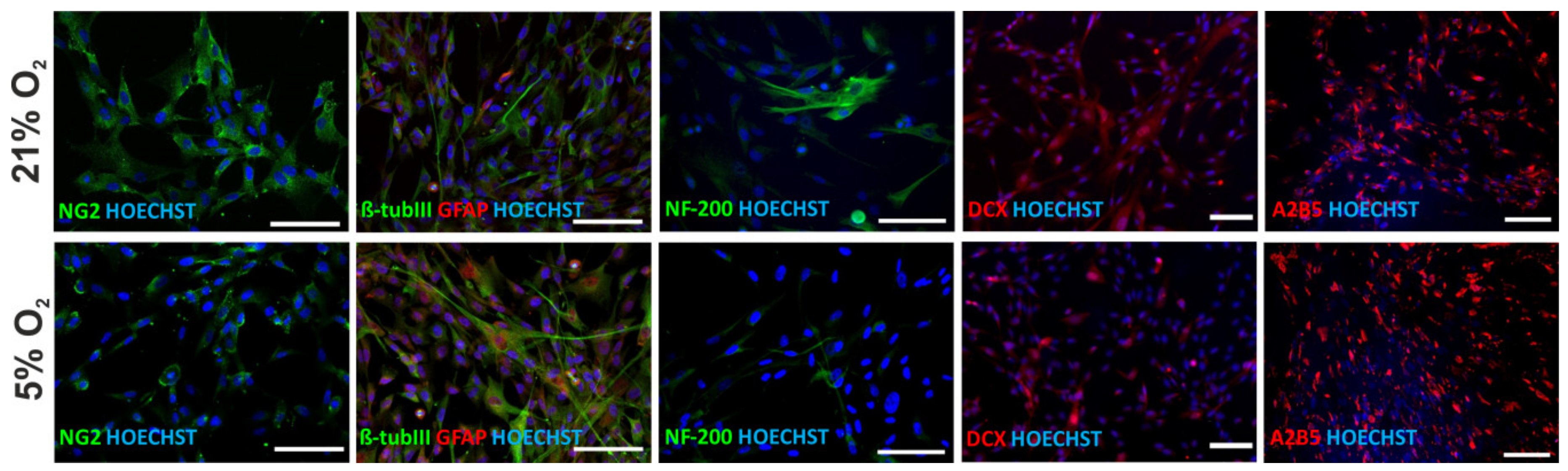
3.7. Neural Differentiation Potential of hWJ-MSCs
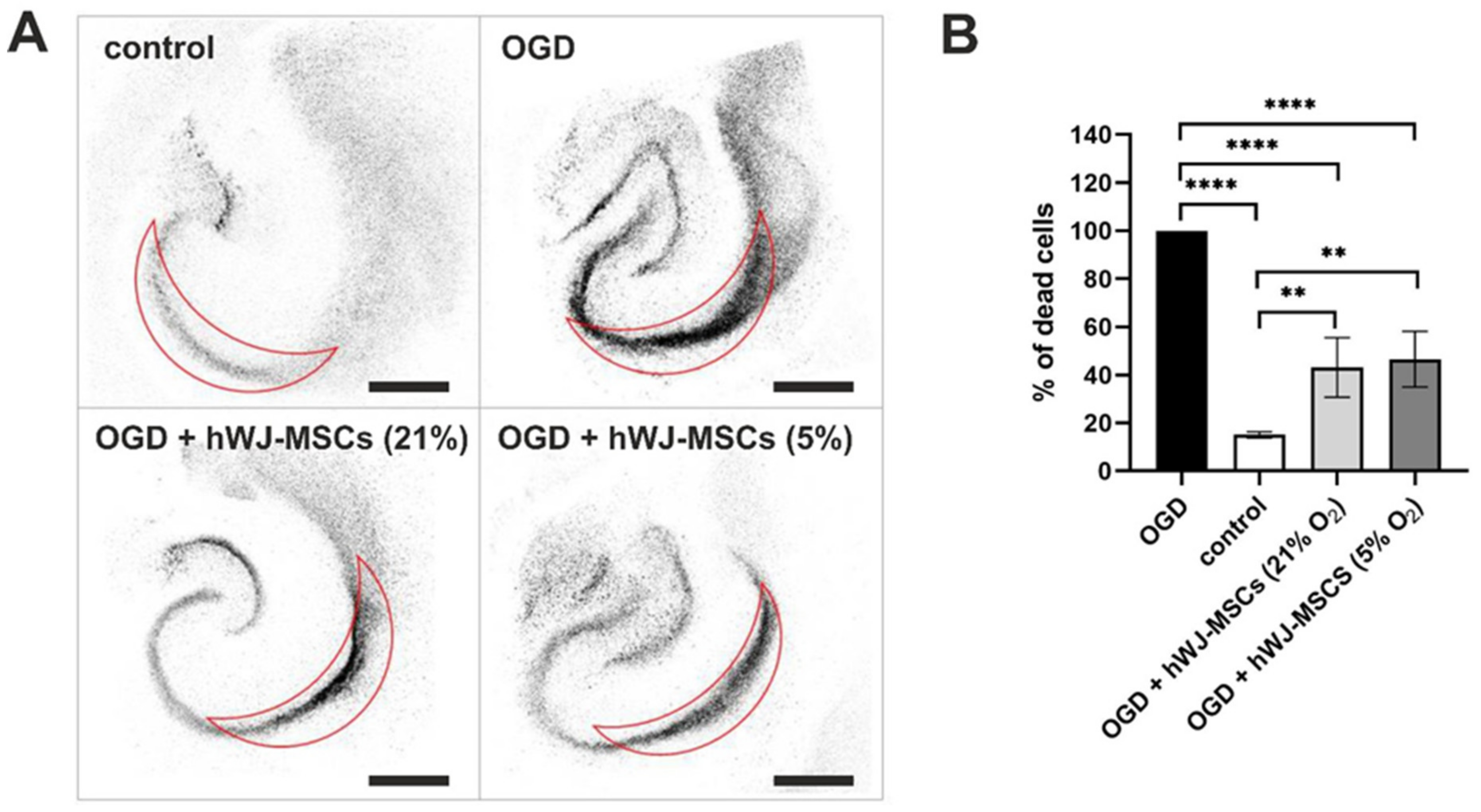
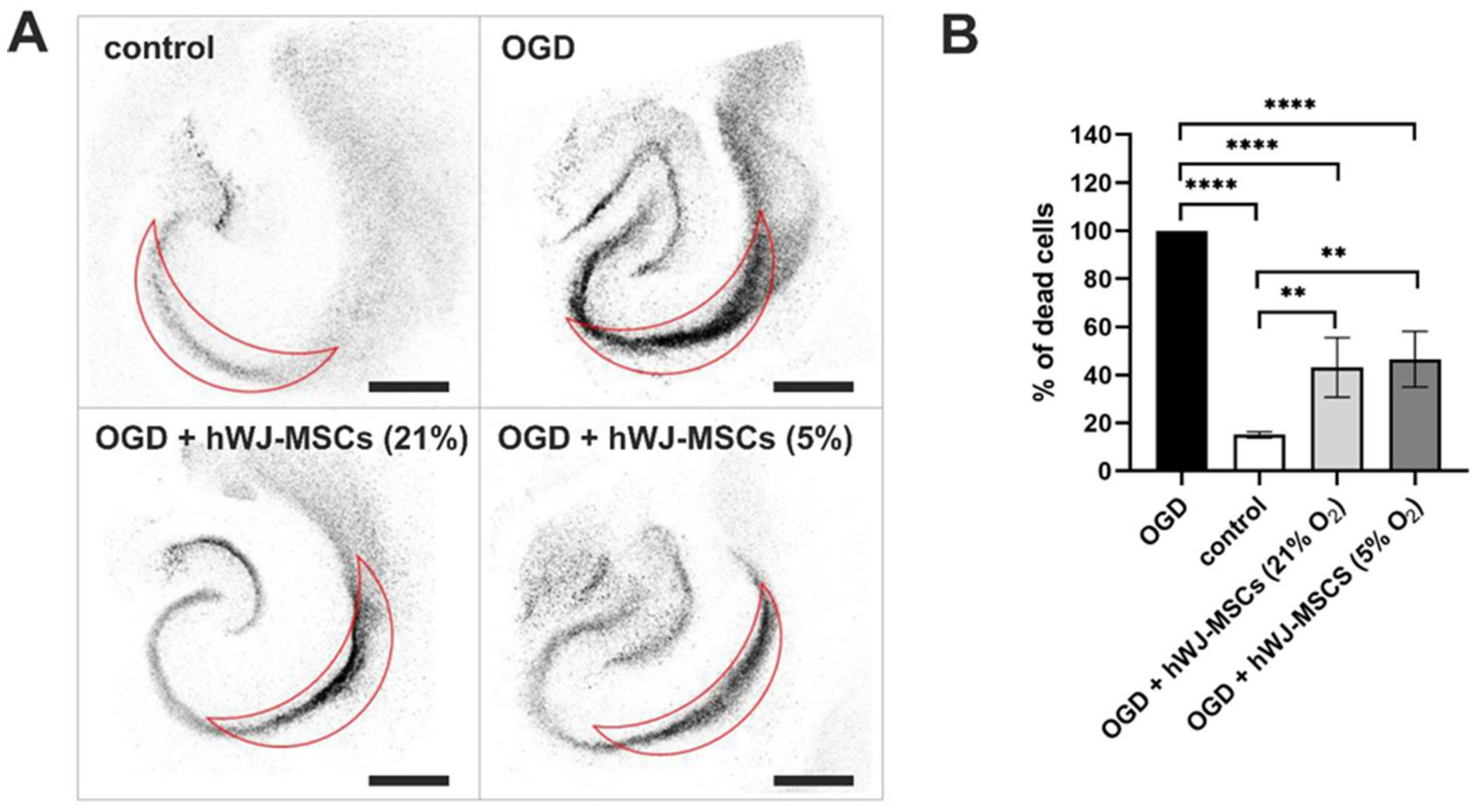
3.8. Neuroprotective Effect of hWJ-MSCs
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dominici, M.; Blanc, K.L.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Widowati, W.; Rihibiha, D.D.; Khiong, K.; Widodo, M.A.; Sumitro, S.B.; Bachtiar, I. Hypoxia in Mesenchymal Stem Cell. In Hypoxia and Human Diseases; InTech: London, UK, 2017; pp. 91–115. [Google Scholar]
- Shuvalova, N.S.; Kordium, V.A. Comparison of proliferative activity of Wharton jelly mesenchymal stem cells in cultures under various gas conditions. Biopolym. Cell 2015, 31, 233–239. [Google Scholar] [CrossRef] [Green Version]
- Abumaree, M.H.; Abomaray, F.M.; Alshehri, N.A.; Almutairi, A.; AlAskar, A.S.; Kalionis, B.; Al Jumah, M.A. Phenotypic and Functional Characterization of Mesenchymal Stem/Multipotent Stromal Cells from Decidua Parietalis of Human Term Placenta. Reprod. Sci. 2016, 23, 1193–1207. [Google Scholar] [CrossRef]
- Berebichez-Fridman, R.; Montero-Olvera, P.R. Sources and Clinical Applications of Mesenchymal Stem Cells: State-of-the-art review. Sultan Qaboos Univ. Med. J. 2018, 18, e264–e277. [Google Scholar] [CrossRef] [Green Version]
- Mushahary, D.; Spittler, A.; Kasper, C.; Weber, V.; Charwat, V. Isolation, cultivation, and characterization of human mesenchymal stem cells. Cytometry 2018, 93, 19–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Yuan, M.; Guo, Q.Y.; Lu, S.B.; Peng, J. Mesenchymal stem cells for treating articular cartilage defects and osteoarthritis. Cell Transplant. 2015, 24, 1661–1678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heidari, B.; Shirazi, A.; Akhondi, M.M.; Hassanpour, H.; Behzadi, B.; Naderi, M.M.; Sarvari, A.; Borjian, S. Comparison of proliferative and multilineage differentiation potential of sheep mesenchymal stem cells derived from bone marrow, liver, and adipose tissue. Avicenna J. Med. Biotechnol. 2013, 5, 104–117. [Google Scholar] [PubMed]
- Squillaro, T.; Peluso, G.; Galderisi, U. Clinical Trials with Mesenchymal Stem Cells: An Update. Cell Transplant. 2016, 25, 829–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, C.; McKee, C.; Bakshi, S.; Walker, K.; Hakman, E.; Halassy, S.; Svinarich, D.; Dodds, R.; Govind, C.K.; Chaudhry, G.R. Mesenchymal stem cells: Cell therapy and regeneration potential. J. Tissue Eng. Regen. Med. 2019, 13, 1738–1755. [Google Scholar] [CrossRef]
- Xu, L.; Liu, Y.; Sun, Y.; Wang, B.; Xiong, Y.; Lin, W.; Wei, Q.; Wang, H.; He, W.; Wang, B.; et al. Tissue source determines the differentiation potentials of mesenchymal stem cells: A comparative study of human mesenchymal stem cells from bone marrow and adipose tissue. Stem Cell Res. Ther. 2017, 8, 275. [Google Scholar] [CrossRef] [Green Version]
- Fossett, E.; Khan, W.S.; Pastides, P.; Adesida, A.B. The Effects of Ageing on Proliferation Potential, Differentiation Potential and Cell Surface Characterisation of Human Mesenchymal Stem Cells. Curr. Stem Cell Res. Ther. 2012, 7, 282–286. [Google Scholar] [CrossRef]
- Zaim, M.; Karaman, S.; Cetin, G.; Isik, S. Donor age and long-term culture affect differentiation and proliferation of human bone marrow mesenchymal stem cells. Ann. Hematol. 2012, 91, 1175–1186. [Google Scholar] [CrossRef] [PubMed]
- Troyer, D.L.; Weiss, M.L. Concise Review: Wharton’s Jelly-Derived Cells Are a Primitive Stromal Cell Population. Stem Cells 2008, 26, 591–599. [Google Scholar] [CrossRef] [Green Version]
- Can, A.; Karahuseyinoglu, S. Concise Review: Human Umbilical Cord Stroma with Regard to the Source of Fetus-Derived Stem Cells. Stem Cells 2007, 25, 2886–2895. [Google Scholar] [CrossRef] [PubMed]
- Hass, R.; Kasper, C.; Böhm, S.; Jacobs, R. Different populations and sources of human mesenchymal stem cells (MSC): A comparison of adult and neonatal tissue-derived MSC. Cell Commun. Signal. 2011, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Eve, D.J.; Sanberg, P.R.; Buzanska, L.; Sarnowska, A.; Domanska-Janik, K. Human Somatic Stem Cell Neural Differentiation Potential. In Human Neural Stem Cells From Generation to Differentiation and Application, 1st ed.; Buzanska, L., Ed.; Springer: Berlin/Heidelberg, Germany, 2018; Volume 66, pp. 21–87. [Google Scholar]
- Drela, K.; Lech, W.; Figiel-Dabrowska, A.; Zychowicz, M.; Mikula, M.; Sarnowska, A.; Domanska-Janik, K. Enhanced neuro-therapeutic potential of Wharton’s Jelly-derived mesenchymal stem cells in comparison with bone marrow mesenchymal stem cells culture. Cytotherapy 2016, 18, 497–509. [Google Scholar] [CrossRef] [PubMed]
- Paladino, F.V.; de Moraes Rodrigues, J.; da Silva, A.; Goldberg, A.C. The Immunomodulatory Potential of Wharton’s Jelly Mesenchymal Stem/Stromal Cells. Stem Cells Int. 2019, 2019, 3548917. [Google Scholar]
- Forraz, N.; Mcguckin, C.P. The umbilical cord: A rich and ethical stem cell source to advance regenerative medicine. Cell Prolif. 2011, 44, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhang, K.; Liu, H.; Yang, T.; Xiao, D.J.; Wang, Y.S. The neuroprotective effect of mesenchymal stem cells is mediated through inhibition of apoptosis in hypoxic ischemic injury. World J. Pediatrics 2020, 16, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Dabrowska, S.; Sypecka, J.; Jablonska, A.; Strojek, L.; Wielgos, M.; Domanska-Janik, K.; Sarnowska, A. Neuroprotective Potential and Paracrine Activity of Stromal Vs. Culture Expanded hMSC Derived from Wharton Jelly under Co-Cultured with Hippocampal Organotypic Slices. Mol. Neurobiol. 2018, 55, 6021–6036. [Google Scholar] [CrossRef] [Green Version]
- Mas-Bargues, C.; Sanz-Ros, J.; Román-Domínguez, A.; Inglés, M.; Gimeno-Mallench, L.; El Alami, M.; Viña-Almunia, J.; Gambini, J.; Viña, J.; Borrás, C. Relevance of oxygen concentration in stem cell culture for regenerative medicine. Int. J. Mol. Sci. 2019, 20, 1195. [Google Scholar] [CrossRef] [Green Version]
- Kolf, C.M.; Cho, E.; Tuan, R.S. Mesenchymal stromal cells. Biology of adult mesenchymal stem cells: Regulation of niche, self-renewal and differentiation. Arthritis Res. Ther. 2007, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Caplan, A.I. Adult mesenchymal stem cells for tissue engineering versus regenerative medicine. J. Cell. Physiol. 2007, 213, 341–347. [Google Scholar] [CrossRef]
- Ward, J.P. Oxygen sensors in context. Biochim. Biophys. Acta Bioenerg. 2008, 1777, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Basciano, L.; Nemos, C.; Foliguet, B.; de Isla, N.; de Carvalho, M.; Tran, N.; Dalloul, A. Long term culture of mesenchymal stem cells in hypoxia promotes a genetic program maintaining their undifferentiated and multipotent status. BMC Cell Biol. 2011, 12, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Dionigi, B.; Ahmed, A.; Pennington, E.C.; Zurakowski, D.; Fauza, D.O. A comparative analysis of human mesenchymal stem cell response to hypoxia in vitro: Implications to translational strategies. J. Pediatric Surg. 2014, 49, 915–918. [Google Scholar] [CrossRef] [PubMed]
- Fotia, C.; Massa, A.; Boriani, F.; Baldini, N.; Granchi, D. Hypoxia enhances proliferation and stemness of human adipose-derived mesenchymal stem cells. Cytotechnology 2015, 67, 1073–1084. [Google Scholar] [CrossRef] [Green Version]
- Adesida, A.B.; Mulet-Sierra, A.; Jomha, N.M. Hypoxia mediated isolation and expansion enhances the chondrogenic capacity of bone marrow mesenchymal stromal cells. Stem Cell Res. Ther. 2012, 3, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, S.P.; Ho, J.H.; Shih, Y.R.V.; Lo, T.; Lee, O.K. Hypoxia promotes proliferation and osteogenic differentiation potentials of human mesenchymal stem cells. J. Orthop. Res. 2012, 30, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Bornes, T.D.; Jomha, N.M.; Mulet-Sierra, A.; Adesida, A.B. Hypoxic culture of bone marrow-derived mesenchymal stromal stem cells differentially enhances in vitro chondrogenesis within cell-seeded collagen and hyaluronic acid porous scaffolds. Stem Cell Res. Ther. 2015, 6, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antebi, B.; Rodriguez, L.A.; Walker, K.P.; Asher, A.M.; Kamucheka, R.M.; Alvarado, L.; Mohammadipoor, A.; Cancio, L.C. Short-term physiological hypoxia potentiates the therapeutic function of mesenchymal stem cells. Stem Cell Res. Ther. 2018, 9, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nekanti, U.; Dastidar, S.; Venugopal, P.; Totey, S.; Ta, M. Increased proliferation and analysis of differential gene expression in human Wharton’s jelly-derived mesenchymal stromal cells under hypoxia. Int. J. Biol. Sci. 2010, 6, 499–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranera, B.; Remacha, A.R.; Álvarez-Arguedas, S.; Romero, A.; Vázquez, F.J.; Zaragoza, P.; Martín-Burriel, I.; Rodellar, C. Effect of hypoxia on equine mesenchymal stem cells derived from bone marrow and adipose tissue. BMC Vet. Res. 2012, 8, 2–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burian, E.; Probst, F.; Palla, B.; Riedel, C.; Saller, M.M.; Cornelsen, M.M.; König, F.; Schieker, M.; Otto, S. Effect of hypoxia on the proliferation of porcine bone marrow-derived mesenchymal stem cells and adipose-derived mesenchymal stem cells in 2- and 3-dimensional culture. J. Cranio-Maxillofac. Surg. 2017, 45, 414–419. [Google Scholar] [CrossRef]
- Cicione, C.; Muiños-López, E.; Hermida-Gómez, T.; Fuentes-Boquete, I.; Díaz-Prado, S.; Blanco, F.J. Effects of severe hypoxia on bone marrow mesenchymal stem cells differentiation potential. Stem Cells Int. 2013, 2013, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Dos Santos, F.; Andrade, P.Z.; Boura, J.S.; Abecasis, M.M.; Da Silva, C.L.; Cabral, J.M.S. Ex vivo expansion of human mesenchymal stem cells: A more effective cell proliferation kinetics and metabolism under hypoxia. J. Cell. Physiol. 2010, 223, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Ranera, B.; Remacha, A.R.; Álvarez-Arguedas, S.; Romero, A.; Vázquez, F.J.; Zaragoza, P.; Martín-Burriel, I.; Rodellar, C. Expansion under hypoxic conditions enhances the chondrogenic potential of equine bone marrow-derived mesenchymal stem cells. Vet. J. 2013, 195, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Reppel, L.; Margossian, T.; Yaghi, L.; Moreau, P.; Mercier, N.; Leger, L.; Hupont, S.; Stoltz, J.-F.; Bensoussan, D.; Huselstein, C. Hypoxic Culture Conditions for Mesenchymal Stromal/Stem Cells from Wharton’s Jelly: A Critical Parameter to Consider in a Therapeutic Context. Curr. Stem Cell Res. Ther. 2014, 9, 306–318. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Park, J.C.; Tae-Wan, K.; Kim, T.W.; Jung, B.J.; Lee, Y.; Shim, E.K.; Park, S.; Choi, E.Y.; Cho, K.S.; et al. Human bone marrow stem cells cultured under hypoxic conditions present altered characteristics and enhanced in vivo tissue regeneration. Bone 2015, 78, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Valorani, M.G.; Montelatici, E.; Germani, A.; Biddle, A.; Alessandro, D.D.; Strollo, R.; Patrizi, M.P.; Lazzari, L.; Nye, E.; Otto, W.R.; et al. Pre-culturing human adipose tissue mesenchymal stem cells under hypoxia increases their adipogenic and osteogenic differentiation potentials. Cell Prolif. 2012, 45, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Zubillaga, V.; Alonso-varona, A.; Fernandes, S.C.M.; Salaberria, A.M.; Palomares, T. Adipose-derived mesenchymal stem cell chondrospheroids cultured in hypoxia and a 3D porous chitosan/chitin nanocrystal scaffold as a platform for cartilage tissue engineering. Int. J. Mol. Sci. 2020, 21, 1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Efimenko, A.; Starostina, E.; Kalinina, N.; Stolzing, A. Angiogenic properties of aged adipose derived mesenchymal stem cells after hypoxic conditioning. J. Transl. Med. 2011, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Pan, Q.; Wang, D.; Chen, D.; Sun, Y.; Feng, X.; Shi, X.; Xu, Y.; Luo, X.; Yu, J.; Li, Y.; et al. Characterizing the effects of hypoxia on the metabolic profiles of mesenchymal stromal cells derived from three tissue sources using chemical isotope labeling liquid chromatography-mass spectrometry. Cell Tissue Res. 2019, 380, 79–91. [Google Scholar] [CrossRef]
- Shell, K.; Raabe, O.; Freitag, C.; Ohrndorf, A.; Christb, H.-J.; Wenisch, S.; Arnhold, S. Comparison of Equine Adipose Tissue-Derived Stem Cell Behavior and Differentiation Potential Under the Influence of 3% and 21% Oxygen Tension. J. Equine Vet. Sci. 2013, 33, 74–82. [Google Scholar] [CrossRef]
- Roemeling-van Rhijn, M.; Mensah, F.K.F.; Korevaar, S.S.; Leijs, M.J.; van Osch, G.J.V.M.; Ijzermans, J.M.N.; Betjes, M.G.H.; Baan, C.C.; Weimar, W.; Hoogduijn, M.J. Effects of hypoxia on the immunomodulatory properties of adipose tissue-derived mesenchymal stem cells. Front. Immunol. 2013, 4, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.R.; Pingguan-Murphy, B.; Abas, W.A.B.W.; Azmi, M.A.N.; Omar, S.Z.; Chua, K.H.; Safwani, W.K.Z.W. Hypoxia Promotes Growth and Viability of Human Adipose-Derived Stem Cells with Increased Growth Factors Secretion. J. Asian Sci. Res. 2014, 4, 328–338. [Google Scholar]
- Choi, J.R.; Pingguan-Murphy, B.; Abas, W.A.B.W.; Azmi, M.A.N.; Omar, S.Z.; Chua, K.H.; Safwani, W.K.Z.W. Impact of low oxygen tension on stemness, proliferation and differentiation potential of human adipose-derived stem cells. Biochem. Biophys. Res. Commun. 2014, 448, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.E.M.B.; Murakami, M.; Kaneko, S.; Nakashima, M. The effects of hypoxia on the stemness properties of human dental pulp stem cells (DPSCs). Sci. Rep. 2016, 6, 35476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iida, K.; Takeda-Kawaguchi, T.; Tezuka, Y.; Kunisada, T.; Shibata, T.; Tezuka, K. Hypoxia enhances colony formation and proliferation but inhibits differentiation of human dental pulp cells. Arch. Oral Biol. 2010, 55, 648–654. [Google Scholar] [CrossRef] [PubMed]
- Aranha, A.M.F.; Zhang, Z.; Neiva, K.G.; Costa, C.A.S.; Hebling, J.; Nör, J.E. Hypoxia Enhances the Angiogenic Potential of Human Dental Pulp Cells. J. Endod. 2010, 36, 1633–1637. [Google Scholar] [CrossRef]
- Peng, L.; Shu, X.; Lang, C.; Yu, X. Effects of hypoxia on proliferation of human cord blood-derived mesenchymal stem cells. Cytotechnology 2016, 68, 1615–1622. [Google Scholar] [CrossRef] [Green Version]
- Kheirandish, M.; Gavgani, S.P.; Samiee, S. The effect of hypoxia preconditioning on the neural and stemness genes expression profiling in human umbilical cord blood mesenchymal stem cells. Transfus. Apher. Sci. 2017, 56, 392–399. [Google Scholar] [CrossRef]
- Lavrentieva, A.; Majore, I.; Kasper, C.; Hass, R. Effects of hypoxic culture conditions on umbilical cord-derived human mesenchymal stem cells. Cell Commun. Signal. 2010, 8, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Naaldijk, Y.; Johnson, A.A.; Ishak, S.; Meisel, H.J.; Hohaus, C.; Stolzing, A. Migrational changes of mesenchymal stem cells in response to cytokines, growth factors, hypoxia, and aging. Exp. Cell Res. 2015, 338, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Huang, S.; Zhao, Z. Comparison of Different Culture Conditions for Mesenchymal Stem Cells from Human Umbilical Cord Wharton’s Jelly for Stem Cell Therapy. Turkish J. Hematol. 2020, 37, 67–69. [Google Scholar] [CrossRef] [PubMed]
- Balgi-Agarwal, S.; Winter, C.; Corral, A.; Mustafa, S.B.; Hornsby, P.; Moreira, A. Comparison of Preterm and Term Wharton’s Jelly-Derived Mesenchymal Stem Cell Properties in Different Oxygen Tensions. Cells Tissues Organs 2018, 205, 137–150. [Google Scholar] [CrossRef]
- Teixeira, F.G.; Panchalingam, K.M.; Anjo, S.I.; Manadas, B.; Pereira, R.; Sousa, N.; Salgado, A.J.; Behie, L.A. Do hypoxia/normoxia culturing conditions change the neuroregulatory profile of Wharton Jelly mesenchymal stem cell secretome? Stem Cell Res. Ther. 2015, 6, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widowati, W.; Wijay, L.; Bachtiar, I.; Gunanegara, R.F.; Sugeng, S.U.; Irawan, Y.A.; Sumitrod, S.B.; Widodoe, M.A. Effect of oxygen tension on proliferation and characteristics of Wharton’s jelly-derived mesenchymal stem cells. Biomark. Genom. Med. 2014, 6, 43–48. [Google Scholar] [CrossRef] [Green Version]
- Obradovic, H.; Krstic, J.; Trivanovic, D.; Mojsilovic, S.; Okic, I.; Kukolj, T.; Ilic, V.; Jaukovic, A.; Terzic, M.; Bugarski, D. Improving stemness and functional features of mesenchymal stem cells from Wharton’s jelly of a human umbilical cord by mimicking the native, low oxygen stem cell niche. Placenta 2019, 82, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, D.; Bhonde, R.; Datta, I. Influence of ischemic microenvironment on human Wharton’s Jelly mesenchymal stromal cells. Placenta 2013, 34, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Sun, J.; Dai, Y.; Cao, P.; Zhang, L.; Peng, S.; Zhou, Y.; Li, G.; Tang, J.; Xiang, J. HIF-1A and C/EBPs transcriptionally regulate adipogenic differentiation of bone marrow-derived MSCs in hypoxia. Stem Cell Res. Ther. 2015, 6, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drela, K.; Sarnowska, A.; Siedlecka, P.; Szablowska-Gadomska, I.; Wielgos, M.; Jurga, M.; Lukomska, B.; Domanska-Janik, K. Low oxygen atmosphere facilitates proliferation and maintains undifferentiated state of umbilical cord mesenchymal stem cells in an hypoxia inducible factor-dependent manner. Cytotherapy 2014, 16, 881–892. [Google Scholar] [CrossRef]
- Musial-Wysocka, A.; Kot, M.; Sukowski, M.; Badyra, B.; Majka, M. Molecular and functional verification of wharton’s jelly mesenchymal stem cells (WJ-MSCs) pluripotency. Int. J. Mol. Sci. 2019, 20, 1807. [Google Scholar] [CrossRef] [Green Version]
- Lech, W.; Figiel-Dabrowska, A.; Sarnowska, A.; Drela, K.; Obtulowicz, P.; Noszczyk, B.H.; Buzanska, L.; Domanska-Janik, K. Phenotypic, Functional, and Safety Control at Preimplantation Phase of MSC-Based Therapy. Stem. Cell Int. 2016, 2016, 2514917. [Google Scholar] [CrossRef] [Green Version]
- Kwon, S.Y.; Chun, S.Y.; Ha, Y.-S.; Kim, D.H.; Kim, J.; Song, P.H.; Kim, H.T.; Yoo, E.S.; Kim, B.S.; Kwon, T.G. Hypoxia Enhances Cell Properties of Human Mesenchymal Stem Cells. Tissue Eng. Regen. Med. 2017, 14, 595–604. [Google Scholar] [CrossRef]
- Kim, J.H.; Song, S.Y.; Park, S.G.; Song, S.U.; Xia, Y.; Sung, J.H. Primary involvement of NADPH oxidase 4 inhypoxia-induced generation of reactive oxygen species in adipose-derived stem cells. Stem. Cells Dev. 2012, 21, 2212–2221. [Google Scholar] [CrossRef] [Green Version]
- Bae, H.C.; Park, H.J.; Wang, S.Y.; Yang, H.R.; Lee, M.C.; Han, H.S. Hypoxic condition enhances chondrogenesis in synovium-derived mesenchymal stem cells. Biomater. Res. 2018, 26, 22–28. [Google Scholar] [CrossRef] [Green Version]
- Hall, B.M.; Balan, V.; Gleiberman, A.S.; Strom, E.; Krasnov, P.; Virtuoso, L.P.; Rydkina, E.; Vujcic, S.; Balan, K.; Gitlin, I.; et al. Aging of mice is associated with p16(Ink4a)- and β-galactosidase-positive macrophage accumulation that can be induced in young mice by senescent cells. Aging 2016, 8, 1294–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, B.M.; Balan, V.; Gleiberman, A.S.; Strom, E.; Krasnov, P.; Virtuoso, L.P.; Rydkina, E.; Vujcic, S.; Balan, K.; Gitlin, I.; et al. p16(Ink4a) and senescence-associated β-galactosidase can be induced in macrophages as part of a reversible response to physiological stimuli. Aging 2017, 9, 1867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.S.; Ko, Y.J.; Lee, M.W.; Park, H.J.; Park, Y.J.; Kim, D.I.; Sung, K.W.; Koo, H.H.; Yoo, K.H. Effect of lowoxygen tension on the biological characteristics of human bone marrow mesenchymal stem cells. Cell Stress Chaperones 2016, 21, 1089–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, C.C.; Chen, Y.J.; Yew, T.L.; Chen, L.L.; Wang, J.Y.; Chiu, C.H.; Hung, S.C. Hypoxia inhibits senescence and maintains mesenchymal stem cell properties through down-regulation of E2A-p21 by HIF-TWIS. Blood 2011, 117, 459–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vono, R.; Garcia, E.J.; Spinetti, G.; Madeddu, P. Oxidative Stress in Mesenchymal Stem Cell Senescence: Regulation by Coding and Noncoding RNAs. Antioxid. Redox Signal. 2018, 29, 864–879. [Google Scholar] [CrossRef]
- Chen, C.; Tang, Q.; Zhang, Y.; Yu, M.; Jing, W.; Tian, W. Physioxia: A more effective approach for culturing human adipose-derived stem cells for cell transplantation. Stem Cell Res. Ther. 2018, 9, 148. [Google Scholar] [CrossRef]
- Ratushnyy, A.; Lobanova, M.; Buravkova, L.B. Expansion of adipose tissue-derived stromal cells at“physiologic” hypoxia attenuates replicative senescence. Cell Biochem. Funct. 2017, 35, 232. [Google Scholar] [CrossRef]
- Safwani, W.K.Z.W.; Choi, J.R.; Yong, K.W.; Ting, I.; Adenan, N.A.M.; Pingguan-Murphy, B. Hypoxia enhances the viability, growth and chondrogenic potential of cryopreserved human adipose-derived stem cells. Cryobiology 2017, 75, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Wagegg, M.; Gaber, T.; Lohanatha, F.L.; Hahne, M.; Strehl, C.; Fangradt, M.; Tran, C.L.; Schonbeck, K.; Hoff, P.; Ode, A.; et al. Hypoxia promotes osteogenesis but suppresses adipogenesis of human mesenchymal stromal cells in a hypoxia-inducible factor-1 dependent manner. PLoS ONE 2012, 7, e46483. [Google Scholar] [CrossRef] [Green Version]
- Markway, B.D.; Tan, G.K.; Brooke, G.; Hudson, J.E.; Cooper-White, J.J.; Doran, M.R. Enhanced chondrogenic differentiation of human bone marrow-derived mesenchymal stem cells in low oxygen environment micropellet cultures. Cell Transplant. 2010, 19, 29–42. [Google Scholar] [CrossRef]
- Kim, J.H.; Yoon, S.M.; Song, S.U.; Park, S.G.; Kim, W.S.; Park, I.G.; Lee, J.; Sung, J.H. Hypoxia suppresses spontaneous mineralization and osteogenic differentiation of mesenchymal stem cells via IGFBP3 Up-regulation. Int. J. Mol. Sci. 2016, 17, E1389. [Google Scholar] [CrossRef] [Green Version]
- Lech, W.; Sarnowska, A.; Kuczynska, Z.; Dabrowski, F.; Figiel-Dabrowska, A.; Domanska-Janik, K.; Buzanska, L.; Zychowicz, M. Biomimetic microenvironmental preconditioning enhance neuroprotective properties of human mesenchymal stem cells derived from Wharton’s Jelly (WJ-MSCs). Sci. Rep. 2020, 10, 16946. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Suda, T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat. Rev. Mol. Cell Biol. 2014, 15, 243–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohyeldin, A.; Garzon-Muvdi, T.; Quinones-Hinojosa, A. Oxygen in stem cell biology: A critical component of the stem cell niche. Cell Stem Cell 2010, 7, 150–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shyh-Chang, N.; Daley, G.Q.; Cantley, L.C. Stem cell metabolism in tissue development and aging. Development 2013, 140, 2535–2547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marino, L.; Castaldi, M.A.; Rosamilio, R.; Ragni, E.; Vitolo, R.; Fulgione, C.; Castaldi, S.G.; Serio, B.; Bianco, R.; Guida, M.; et al. Mesenchymal Stem Cells from the Wharton’s Jelly of the Human Umbilical Cord: Biological Properties and Therapeutic Potential. Int. J. Stem Cells 2019, 12, 218–226. [Google Scholar] [CrossRef]
- Puig-Pijuan, T.; de Godoy, M. A, Pinheiro Carvalho, L.R.; Bodart-Santos, V.; Soares Lindoso, R.; Moreno Pimentel-Coelho, P.; Mendez-Otero, R. Human Wharton’s jelly mesenchymal stem cells protect neural cells from oxidative stress through paracrine mechanisms. Future Sci. OA 2020, 6, FSO627. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tomecka, E.; Lech, W.; Zychowicz, M.; Sarnowska, A.; Murzyn, M.; Oldak, T.; Domanska-Janik, K.; Buzanska, L.; Rozwadowska, N. Assessment of the Neuroprotective and Stemness Properties of Human Wharton’s Jelly-Derived Mesenchymal Stem Cells under Variable (5% vs. 21%) Aerobic Conditions. Cells 2021, 10, 717. https://doi.org/10.3390/cells10040717
Tomecka E, Lech W, Zychowicz M, Sarnowska A, Murzyn M, Oldak T, Domanska-Janik K, Buzanska L, Rozwadowska N. Assessment of the Neuroprotective and Stemness Properties of Human Wharton’s Jelly-Derived Mesenchymal Stem Cells under Variable (5% vs. 21%) Aerobic Conditions. Cells. 2021; 10(4):717. https://doi.org/10.3390/cells10040717
Chicago/Turabian StyleTomecka, Ewelina, Wioletta Lech, Marzena Zychowicz, Anna Sarnowska, Magdalena Murzyn, Tomasz Oldak, Krystyna Domanska-Janik, Leonora Buzanska, and Natalia Rozwadowska. 2021. "Assessment of the Neuroprotective and Stemness Properties of Human Wharton’s Jelly-Derived Mesenchymal Stem Cells under Variable (5% vs. 21%) Aerobic Conditions" Cells 10, no. 4: 717. https://doi.org/10.3390/cells10040717
APA StyleTomecka, E., Lech, W., Zychowicz, M., Sarnowska, A., Murzyn, M., Oldak, T., Domanska-Janik, K., Buzanska, L., & Rozwadowska, N. (2021). Assessment of the Neuroprotective and Stemness Properties of Human Wharton’s Jelly-Derived Mesenchymal Stem Cells under Variable (5% vs. 21%) Aerobic Conditions. Cells, 10(4), 717. https://doi.org/10.3390/cells10040717