
The Nigral Coup in Parkinson’s Disease by α-Synuclein and Its Associated Rebels

{kind=link}
Abstract
1. Introduction
2. Typical Features of Parkinson’s Disease
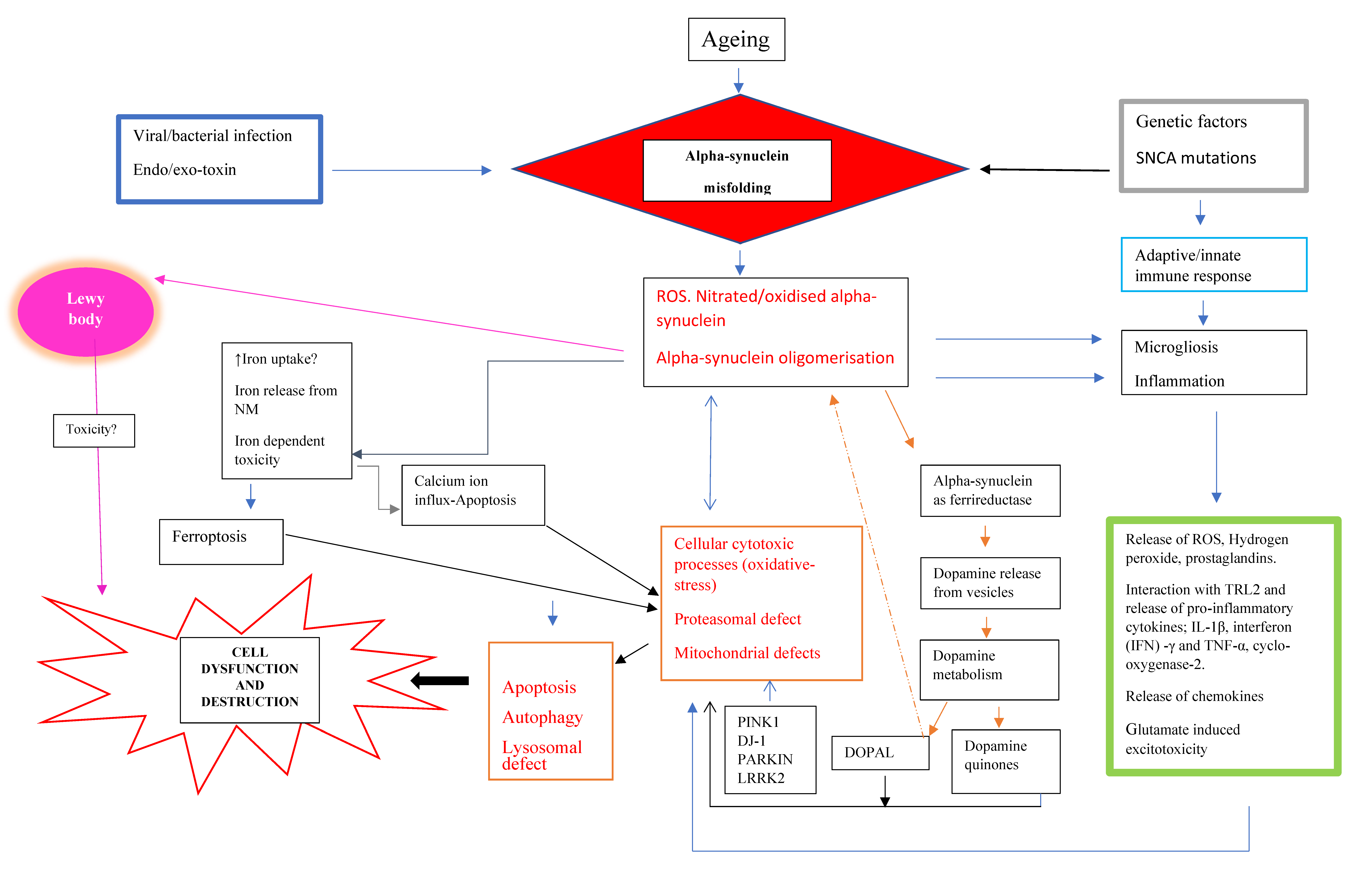
2.1. Why Dopaminergic Neurons Die?
2.2. Lewy Body Pathology
| DOPAL | 3,4 dihyroxyphenylacetaldehyde |
| NM | Neuromelanin |
| ROS | Reactive oxygen species |
| TRL-2 | Toll-like receptor 2 |
| TNF-α | Tumor necrosis factor |
| SNCA | Synuclein Alpha |
| PINK 1 | PTEN-induced kinase 1 |
| DJ-1 | Protein deglycase |
| PARKIN | (PARK2), an E3 ubiquitin ligase |
| LRRK2 | Leucine-rich repeat kinase 2 |
2.3. Disturbed Metabolic and Physiological Mechanisms
2.4. Immunological Aspects
3. Risk Factors for PD
3.1. Aging
3.2. Genetic and α-Synuclein-Related Pathological Processes in PD
3.3. Neurotoxins
3.4. Infectious Agents
3.5. Neuroinflammation
4. Conclusions
- -
- ROS-mediated OS
- -
- inflammation via pro-inflammatory cytokines (IL-1β, TNF-α and others)
- -
- microgliosis
- -
- iron dyshomeostasis by operating as ferrireductase
- -
- ferroptosis
- -
- release of dopamine from vesicles, thus augmenting dopamine metabolism resulting in OS
- -
- mitochondrial dysfunction
- -
- immunological alterations
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Twelves, D.; Perkins, K.S.; Counsell, C. Systematic review of incidence studies of Parkinson’s disease. Mov. Disord. 2003, 18, 19–31. [Google Scholar] [CrossRef]
- De Pablo-Fernandez, E.; Lees, A.J.; Holton, J.L.; Warner, T.T. Prognosis and neuropathologic correlation of clinical subtypes of Parkinson’s disease. JAMA Neurol. 2019, 76, 470–479. [Google Scholar] [CrossRef]
- Ehringer, H.; Hornykiewicz, O. Verteilung von Noradrenalin und Dopamin (3-Hydroxytyramin) im Gehirn des Menschen und ihr Verhalten bei Erkrankungen des extrapyramidalen Systems. Klin. Wochenschr. 1960, 38, 1236–1239. [Google Scholar] [CrossRef] [PubMed]
- Dijkstra, A.A.; Voorn, P.; Berendse, H.W.; Groenewegen, H.J.; Rozemuller, A.J.; van de Berg, W.D. Stage dependent nigral neuronal loss in incidental Lewy body and Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2014, 29, 1244–1251. [Google Scholar] [CrossRef] [PubMed]
- Nemani, V.; Lu, W.; Berg, V.; Nakamura, K.; Onoa, B.; Lee, M.K.; Chaudhry, F.A.; Nicoll, R.A.; Edwards, R.H. Increased expression of α-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron 2010, 65, 66–79. [Google Scholar] [CrossRef]
- Urban, P.; Falkenburger, B.; Jost, W.H.; Ransmayr, G.; Riederer, P.; Winkler, C. Struktur und Efferenzen der Substantia nigra pars compacta beim idiopathischen Parkinson-Syndrom.Structure and efferences of the substantia nigra pars compacta in Parkinson’s disease. Fortschr. Neurol. Psychiatr. 2020, 88, 591–599. [Google Scholar] [CrossRef]
- Cheng, H.C.; Ulane, C.M.; Burke, R.E. Clinical progression in Parkinson disease and the neurobiology of axons. Ann. Neurol. 2010, 67, 715–725. [Google Scholar] [CrossRef]
- Sian, J.; Dexter, D.T.; Lees, A.J.; Daniel, S.; Agid, Y.; Javoy-Agid, F.; Jenner, P.; Marsden, C.D. Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann. Neurol. 1994, 36, 348–355. [Google Scholar] [CrossRef]
- Pearce, R.K.B.; Hawkes, C.H.; Daniel, S.E. The anterior olfactory nucleus in Parkinson’s disease. Mov. Disord. 1995, 10, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Mueller, A.; Abolmaali, N.D.; Hakimi, A.R.; Gloeckler, T.; Herting, B.; Reichmann, H.; Hummel, T. Olfactory bulb volumes in patients with idiopathic Parkinson’s disease a pilot study. J. Neural Transm. 2005, 112, 1363–1370. [Google Scholar] [CrossRef] [PubMed]
- Gibb, W.R.G.; Lees, A.J. The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 1988, 51, 745–752. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-Synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Riess, O.; Kruger, R. Parkinson’s disease-a multifactorial neurodegenerative disorder. In Diagnosis and Treatment of Parkinson’s Disease—State of the Art.Journal of Neural Transmission; Przuntek, H., Müller, T., Eds.; Springer Science & Business Media: New York, NY, USA, 1999; Volume 56, pp. 113–125. [Google Scholar] [CrossRef]
- Migliore, L.; Coppedé, F. Environmental induced oxidative stress in neurodegenerative disorders and aging. Mutat. Res. 2009, 674, 73–84. [Google Scholar] [CrossRef]
- Giguère, N.; Delignat-Lavaud, B.; Herborg, F.; Voisin, A.; Li, Y.; Jacquemet, V. Increased vulnerability of nigral dopamine neurons after expansion of their axonal arborization size through D2 dopamine receptor conditional knockout. PLoS Genet. 2019, 15, e1008352. [Google Scholar] [CrossRef]
- Collier, T.J.; Kanaan, N.M.; Kordower, J.H. Ageing as a primary risk factor for Parkinson’s disease: Evidence from studies of non-human primates. Nat. Rev. Neurosci. 2011, 12, 359–366. [Google Scholar] [CrossRef]
- Cohen, G. Oxidative Stress and Parkinson’s Disease. In Reactive Oxygen Species in Biological Systems; Springer: Boston, MA, USA, 2002. [Google Scholar] [CrossRef]
- Goedert, M.; Jakes, R.; Spillantini, M.G. The Synucleinopathies: Twenty Years On. J. Parkinson’s Dis. 2017, 7, S53–S71. [Google Scholar] [CrossRef]
- Surmeier, D.J.; Obeso, J.A.; Halliday, G.M. Selective neuronal vulenerability in Parkinson’s disease. Nat. Rev. Neurosci. 2017, 18, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Yavich, L.; Tanila, H.; Vepsalainen, S.; Jakala, P. Role of α-synuclein in presynaptic dopamine recruitment. J. Neurosci. 2004, 24, 11165–11170. [Google Scholar] [CrossRef] [PubMed]
- Covell, D.J.; Robinson, J.L.; Aktrar, R.S.; Grossman, M.; Weintraub, D.; Bucklin, H.M.; Pitkin, R.M.; Riddle, D.; Yousef, A.; TroJanowski, J.Q.; et al. Novel conformation-selective alpha- synuclein antibodies raised against different in vitro fibril forms show distinct patterns of Lewy pathology in Parkinson’s disease. Neuropathol. Appl. Neurobiol. 2017, 43, 604–620. [Google Scholar] [CrossRef]
- Yamagata, Y.; Nairn, A.C. Contrasting features of ERK1/2 activity and synapsin I phosphorylation at the ERK1/2- dependent site in the rat brain in status epilepticus induced by kainic acid in vivo. Brain Res. 2015, 1625, 314–323. [Google Scholar] [CrossRef]
- Shahmoradian, S.H.; Lewis, A.J.; Hench, J.; Moors, T.E.; Navarro, P.P. Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat. Neurosci. 2019, 22, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Sian-Hulsmann, J.; Monoranu CStrobel, S.; Riederer, P. Lewy bodies: A spectator or salient killer? CNS Neurol. Disord. Drug Targets 2015, 14, 947–955. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, K.; Tanji, K.; Mori, F.; Takahashi, H. The Lewy body in Parkinson’s disease: Molecules implicated in the formation and degradation of α-synuclein aggregates. Neuropathology 2007, 27, 494–506. [Google Scholar] [CrossRef]
- Bossy-Wetzel ESchwarzenbacher, R.; Lipton, S.A. Molecular pathways to neurodegeneration. Nat. Med. 2004, 10, S2–S9. [Google Scholar] [CrossRef] [PubMed]
- Shults, C.W. Lewy bodies. Proc. Natl. Acad. Sci. USA 2006, 103, 1661–1668. [Google Scholar] [CrossRef]
- Kuusisto, E.; Parkkinen, L.; Alafuzoff, I. Morphogenesis of Lewy bodies: Dissimilar incorporation of α-synuclein, ubiquitin, and p62. J. Neuropathol. Exp. Neurol. 2003, 62, 1241–1253. [Google Scholar] [CrossRef]
- Beyer, K.; Domingo-Sàbat, M.; Ariza, A. Molecular pathology of Lewy body diseases. Int J. Mol. Sci. 2009, 10, 724–745. [Google Scholar] [CrossRef]
- Ruf, V.C.; Nübling, G.S.; Willikens, S.; Shi, S.; Schmidt, F.; Levin, J.; Bötzel, K.; Kamp, F.; Giese, A. Different Effects of α-Synuclein Mutants on Lipid Binding and Aggregation Detected by Single Molecule Fluorescence Spectroscopy and ThT Fluorescence-Based Measurements. ACS Chem. Neurosci. 2019, 10, 1649–1659. [Google Scholar] [CrossRef]
- Riederer, P.; Berg, D.; Casadei, N.; Cheng, F.; Classen, J.; Dresel, C.; Jost, W.; Krüger, R.; Müller, T.; Reichmann, H.; et al. α-Synuclein in Parkinson’s disease:causal or bystander? J. Neural Transm. 2019, 126, 815–840. [Google Scholar] [CrossRef]
- Erskine, D.; Koss, D.; Korolchuk, V.I.; Outeiro, T.F.; Attems, J.; McKeith, I. Lipids, lysosomes and mitochondria:insights into Lewy body formation from rare monogenic disorders. Acta Neuropathol. 2021. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. α-synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar] [CrossRef]
- Schlossmacher, M.G.; Frosch, M.P.; Gai, W.P.; Medina, M.; Sharma, N.; Forno, L.; Ochiishi, T.; Shimura, H.; Sharon, R.; Hattori, N.; et al. Parkin localizes to the Lewy bodies of Parkinson disease and dementia with Lewy bodies. Am. J. Pathol. 2002, 160, 1655–1667. [Google Scholar] [CrossRef]
- Lashuel, H.A. Do Lewy bodies contain α-synuclein fibrils? and Does it matter? A brief history and critical analysis of recent reports. Neurobiol. Dis. 2020, 141, 104876. [Google Scholar] [CrossRef]
- Mahul-Mellier, A.L.; Burtscher, J.; Maharjan, N.; Weerens, L.; Croisier, M.; Kuttler, F.; Leleu, M.; Knott, G.W.; Laushuel, H.A. The process of Lewy body formation, rather than simply α-synuclein fibrillization, is one of the major drivers of neurodegeneration. Proc. Natl. Acad. Sci. USA 2020, 117, 4971–4982. [Google Scholar] [CrossRef] [PubMed]
- Roberts, H.L.; Brown, D.R. Seeking a mechanism for the toxicity of oligomeric α-synuclein. Biomolecules 2015, 5, 282–305. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, J.; Bains, Y.; Guha, S.; Kahn, A.; Hall, D.; Bose, N.; Gugliucci, A.; Kapahi, P. The role of advanced glycation end products in aging and metabolic diseases: Bridging association and causality. Cell Metab. 2018, 28, 337–352. [Google Scholar] [CrossRef]
- Münch, G.; Gerlach, M.; Sian, J.; Wong, A.; Riederer, P. Advanced glycation end products in neurodegeneration: More than early markers of oxidative stress? Ann. Neurol. 1998, 44 (Suppl. S1), S85–S88. [Google Scholar] [CrossRef] [PubMed]
- Munch, G.; Luth, H.J.; Wong, A.; Arendt, T.; Hirsch, E.; Ravid, R.; Riederer, P. Crossing linking of α-synuclein by advanced glycation end products-an early pathophysiological step in Lewy body formation? J. Chem. Neuroanat. 2000, 253–257. [Google Scholar] [CrossRef]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Jenner, P.; Clark, J.B.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet 1989, 333, 1269. [Google Scholar] [CrossRef]
- Mizuno, Y.; Ohta, S.; Tanaka, M.; Takamiya, S.; Suzuki, K.; Sato, T.; Oya, H.; Ozawa, T.; Kagawa, Y. Deficiences in complex I subunits of the respiratory chain in Parkinsons. Biochem. Biophys. Res. Commun. 1989, 163, 1450–1455. [Google Scholar] [CrossRef]
- Reichmann, H.; Riederer, P. Biochemische Analyse der Atmungskettenkomplexe verschiedener Hirnregionen von Patienten mit M.Parkinson. In Morbus Parkinson und andere Basalganglienerkrankungen. Symposium des BMBF, Bad Kissingen; BMBF: Bonn, Germany, 1989; p. 44. [Google Scholar]
- Cohen, G. The pathobiology of Parkinson’s disease: Biochemical aspects of dopamine neuron senescence. J. Neural Transm. Suppl. 1983, 9, 89–103. [Google Scholar]
- Guzman, J.N.; Ilijic, E.; Yang, B.; Sanchez-Padilla, J.; Wokosin, D.; Galtieri, D.; Kondapalli, J.; Schumacker, P.T.; Surmeier, D.J. Systemic isradipine treatment diminishes calcium-dependent mitochondrial oxidant stress. J. Clin. Investig. 2018, 128, 2266–2280. [Google Scholar] [CrossRef]
- Riederer, P.; Sofic, E.; Rausch, W.D.; Schmidt, B.; Reynolds, G.P.; Jellinger, K.; Youdim, M.B. Transition metals, ferritin, glutathione, and ascorbic acid in parkinsonian brains. J. Neurochem. 1989, 52, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Freed, C.R. DJ-1 up-regulates glutathione synthesis during oxidative stress and inhibits A53T α-synuclein toxicity. J. Biol. Chem. 2005, 280, 43150–43158. [Google Scholar] [CrossRef]
- Sofic, E.; Riederer, P.; Heinsen, H.; Beckmann, H.; Reynolds, G.P.; Hebenstreit, G.; Youdim, M.B. Increased iron (III) and total iron content in post mortem substantia nigra of parkinsonian brain. J. Neural Transm. 1988, 74, 199–205. [Google Scholar] [CrossRef]
- Dexter, D.T.; Carter, C.J.; Wells, F.R.; Javoy-Agid, F.; Agid, Y.; Lees, A.; Jenner, P.; Marsden, C.D. Alterations in the levels of iron, ferritin and other trace metals in Parkinson’s disease and other neurodegenerative diseases affecting the basal ganglia. Brain 1991, 114, 1953–1975. [Google Scholar] [CrossRef]
- Belarbi, K.; Cuvelier, E.; Destée, A.; Gressier, B.; Chartier-Harlin, M.-C. NADPH oxidases in Parkinson’s disease: A systematic review. Mol. Neurodegener. 2017, 12, 84. [Google Scholar] [CrossRef] [PubMed]
- Dexter, D.T.; Carter, C.J.; Wells, F.R.; Javoy-Agid, F.; Agid, Y.; Lees, A.; Jenner, P.; Marsden, C.D. Basal Lipid Peroxidation in Substantia Nigra Is Increased in Parkinson’s Disease. J. Neurochem. 1989, 52, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Kruman, I.; Annadora JBruce-Keller Bredesen, D.; Waeg, G.; Mattson, M.P. Evidence that 4-Hydroxynonenal mediates Oxidative Stress-Induced Neuronal Apoptosis. J. Neurosci. 1997, 17, 5089–5100. [Google Scholar] [CrossRef]
- Ren, J.-X.; Sun, X.; Yan, X.L.; Guo, Z.-N.; Yang, Y. Ferroptosis in Neurological Diseases. Front. Cell Neurosci. 2020, 14, 218. [Google Scholar] [CrossRef]
- Mahoney-Sánchez, L.; Bouchaoui, H.; Ayton, S.; Devos, D.; Duce, J.A.; Devedjian, J.C. Ferroptosis and its potential role in the physiopathology of Parkinson’s Disease. Prog. Neurobiol. 2020, 101890. [Google Scholar] [CrossRef]
- Liu, B.; Moloney, A.; Meehan, S.; Morris, K.; Thomas, S.E.; Serpell, L.C.; Hider, R.; Marciniak, S.J.; Lomas, D.A.; Crowther, D.C. Iron promotes the toxicity of amyloid β peptide by impeding its ordered aggregation. J. Biol. Chem. 2011, 286, 4248–4256. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef]
- Zecca, L.; Youdim, M.B.H.; Riederer, P.; Connor, J.R.; Crichton, R.R. Iron, brain ageing and neurodegenerative disorders. Nat. Rev. Neurosci. 2004, 11, 863–873. [Google Scholar] [CrossRef]
- Gerlach, M.; Double, K.L.; Riederer, P. Iron-Induced Dopaminergic Cell Death In Vivo as a Model of Parkinson’s Disease. In Handbook of Neurotoxicity; Kostrzewa, R., Ed.; Springer: Berlin/Heidelberg, Germany, 2015; pp. 2065–2073. [Google Scholar]
- Sian-Hulsmann, J.; Riederer, P. The role of α-synuclein as ferrireductase in neurodegeneration associated with Parkinson’s disease. J. Neural Transm. Neurol. Preclin. Neurol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Souza, J.M.; Giasson, B.I.; Chen, Q.; Lee, V.M.Y.; Ischiropoulos, H. Dityrosine Cross-linking Promotes Formation of Stable a-Synuclein Polymers imnplication of nitrative and oxidative stress in the pathogenesis of neurodegenerative synucleinopathies. J. Biol. Chem. 2000, 275, 18344–18349. [Google Scholar] [CrossRef] [PubMed]
- Puspita, L.; Chung, S.Y.; Shim, J.W. Oxidative stress and cellular pathologies in Parkinson’s disease. Mol. Brain 2017, 10, 53. [Google Scholar] [CrossRef]
- Angelova, P.R.; Choi, M.L.; Berezhnov, A.V.; Horrocks, M.H.; Hughes, C.D.; De, S.; Rodrigues, M.; Yapom, R.; Little, D.; Dolt, K.S.; et al. Alpha synuclein aggregation drives ferroptosis: An interplay of iron, calcium and lipid peroxidation. Cell Death Differ. 2020, 27, 1–2796. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.R. A Synuclein as a ferrireductase. Biochem. Soc. Trans. 2013, 41, 1513–1517. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Ueda, K.; Chan, P. α-synuclein and dopamine metabolism. Mol. Neurobiol. 2005, 31, 243–254. [Google Scholar] [CrossRef]
- Jinsmaa, Y.; Sullivan, P.; Gross, D.; Cooney, A.; Sharabi, Y.; Goldstein, D. Divalent metal ions enhance DOPAL-induced oligomerization of α-synuclein. Neurosci. Lett. 2014, 569, 27–32. [Google Scholar] [CrossRef]
- Jinsmaa, Y.; Isonaka, R.; Sharabi, Y.; Goldstein, D. 3,4-Dihydroxyphenylacetaldehyde Is More Efficient than Dopamine in Oligomerizing and Quinonizing α-Synuclein. J. Pharmacol. Exp. Ther. 2020, 372, 157–165. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; Itagaki, S.; Boyes, B.E.; McGeer, E.G. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 1988, 8, 1285–1291. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Suk, J.E.; Bae, E.J.; Lee, J.H.; Paik, S.R.; Lee, S.J. Assembly-dependent endocytosis and clearance of extracellular α-synuclein. Int. J. Biochem. Cell Biol. 2008, 40, 1835–1849. [Google Scholar] [CrossRef] [PubMed]
- Block, M.L.; Zecca, L.; Hong, J.S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef]
- Liu, Y.; Ye, Z.; Li, X.; Anderson, J.L.; Khan, M.; DaSilva, D.; Baron, M.; Wilson, D.; Bocoun, V.; Ivacic, L.C.; et al. Genetic and Functional Associations with Decreased Anti- inflammatory Tumor Necrosis Factor Alpha Induced Protein 3 in Macrophages from Subjects with Axial Spondyloarthritis. Front. Immunol. 2017, 8, 860. [Google Scholar] [CrossRef] [PubMed]
- Arlehamn, C.S.L.; Dhanwani, R.; Kuan, R.; Frazier, A.; Rezende Dutra, J.R.; Phillips, E.; Mallal, S.; Roederer, M.; Marder, K.S.; Amara, A.W.; et al. α-Synuclein-specific T cell reactivity is associated with preclinical and early Parkinson’s disease. Nat. Commun. 2020, 11, 1875. [Google Scholar] [CrossRef]
- Mogi, M.; Harada, M.; Kondob, T.; Riederer, P.; Inagaki, H.; Minami, M.; Nagatsu, T. Interleukin-1β, interleukin-6, epidermal growth factor and transforming growth factor-α are elevated in the brain from parkinsonian patients. Neurosci. Lett. 1994, 180, 147–150. [Google Scholar] [CrossRef]
- Nagatsu, T.; Mogi, M.; Ichinose, H.; Togari, A. Changes in cytokines and neurotrophins in Parkinson’s disease. J. Neural Transm. 2000, 60, 277–290. [Google Scholar] [CrossRef]
- Mount, M.P.; Lira, A.; Grimes, D.; Smith, P.D.; Faucher, S.; Slack, R.; Anisman, H.; Hayley, S.; Park, D.S. Involvement of interferon-γ in microglial-mediated loss of dopaminergic neurons. J. Neurosci. 2007, 27, 3328–3337. [Google Scholar] [CrossRef] [PubMed]
- Chao, Y.; Wong, S.W.; Tan, E.K. Evidence of Inflammatory System Involvement in Parkinson’s Disease. Biomed. Res. Int. 2014, 2014. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Hunot, S. Neuroinflammation in Parkinson’s disease: A target for neuroprotection? Lancet Neurol. 2009, 8, 382–397. [Google Scholar] [CrossRef]
- Jin, F.; Wu, Q.; Lu, Y.F.; Gong, Q.H.; Shi, J.S. Neuroprotective effect of resveratrol on 6-OHDA-induced Parkinson’s disease in rats. Eur. J. Pharmacol. 2008, 600, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Reeve ASimcox, E.; Turnbull, D. Ageing and Parkinson’s disease:why is advancing age the biggest risk factor? Ageing Res. Rev. 2014, 14, 19–30. [Google Scholar] [CrossRef]
- Pringsheim, T.; Jette, N.; Frolkis, A.; Steeves, T.D.L. The prevalence of Parkinson’s disease: A systematic review and meta analysis. Mov. Disord. 2014, 29, 1583–1590. [Google Scholar] [CrossRef] [PubMed]
- Zucca, F.A.; Segura-Aguilar, J.; Ferrari, E.; Muñoz PIrmgard Paris Sulzer, D.; Sarna, T.; Casella, L.; Zecca, L. Interactions of Iron, dopamine and neuromelanin pathways in brain aging and Parkinson’s disease. Prog. Neurobiol. 2017, 155, 96–119. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, M.; Double, K.L.; Ben-Shachar, D.; Zecca, L.; Youdim, M.B.H.; Riederer, P. Neuromelanin and its interaction with iron as a potential risk factor for dopaminergic neurodegeneration underlying Parkinson’s disease. Neurotox. Res. 2003, 5, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Oberländer, U.; Pletinckx, K.; Döhler, A.; Müller, N.; Lutz, M.B.; Arzberger, T.; Riederer, P.; Gerlach, M.; Koutsilieri, E.; Scheller, C. Neuromelanin is an immune stimulator for dendritic cells in vitro. BMC Neurosci. 2011, 12, 116. [Google Scholar] [CrossRef]
- Gibb, W.R.G.; Lees, A.J. Anatomy, pigmentation, ventral and dorsal subpopulations of the substantia nigra, and differential cell death in Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 1991, 54, 388–396. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef]
- Killinger, B.A.; Kordower, J.H. Spreading of α-synuclein-relevant or epiphenomenon? J. Neurochem. 2019, 5, 605–611. [Google Scholar] [CrossRef]
- Brundin, P.; Melki, R. Prying into the Prion hypothesis for Parkinson’s disease. J. Soc. Neurosci. 2017, 37, 9808–9818. [Google Scholar] [CrossRef]
- Li, J.Y.; Englund, E.; Holton, J.L.; Soulet, D.; Hagell, P.; Lees, A.J.; Lashley, T.; Quinn, N.P.; Rehncrona, S.; Björklund, A.; et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat. Med. 2008, 14, 501–503. [Google Scholar] [CrossRef]
- Vargas, J.Y.; Grudina, C.; Zurzolo, C. The prion-like spreading of α-synuclein:from in vitro to in vivo models of Parkinson’s disease. Ageing Res. 2019, 50, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Abounit, S.; Bousset, L.; Loria, F.; Zhu, S.; De Chaumont, F.; Pieri, L.; Olivo-Marin, J.-C.; Melki, R.; Zurzolo, C. Tunneling nanotubes spread fibrillar α-synuclein by intercellular trafficking of lysosomes. EMBO J. 2016, 35, 2120–2138. [Google Scholar] [CrossRef] [PubMed]
- Dieriks, B.V.; Park, T.I.; Fourie, C.; Faull, R.L.; Dragunow, M.; Curtis, M.A. α-synuclein transfer through tunneling nanotubes occurs in SH-SY5Y cells and primary brain pericytes from Parkinson’s disease patients. Sci. Rep. 2017, 7, 42984. [Google Scholar] [CrossRef]
- Jeong, G.R.; Lee, B.D. Pathological functions of LRRK2 in Parkinson’s disease. Cells 2020, 9, 2565. [Google Scholar] [CrossRef]
- Bieri, G.; Brahic, M.; Bousset, L.; Couthouis, J.; Kramer, N.J.; Ma, R.; Nakayama, L.; Monbureau, M.; Defensor, E.; Schule, B.; et al. LRRK2 modifies α-synuclein pathology and spread in mouse models and human neurons. Acta Neuropathol. 2019, 137, 961–980. [Google Scholar] [CrossRef] [PubMed]
- Alkanli, N.; Ay, A. The relationship between α-synuclein (SNCA) gene polymorphisms and then development risk of Parkinson’s disease. Synucleins-Biochem. Role Dis. 2019. [Google Scholar] [CrossRef]
- Flagmeier, P.; Meisl, G.; Vendruscolo, M.; Knowles, P.J.T.; Dobson, C.M.; Buell, A.K.; Galvagnion, C. Mutations associated with familial Parkinson’s disease alter the initiation and amplification steps of α-synuclein aggregation. Proc. Natl. Acad. Sci. USA 2016, 113, 10328–10333. [Google Scholar] [CrossRef]
- Tofaris, G.K.; Kim, H.T.; Hourez, R.; Jung, J.W.; Kim, K.P.; Goldberg, A.L. Ubiquitin ligase Nedd4 promotes α-synuclein degradation by the endosomal-lysosomal pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 17004–17009. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.; Stefanis, L.; Fredenburg, R.; Lansbury, P. Impaired degradation of mutant α-synuclein by chaperone-mediated autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, I.; Pervaiz, N.; Abbasi, A. The Parkinson Disease gene SNCA: Evolutionary and structural insights with pathological implication. Sci. Rep. 2016, 6, 24475. [Google Scholar] [CrossRef]
- Meade, R.M.; Fairlie, D.P.; Mason, J.M. α-synuclein structure and Parkinson’s disease–lessons and emerging principles. Mol. Neurodegener. 2019, 14, 29. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Park, H.-J.; Zhang, J.; Junn, E.; Andrews, R.J.; Velagapudi, S.P.; Abegg, D.; Vishnu, K.; Costales, M.G.; Child-Disney, J.L.; et al. Translation of the intrinsically disordered protein α-synuclein is inhibited by a small molecule targeting its structured mRNA. Proc. Natl. Acad. Sci. USA 2020, 117, 1457–1467. [Google Scholar] [CrossRef]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Cookson, M.R. Mechanisms of Mutant LRRK2 Neurodegeneration. Adv. Neurobiol. 2017, 14, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Daher, J.P. Interaction of LRRK2 and α-Synuclein in Parkinson’s Disease. Adv. Neurobiol. 2017, 14, 209–226. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Aliaga, L.; Cai, H. α-synuclein, LRRK2 and their interplay in Parkinson’s disease. Future Neurol. 2012, 7, 145–153. [Google Scholar] [CrossRef]
- Bae, E.J.; Kim, D.K.; Kim, C.; Mante, M.; Adame, A.; Rockenstein, E.; Ulusoy, A.; Klinkenberg, M.; Jeong, G.R.; Bae, J.R.; et al. LRRK2 kinase regulates α-synuclein propagation via RAB35 phosphorylation. Nat. Commun. 2018, 9, 3465. [Google Scholar] [CrossRef]
- O’Hara, D.M.; Pawar, G.; Kalia, S.K.; Kalia, L.V. LRRK2 and α-Synuclein: Distinct or Synergistic Players in Parkinson’s Disease? Front. Neurosci. 2020, 14, 577. [Google Scholar] [CrossRef]
- Jellinger, K. Neuropathology and pathogenesis of extrapyramidal movement disorders: A critical update—I. Hypokinetic-rigid movement disorders. J. Neural Transm. 2019, 126, 933–995. [Google Scholar] [CrossRef] [PubMed]
- Henderson, M.X.; Peng, C.; Trojanowski, J.Q.; Lee, V.M.Y. LRRK2 activity does not dramatically alter α-synuclein pathology in primary neurons. Acta Neuropathol. Commun. 2018, 6, 45. [Google Scholar] [CrossRef] [PubMed]
- Henderson, M.X.; Sengupta, M.; McGeary, I.; Zhang, B.; Olufemi, M.F.; Brown, H.; Trojanowski, J.Q.; Lee, V.M.Y. LRRK2 inhibition does not impart protection from α-synuclein pathology and neuron death in non-transgenic mice. Acta Neuropathol. Commun. 2019, 7, 28. [Google Scholar] [CrossRef] [PubMed]
- West, A.B. Achieving neuroprotection with LRRK2 kinase inhibitors in Parkinson disease. Exp. Neurol. 2017, 298, 236–245. [Google Scholar] [CrossRef]
- Tolosa, E.; Vila, M.; Klein, C.; Rascol, O. LRRK2 in Parkinson disease: Challenges of clinical trials. Nat. Rev. Neurol. 2020, 16, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Wang, G. Mitochondrial dysfunction in Parkinson’s disease. Transl. Neurodegener. 2016, 5, 14. [Google Scholar] [CrossRef]
- Goedert, M. α-synuclein and neurodegenerative diseases. Nat. Rev. Neurosci. 2001, 2, 492–501. [Google Scholar] [CrossRef]
- Ge, P.; Dawson, V.L.; Dawson, T.M. PINK1 and Parkin mitochondrial quality control: A source of regional vulnerability in Parkinson’s disease. Mol. Neurodegeneration. 2020, 15, 20. [Google Scholar] [CrossRef]
- Liu, J.; Wang, X.; Lu, Y.; Duann, C.; Lu, L.; Yang, H. Pink1 interacts with α-synuclein and abrogates α-synuclein-induced neurotoxicity by activating autophagy. Cell Death Dis. 2017, 8, e3056. [Google Scholar] [CrossRef]
- Hsu, L.J.; Sagara, T.; Arroyo, A.; Rockstein, E.; Sisk, A.; Mallory, M.; Wong, J.; Takenouchi, T.; Hashimoto, M.; Masliah, E. Alpha-synuclein promotes mitochondrial deficit and oxidative stress. Am. J Pathol. 2000, 157, 401–410. [Google Scholar] [CrossRef]
- Liu, W.; Vives-Bauza, C.; Acín-Peréz-, R.; Yamamoto, A.; Tan, Y.; Li, Y.; Magrané, J.; Stavarache, M.A.; Shaffer, S.; Chang, S.; et al. PINK1 defect causes mitochondrial dysfunction, proteasomal deficit and α-synuclein aggregation in cell culture models of Parkinson’s disease. PLoS ONE 2009, 4, e4597. [Google Scholar] [CrossRef]
- Creed, R.B.; Goldberg, M.S. Analysis of α-Synuclein Pathology in PINK1 Knockout Rat Brains. Front. Neurosci. 2019, 12, 1034. [Google Scholar] [CrossRef]
- Oliveras-Salvá, M.; Macchi, F.; Coessens, V.; Deleersnijder, A.; Gérard, M.; Van der Perren, A.; Van den Haute, C.; Baekelandt, V. α-synuclein-induced neurodegeneration is exacerbated in PINK1 knockout mice. Neurobiol. Aging 2014, 35, 2625–2636. [Google Scholar] [CrossRef]
- Kamp, F.; Exner, N.; Lutz, A.K.; Wender, N.; Hegermann, J.; Brunner, B.; Nuscher, B.; Bartels, T.; Giese, A.; Beyer, K.; et al. Inhibition of mitochondrial fusion by α-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J. 2010, 29, 3571–3589. [Google Scholar] [CrossRef]
- Dolgacheva, L.P.; Berezhnov, A.V.; Fedotova, E.I.; Zinchenko, V.P.; Abramov, A.Y. Role of DJ-1 in the mechanism of pathogenesis of Parkinson’s disease. J. Bioenerg. Biomembr. 2019, 51, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Zondler, L.; Miller-Fleming, L.; Repici, M.; Goncalves, S.; Tenreiro, S.; Rosado-Ramos, R.; Betzer, C.; Straatman, K.R.; Jensen, P.H.; Giorgini, F.; et al. DJ-1 interactions with α-synuclein attenuate aggregation and cellular toxicity in models of Parkinson’s disease. Cell Death Dis. 2014, 5, e1350. [Google Scholar] [CrossRef]
- Xu, C.Y.; Kang, W.Y.; Chen, Y.M.; Jiang, T.F.; Zhang, J.; Zhang, L.N.; Ding, J.Q.; Liu, J.; Chen, S.D. DJ-1 Inhibits α-Synuclein Aggregation by Regulating Chaperone-Mediated Autophagy. Front. Aging Neurosci. 2017, 9, 308. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Rao, S.P.; Kalivendi, S.V. The deglycase activity of DJ-1 mitigates α-synuclein glycation and aggregation in dopaminergic cells: Role of oxidative stress mediated downregulation of DJ-1 in Parkinson’s disease. Free Radic. Biol. Med. 2019, 135, 28–37. [Google Scholar] [CrossRef]
- Kumar, R.; Kumar, S.; Hanpude, P.; Singh, A.B.; Johari, T.; Majumder, S.; Maiti, T.K. Partially oxidized DJ-1 inhibits α-synuclein nucleation and remodels mature α-synuclein fibrils in vitro. Commun. Biol. 2019, 2, 395. [Google Scholar] [CrossRef]
- Chung, K.K.K.; Zhang, Y.; Lim, K.L.; Tanaka, Y.; Huang, H.; Gao, J.; Ross, C.A.; Dawson, V.L.; Dawson, T.M. Parkin ubiquitinates the α-synuclein-interacting protein, synphilin-1: Implications for Lewy-body formation in Parkinson disease. Nat. Med. 2001, 7, 1144–1150. [Google Scholar] [CrossRef] [PubMed]
- Petrucelli, L.; O’Farrell, C.; Lockhart, P.J.; Baptista, M.; Kehoe, K.; Vink, L.; Choi, P.; Wolozin, B.; Farrer, M.; Hardy, J.; et al. Parkin protects against the toxicity associated with mutant α-synuclein: Proteasome dysfunction selectively affects catecholaminergic neurons. Neuron 2002, 36, 1007–1019. [Google Scholar] [CrossRef]
- Norris, K.L.; Hao, R.; Chen, L.F.; Lai, C.H.; Kapur, M.; Shaughnessy, P.J.; Chou, D.; Yan, J.; Taylor, J.P.; Engelender, S.; et al. Convergence of Parkin, PINK1, and α-Synuclein on Stress-induced Mitochondrial Morphological Remodeling. J. Biol. Chem. 2015, 290, 13862–13874. [Google Scholar] [CrossRef]
- Cartier, A.E.; Ubhi, K.; Spencer, B.; Vazquez-Roque, R.A.; Kosberg, K.A.; Fourgeaud, L.; Kanayson, P.; Patrick, C.; Rockenstein, E.; Patrick, G.N.; et al. Differential effects of UCHL1 modulation on α-synuclein in PD-like models of α-synucleinopathy. PLoS ONE 2012, 7, e34713. [Google Scholar] [CrossRef]
- Liu, Z.; Meray, R.K.; Grammatopoulos, T.N.; Fredenburg, R.A.; Cookson, M.R.; Liu, Y.; Logan, T.; Lansbury, P.T., Jr. Membrane-associated farnesylated UCH-L1 promotes α-synuclein neurotoxicity and is a therapeutic target for Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 4635–4640. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Fallon, L.; Lashuel, H.A.; Liu, Z.; Lansbury, P.T., Jr. The UCH-L1 gene encodes two opposing enzymatic activities that affect α-synuclein degradation and Parkinson’s disease susceptibility. Cell 2002, 111, 209–218. [Google Scholar] [CrossRef]
- Kumar, R.; Jangir, D.; Verma, G.; Shekhar, S.; Hanupude, P.; Kumar, S.; Kumari, R.; Singh, N.; Bhavesh, N.S.; Jana, N.J.; et al. S-nitrosylation of UCHL1 induces its structural instability and promotes α-synuclein aggregation. Sci. Rep. 2017, 7, 44558. [Google Scholar] [CrossRef] [PubMed]
- Fornai, F.; Schlüter, O.M.; Lenzi, P.; Gesi, M.; Ruffoli, R.; Ferrucci, M.; Lazzeri, G.; Busceti, C.L.; Pontarelli, F.; Battaglia, G.; et al. Parkinson-like syndrome induced by continuous MPTP infusion: Convergent roles of the ubiquitin-proteasome system and α-synuclein. Proc. Natl. Acad. Sci. USA 2005, 102, 3413–3418. [Google Scholar] [CrossRef]
- Manning-Bog, A.B.; McCormack, A.L.; Purisai, M.G.; Bolin, L.M.; Di Monte, D.A. α-synuclein overexpression protects against paraquat-induced neurodegeneration. J. Neurosci. 2003, 23, 3095–3099. [Google Scholar] [CrossRef]
- Jethva, P.N.; Kardani, J.R.; Roy, I. Modulation of α-synuclein aggregation by dopamine in the presence of MPTP and its metabolite. FEBS J. 2011, 278, 1688–1698. [Google Scholar] [CrossRef]
- Vila, M.; Vukosavic, S.; Jackson-Lewis, V.; Neystat, M.; Jakowec, M.; Przedborski, S. α-synuclein up-regulation in substantia nigra dopaminergic neurons following administration of the parkinsonian toxin MPTP. J. Neurochem. 2000, 74, 721–729. [Google Scholar] [CrossRef]
- Kowall, N.W.; Hantraye, P.; Brouillet, E.; Beal, M.F.; McKee, A.C.; Ferrante, R.J. MPTP induces α-synuclein aggregation in the substantia nigra of baboons. Neuroreport 2000, 11, 211–213. [Google Scholar] [CrossRef] [PubMed]
- Dauer, W.; Kholodilov, N.; Vila, M.; Trillat, A.C.; Goodchild, R.; Larsen, K.E.; Staal, R.; Tieu, K.; Schmitz, Y.; Yuan, C.A.; et al. Resistance of alpha-synuclein null mice to the parkinsonian neurotoxin MPTP. Proc. Natl. Acad. Sci. USA 2002, 99, 14524–14529. [Google Scholar] [CrossRef]
- Klivenyi, P.; Siwek, D.; Gardian, G.; Yang, L.; Starkov, A.; Cleren, C.; Ferrante, R.J.; Kowall, N.W.; Abeliovich, A.; Beal, M.F. Mice lacking α-synuclein are resistant to mitochondrial toxins. Neurobiol. Dis. 2006, 21, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Shimoji, M.; Zhang, L.; Mandir, A.S.; Dawson, V.L.; Dawson, T.M. Absence of inclusion body formation in the MPTP mouse model of Parkinson’s disease. Brain Res. Mol. Brain Res. 2005, 134, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Vermilyea, S.C.; Guthrie, S.; Hernandez, I.; Bondarenko, V.; Emborg, M.E. α-Synuclein Expression Is Preserved in Substantia Nigra GABAergic Fibers of Young and Aged Neurotoxin-Treated Rhesus Monkeys. Cell Transplant. 2019, 28, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Forno, L.S.; DeLanney, L.E.; Irwin, I.; Langston, J.W. Similarities and differences between MPTP-induced parkinsonsim and Parkinson’s disease. Neuropathologic considerations. Adv. Neurol. 1993, 60, 600–608. [Google Scholar] [PubMed]
- Heikkila, R.E.; Hess, A.; Duvoisin, R. Dopaminergic neurotoxicity of 1-methyl-4-phenyl-l,2,3,6-tetrahydropyridine in mice. Science 1984, 224, 1451–1453. [Google Scholar] [CrossRef]
- Drolet, R.E.; Behrouz, B.; Lookingland, K.J.; Goudreau, J.L. Mice lacking α-synuclein have an attenuated loss of striatal dopamine following prolonged chronic MPTP administration. Neurotoxicology 2004, 25, 761–769. [Google Scholar] [CrossRef]
- Canerina-Amaro, A.; Pereda, D.; Diaz, M.; Rodriguez-Barreto, D.; Casañas-Sánchez, V.; Heffer, M.; Garcia-Esparcia, P.; Ferrer, I.; Puertas-Avendaño, R.; Marin, R. Differential Aggregation and Phosphorylation of Alpha Synuclein in Membrane Compartments Associated With Parkinson Disease. Front. Neurosci. 2019, 13, 382. [Google Scholar] [CrossRef]
- Hoffman, L.A.; Vilensky, J.A. Encephalitis lethargica: 100 years after the epidemic. Brain 2017, 140, 2246–2251. [Google Scholar] [CrossRef]
- Ravenholt, R.; Foege, W. 1918 influenza, encephalitis lethargica, parkinsonism. Lancet N. Am. Ed. 1982, 320, 860–864. [Google Scholar] [CrossRef]
- Foley, P.B. Encephalitis lethargica and influenza. I. The role of the influenza virus in the influenza pandemic of 1918/1919. J. Neural Transm. 2009, 116, 143–150. [Google Scholar]
- Mattos, J.P.; Rosso, A.L.Z.; Corrêa, R.B.; Novis, S.A. Movement disorders in 28 HIV-infected patients. Arq. Neuro-Psiquiatr. 2002, 60, 525–530. [Google Scholar] [CrossRef]
- Vlajinac, H.; Dzoljic, E.; Maksimovic, J.; Marinkovic, J.; Sipetic, S.; Kostic, V. Infections as a risk factor for Parkinson’s disease: A case–control study. J. Neurosci. 2013, 123, 329–332. [Google Scholar] [CrossRef]
- Jellinger, K. Absence of α-synuclein pathology in postencephalitic parkinsonism. Acta Neuropathol. 2009, 118, 371–379. [Google Scholar] [CrossRef]
- Jang, H.; Boltz, D.; Sturm-Ramirez, K.; Shepherd, K.R.; Jiao, Y.; Webster, R.; Smeyne, R.J. Highly pathogenic H5N1 influenza virus can enter the central nervous system and induce neuroinflammation and neurodegeneration. Proc. Natl. Acad. Sci. USA 2009, 106, 14063–14068. [Google Scholar] [CrossRef] [PubMed]
- Bantle, C.M.; Philips, A.T.; Smeyne, R.K.; Rocha, S.M.; Olson, K.E.; Tjalken, R.B. Infection with mosquito-borne alphavirus induces selective loss of dopaminergic neurons, neuroinflammation and widespread protein aggregation. NPJ Parkinsons Dis. 2019, 5, 20. [Google Scholar] [CrossRef]
- Khanlou, N.; Moore, D.J.; Chana, G.; Cherner, M.; Lazzaretto, D.; Dawes, S.; Grant, I.; Masliah, E.; Everall, I.P. Increased frequency of alpha synuclein in the substantia nigra increased frequency of -synuclein in the substantia nigra in human immunodeficiency virus infection. J. Neurovirol. 2009, 15, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Koutsilieri, E.; Sopper, S.; Scheller, C.; Ter Meulen, V.; Riederer, P. Involvement of dopamine in the progression of AIDS Dementia Complex. J. Neural Transm. 2002, 109, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Koutsilieri, E.; Sopper, S.; Scheller, C.; Ter Meulen, V.; Riederer, P. Parkinsonism in HIV dementia. J. Neural Transm. 2002, 109, 767–775. [Google Scholar] [CrossRef]
- Wang, G.J.; Chang, L.; Volkow, N.D.; Telang, F.; Logan, J.; Ernst, T.; Fowler, J.S. Decreased brain dopaminergic transporters in HIV-associated dementia patients. Brain 2004, 127, 2452–2458. [Google Scholar] [CrossRef]
- Helms, J.; Kremer, S.; Merdji, H.; Clere-jehl, R.; Schenck, M.; Kummerlen, C. Neurologic features in severe SARS-CoV-2 infection. N. Engl. J. Med. 2020, 382, 2268–2270. [Google Scholar] [CrossRef]
- Khan, S.H.; Lindroth, H.; Hendrie, K.; Wang, S.; Imran, S.; Perkins, A.J.; Gao, S.; Vahidy, F.S.; Boustani, M.; Khan, B.A. Time trends of delirium rates in the intensive care unit. Heart Lung 2020, 49, 572–577. [Google Scholar] [CrossRef] [PubMed]
- Dreher, M.; Kersten, A.; Bickenbach, J.; Balfanz, P.; Hartmann, B.; Cornelissen, C.; Stohr, R.; Kleines, M.; Lemmen, S.W.; Brokmann, J.C.; et al. Charakteristik von 50 hospitalisierten COVID-19-Patienten mit und ohne ARDS (The characteristics of 50 hospitalized COVID-19 patients with and without ARDS). Dtsch. Arztebl. Int. 2020, 117, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Fazzani, E.; Fleming, J.; Fahn, S. Cerebrospinal fluid antibodies to coronavirus in patients with Parkinson’s disease. Mov. Disord. 1992, 7, 153–158. [Google Scholar] [CrossRef]
- Lou, J.J.; Movassaghi, M.; Gordy, D.; Olson, M.G.; Zhang, T.; Khurana, M.S.; Chen, Z.; Perez-Rosendahl, M.; Thammachachantha, S.; Singer, E.J.; et al. Neuropathology of COVID-19(neuro-COVID):clincopathological update. Free Neuropathol. 2021, 2, 2. [Google Scholar] [CrossRef] [PubMed]
- Al-Sarraj, S.; Troakes, C.; Hanley, B.; Richardson, M.P.; Hotopf, M.; Bullmore, E.; Everall, I.P. Invited review: The spectrum of neuropathology in COVID-19. Neuropathol. Appl. Neurobiol. 2021, 47, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Benezit, F.; Turnier, P.L.; Declerck, C.; Paille, C.; Revest, M.; Dubee, V.; Tattevin, P. Utility of hyposmia and hypogeusia for the diagnosis of COVID-19. Lancet, Infect. Dis. 2020, 20, 1014–1015. [Google Scholar] [CrossRef]
- Goldstein, D.S.; Sewell, L.T.; CRNP; Courtney Holmes, C. Association of anosmia with autonomic failure in Parkinson disease. Neurology 2010, 74, 245–251. [Google Scholar] [CrossRef]
- Matschke, J.; Lutgehetmann, M.; Hagel, C.; Sperhake, J.P.; Schroder, A.S.; Edler, C.; Mushumba, H.; Fitzek, A.; Allweiss, L.; Dandri, M.; et al. Neuropathology of patients with COVID-19 in Germany: A postmortem case series. Lancet Neurol. 2020, 19, 919–929. [Google Scholar] [CrossRef]
- Anderson, G.; Reiter, R.J. Melatonin: Roles in influenza, Covid-19, and other viral infections. Rev. Med. Virol. 2020, 30, e2109. [Google Scholar] [CrossRef]
- Abderrahmane, A.; Hasnaa, S.; Aziz Naciri, M.; Mohamed, A.B.; Ahmed, K.; Youssef, B.; Mohamed, N. Can the 2019 novel coronavirus cause Parkinson’s disease? Mov. Disord. 2020. [Google Scholar] [CrossRef]
- Haddadi, K.; Ghasemian, R.; Shafizad, M. Basal ganglia involvement and altered mental status: A unique neurological manifestation of coronavirus disease. Cureus 2020, 12, e7869. [Google Scholar] [CrossRef]
- Lippi, A.; Domingues, R.; Setz, C.; Outeiro, T.F.; Krisko, A. SARS-CoV-2: At the crossroad between aging and neurodegeneration. Mov. Disord. 2020, 35, 716–720. [Google Scholar] [CrossRef]
- Riederer, P.; Ter Meulen, V. Coronaviruses: A challenge of today and a call for extended human postmortem brain analysis. J. Neural Transm. Neurol. Preclin. Neurol. Stud. Rev. Artic. 2020, 127, 1–12. [Google Scholar] [CrossRef]
- Nataf, S. An alteration of the dopamine synthetic pathway is possibly involved in the pathophysiology of COVID-19. J. Med. Virol. 2020, 92, 1743–1744. [Google Scholar] [CrossRef] [PubMed]
- Antonini, A.; Leta, V.; Teo, J.; Chaudhuri, K.R. Outcome of Parkinson’s Disease Patients Affected by COVID 19. Mov. Disord. Lett. 2020. [Google Scholar] [CrossRef]
- Zhai, H.; Lv, Y.; Xu, Y.; Wu, Y.; Zeng, W.; Wang, T.; Cao, X.; Xu, Y. Characteristics of Parkinson’s disease with severe COVID-19: A study of 10 cases from Wuhan. J. Neural Transm. 2021, 128, 37–48. [Google Scholar] [CrossRef]
- Chapman, M.R.; Robinson, L.S.; Pinkner, J.S.; Roth, R.; Heuser, J.; Hammar, M.; Nomark, S.; Hultgren, S.J. Role of Escherichia coli curli operons in directing amyloid fiber formation. Science 2002, 295, 851–855. [Google Scholar] [CrossRef]
- Braak, H.; Rüb, U.; Gai, W.P.; Del Tredici, K. Idiopathic Parkinson’s disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J. Neural Transm. 2003, 110, 517–536. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Foffani, G.; Obeso, J.A.A. Cortical Pathogenic Theory of Parkinson’s Disease. Neuron 2018, 99, 1116–1128. [Google Scholar] [CrossRef] [PubMed]
- Engelender, S.; Isacson, O. The Threshold Theory for Parkinson’s Disease. Trends Neurosci. 2017, 40, 4–14. [Google Scholar] [CrossRef]
- Laperle, A.H.; Sances, S.; Yucer, N.; Dardov, V.J.; Garcia, V.J.; Ho, R.; Fulton, A.N.; Jones, M.R.; Roxas, K.M.; Avalos, P.; et al. iPSC modeling of young-onset Parkinson’s disease reveals molecular signature of disease and novel therapeutic candidates. Nat. Med. 2020, 26, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Riederer, P.; Jellinger, K.A.; Kolber, P.; Hipp, G.; Sian-Hülsmann, J.; Krüger, R. Lateralisation in Parkinson disease. Cell Tissue Res. 2018, 373, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.T.; Woulfe, J.M. Striatal blood–brain barrier permeability in Parkinson’s disease. J. Cereb. Blood Flow Metab. 2015, 35, 747–750. [Google Scholar] [CrossRef]
- Saunders, J.A.; Estes, K.A.; Kosloski, L.M.; Allen, H.E.; Dempsey, K.M.; Torres-Russotto, D.R.; Meza, J.L.; Santamaria, P.M.; Bertoni, J.M.; Murman, D.L.; et al. CD4+ regulatory and effector/memory T cell subsets profile motor dysfunction in Parkinson’s disease. J. Neuroimmune Pharmacol. 2012, 7, 927–938. [Google Scholar] [CrossRef] [PubMed]
- Kustrimovic, N.; Comi, C.; Magistrelli, L.; Rasini, E.; Legnaro, M.; Bombelli, R.; Aleksic, I.; Blandini, F.; Minafra, B.; Riboldazzi, G.; et al. Parkinson’s disease patients have a complex phenotypic and functional Th1 bias: Cross-sectional studies of CD4+ Th1/Th2/T17 and Treg in drug-naive and drug-treated patients. J. Neuroinflamm. 2018, 15, 205. [Google Scholar] [CrossRef] [PubMed]
- Manzanillo, P.S.; Ayres, J.S.; Watson ROCollins, A.C.; Souza, G.; Rae, C.S.; Schneider, D.S.; Nakamura, K.; Shiloh, M.U.; Cox, J.S. The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature 2013, 501–512. [Google Scholar] [CrossRef]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Sian-Hulsmann, J. The star studded protein and its chaperone mediated pathways:Faulty lysosomal autophagic system, alpha aggregates in Parkinson’s disease and its relevance to therapeutic intervention. EC Neurol. 2020, 12, 46–49. [Google Scholar]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sian-Hulsmann, J.; Riederer, P. The Nigral Coup in Parkinson’s Disease by α-Synuclein and Its Associated Rebels. Cells 2021, 10, 598. https://doi.org/10.3390/cells10030598
Sian-Hulsmann J, Riederer P. The Nigral Coup in Parkinson’s Disease by α-Synuclein and Its Associated Rebels. Cells. 2021; 10(3):598. https://doi.org/10.3390/cells10030598
Chicago/Turabian StyleSian-Hulsmann, Jeswinder, and Peter Riederer. 2021. "The Nigral Coup in Parkinson’s Disease by α-Synuclein and Its Associated Rebels" Cells 10, no. 3: 598. https://doi.org/10.3390/cells10030598
APA StyleSian-Hulsmann, J., & Riederer, P. (2021). The Nigral Coup in Parkinson’s Disease by α-Synuclein and Its Associated Rebels. Cells, 10(3), 598. https://doi.org/10.3390/cells10030598