
Astrocytes in Alzheimer’s Disease: Pathological Significance and Molecular Pathways

 ,
,  and
and
Abstract
1. Introduction
2. Astrocytes in the Healthy Brain
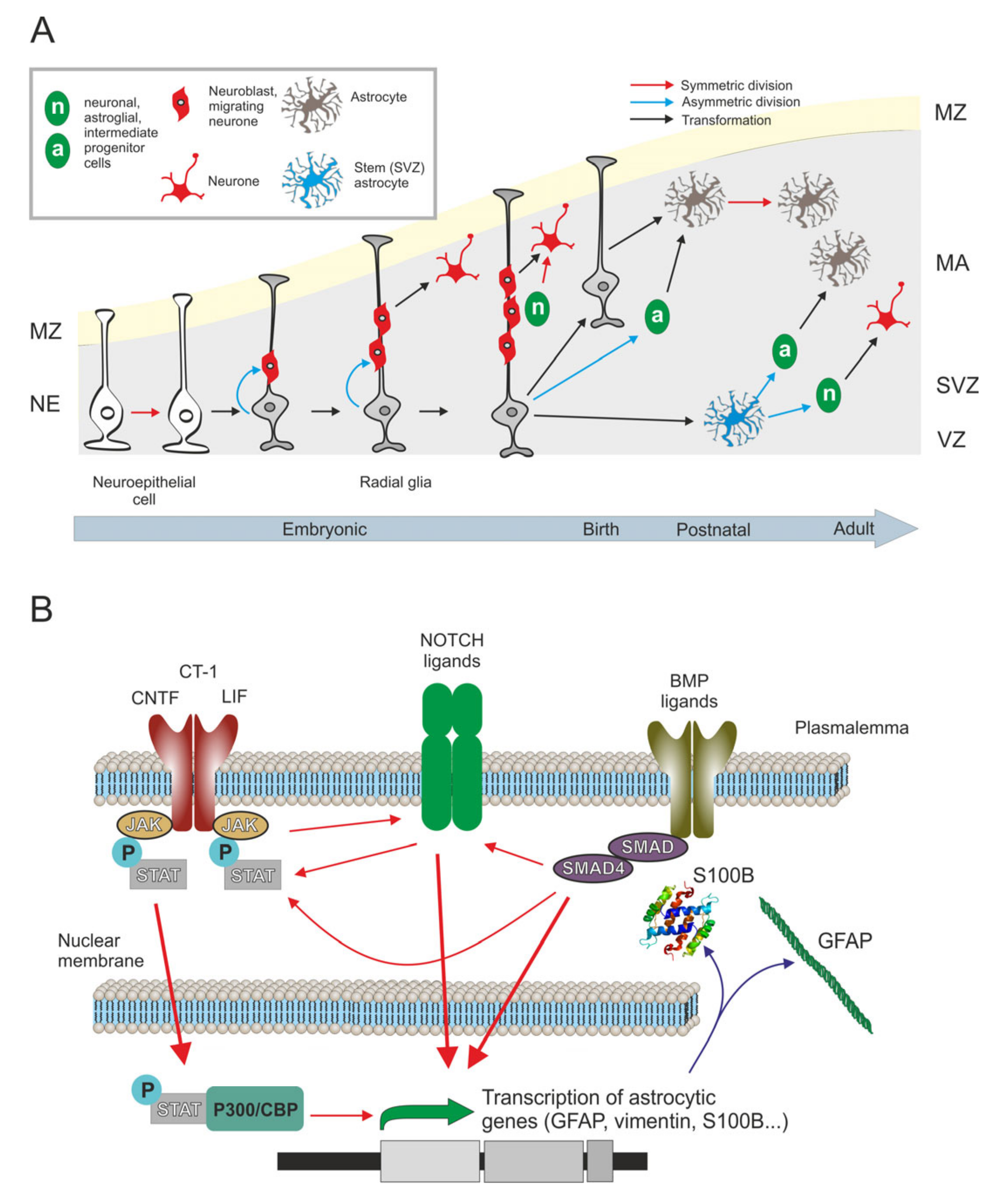
2.1. Origin, Development and Numbers
2.1.1. Origin
2.1.2. Prenatal Astrogenesis
2.1.3. Postnatal Astrogenesis
2.1.4. Astrocyte Numbers
2.2. Astrocyte Functions in Healthy Brain
2.3. Astrocyte Diversity
2.3.1. Morphological Subtypes of Cortical Astrocytes: Protoplasmic, Interlaminar, Varicose-Projection and Fibrous Astrocytes
2.3.2. Molecular Diversity and Functional Implications
3. Astrocytes in Alzheimer’s Disease
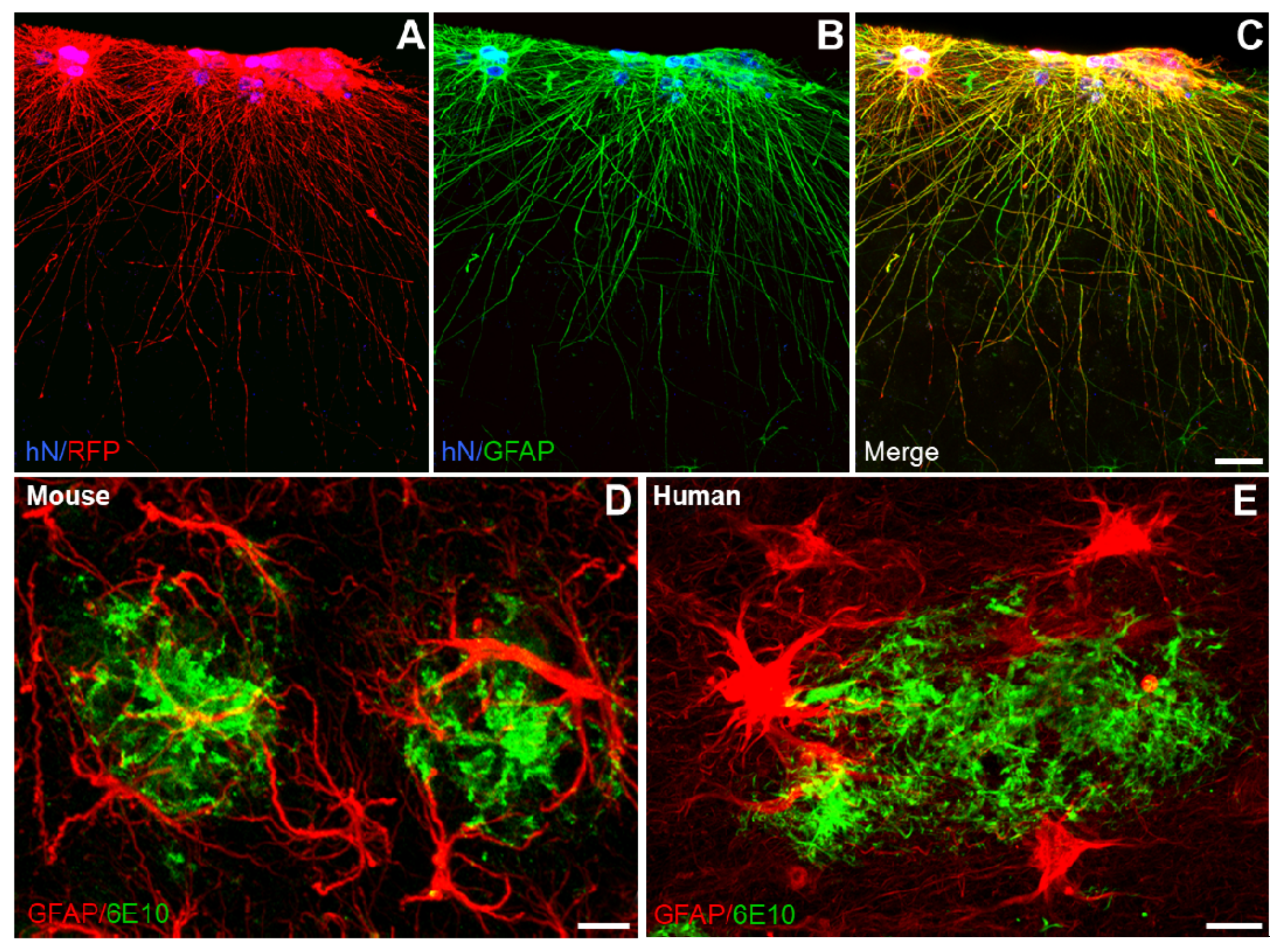
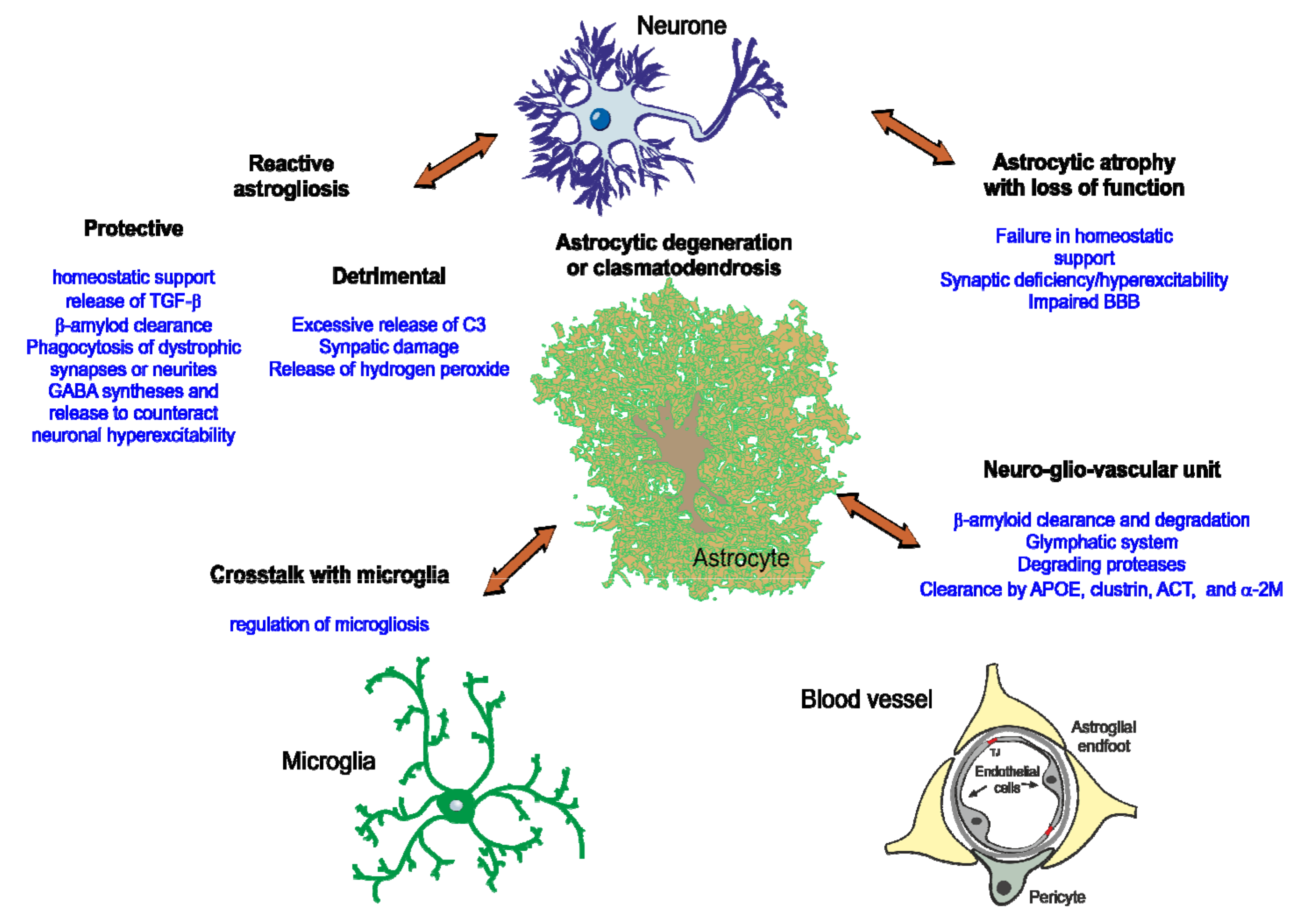
3.1. Major Roles of Astrocytes in Alzheimer´s Disease
3.2. Astrocyte Genes and Altered Molecular Pathways in AD
4. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Virchow, R. Gesammelte Abhandlungen Zyr Wissenschaftlischen Medizin; Verlag von Meidinger Sohn & Comp: Frankfurt, Germany, 1856. [Google Scholar]
- Chvátal, A.; Verkhratsky, A. An Early History of Neuroglial Research: Personalities. Neuroglia 2018, 1, 245–281. [Google Scholar] [CrossRef]
- von Lenhossék, M. Der Feinere Bau Des Nervensystems Im Lichte Neuester Forschung, 2nd ed.; Kornfield, H., Ed.; Fischer’s Medicinische Buchhandlung: Berlin, Germany, 1895. [Google Scholar]
- Ramón y Cajal, S. Contribución Al Conocimiento de La Neuroglia Del Cerebro Humano. Trab. Lab. Invest. Biol. Univ. Madr. 1913, 11, 255–315. [Google Scholar]
- Molofsky, A.V.; Deneen, B. Astrocyte development: A Guide for the Perplexed. Glia 2015, 63, 1320–1329. [Google Scholar] [CrossRef]
- Schitine, C.; Nogaroli, L.; Costa, M.R.; Hedin-Pereira, C. Astrocyte Heterogeneity in the Brain: From Development to Disease. Front. Cell. Neurosci. 2015, 9. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Nedergaard, M. Physiology of Astroglia. Physiol. Rev. 2018, 98, 239–389. [Google Scholar] [CrossRef]
- Guillemot, F. Cell fate specification in the mammalian telencephalon. Prog. Neurobiol. 2007, 83, 37–52. [Google Scholar] [CrossRef]
- He, F.; Ge, W.; Martinowich, K.; Becker-Catania, S.; Coskun, V.; Zhu, W.; Wu, H.; Castro, D.; Guillemot, F.; Fan, G.; et al. A positive autoregulatory loop of Jak-STAT signaling controls the onset of astrogliogenesis. Nat. Neurosci. 2005, 8, 616–625. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, K.; Yanagisawa, M.; Arakawa, H.; Taga, T. Astrocyte differentiation mediated by LIF in cooperation with BMP2. FEBS Lett. 1999, 457, 43–46. [Google Scholar] [CrossRef]
- Freeman, M.R. Specification and Morphogenesis of Astrocytes. Science 2010, 330, 774–778. [Google Scholar] [CrossRef] [PubMed]
- Kanski, R.; Van Strien, M.E.; Van Tijn, P.; Hol, E.M. A star is born: New insights into the mechanism of astrogenesis. Cell. Mol. Life Sci. 2013, 71, 433–447. [Google Scholar] [CrossRef]
- Urayama, S.; Semi, K.; Sanosaka, T.; Hori, Y.; Namihira, M.; Kohyama, J.; Takizawa, T.; Nakashima, K. Chromatin Accessibility at a STAT3 Target Site Is Altered Prior to Astrocyte Dif-ferentiation. Cell Struct. Funct. 2013, 38, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Gross, R.E.; Mehler, M.F.; Mabie, P.C.; Zang, Z.; Santschi, L.; Kessler, J.A. Bone Morphogenetic Proteins Promote Astroglial Lineage Commitment by Mammalian Subventricular Zone Progenitor Cells. Neuron 1996, 17, 595–606. [Google Scholar] [CrossRef]
- Krencik, R.; van Asperen, J.V.; Ullian, E.M. Human astrocytes are distinct contributors to the complexity of synaptic function. Brain Res. Bull. 2017, 129, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Niu, W.; Iqbal, N.; Smith, D.K.; Zhang, C.L. Orphan Nuclear Receptor TLX Regulates Astrogenesis by Modulating BMP Signaling. Front. Neurosci. 2014, 8, 74. [Google Scholar] [CrossRef][Green Version]
- Takizawa, T.; Ochiai, W.; Nakashima, K.; Taga, T. Enhanced gene activation by Notch and BMP signaling cross-talk. Nucleic Acids Res. 2003, 31, 5723–5731. [Google Scholar] [CrossRef]
- Ge, W.P.; Miyawaki, A.; Gage, F.H.; Jan, Y.N.; Jan, L.Y. Local Generation of Glia Is a Major Astrocyte Source in Postnatal Cortex. Nature 2012, 484, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Dimou, L.; Simon, C.; Kirchhoff, F.; Takebayashi, H.; Götz, M. Progeny of Olig2-Expressing Progenitors in the Gray and White Matter of the Adult Mouse Cerebral Cortex. J. Neurosci. 2008, 28, 10434–10442. [Google Scholar] [CrossRef]
- Du, X.; Zhang, Z.; Zhou, H.; Zhou, J. Differential Modulators of NG2-Glia Differentiation into Neurons and Glia and Their Crosstalk. Cell. Mol. Neurobiol. 2021, 41, 1–15. [Google Scholar] [CrossRef]
- Huang, W.; Guo, Q.; Bai, X.; Scheller, A.; Kirchhoff, F. Early Embryonic NG2 Glia Are Ex-clusively Gliogenic and Do Not Generate Neurons in the Brain. GLIA 2019, 67, 1094–1103. [Google Scholar] [CrossRef]
- Gomes, W.A.; Mehler, M.F.; Kessler, J.A. Transgenic Overexpression of BMP4 Increases Astroglial and Decreases Oligodendroglial Lineage Commitment. Dev. Biol. 2003, 255, 164–177. [Google Scholar] [CrossRef]
- Von Bartheld, C.S.; Bahney, J.; Herculano-Houzel, S. The search for true numbers of neurons and glial cells in the human brain: A review of 150 years of cell counting. J. Comp. Neurol. 2016, 524, 3865–3895. [Google Scholar] [CrossRef]
- Sherwood, C.C.; Stimpson, C.D.; Raghanti, M.A.; Wildman, D.E.; Uddin, M.; Grossman, L.I.; Goodman, M.; Redmond, J.C.; Bonar, C.J.; Erwin, J.M.; et al. Evolution of In-creased Glia-Neuron Ratios in the Human Frontal Cortex. Proc. Natl. Acad. Sci. USA 2006, 103, 13606–13611. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, F.A.; Carvalho, L.R.; Grinberg, L.T.; Farfel, J.M.; Ferretti, R.E.; Leite, R.E.; Jacob Filho, W.J.; Lent, R.; Herculano-Houzel, S. Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. J. Comp. Neurol 2009, 513, 532–541. [Google Scholar] [CrossRef] [PubMed]
- Pelvig, D.P.; Pakkenberg, H.; Stark, A.K.; Pakkenberg, B. Neocortical Glial Cell Numbers in Human Brains. Neurobiol. Aging 2008, 29, 1754–1762. [Google Scholar] [CrossRef]
- Keller, D.; Erö, C.; Markram, H. Cell Densities in the Mouse Brain: A Systematic Review. Front. Neuroanat. 2018, 12, 83. [Google Scholar] [CrossRef]
- Verkhratsky, A. Physiology of neuronal–glial networking. Neurochem. Int. 2010, 57, 332–343. [Google Scholar] [CrossRef]
- Rose, C.R.; Verkhratsky, A. Principles of sodium homeostasis and sodium signalling in astroglia. Glia 2016, 64, 1611–1627. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Rose, C.R. Na+-dependent transporters: The backbone of astroglial homeostatic function. Cell Calcium 2020, 85, 102136. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Semyanov, A.; Zorec, R. Physiology of Astroglial Excitability. Function 2020, 1, 16. [Google Scholar] [CrossRef]
- Vasile, F.; Dossi, E.; Rouach, N. Human astrocytes: Structure and functions in the healthy brain. Anat. Embryol. 2017, 222, 2017–2029. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Nedergaard, M. Astroglial cradle in the life of the synapse. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130595. [Google Scholar] [CrossRef]
- Allen, N.J.; Eroglu, C. Cell Biology of Astrocyte-Synapse Interactions. Neuron 2017, 96, 697–708. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, K.T.; Eroglu, C. Molecular mechanisms of astrocyte-induced synaptogenesis. Curr. Opin. Neurobiol. 2017, 45, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Kim, J.-Y.; Noh, S.; Lee, H.; Lee, S.Y.; Mun, J.Y.; Park, H.; Chung, W.-S. Astrocytes phagocytose adult hippocampal synapses for circuit homeostasis. Nat. Cell Biol. 2020, 1–6. [Google Scholar] [CrossRef]
- Jung, Y.-J.; Chung, W.-S. Phagocytic Roles of Glial Cells in Healthy and Diseased Brains. Biomol. Ther. 2018, 26, 350–357. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-Brain Barrier: From Physiology to Disease and Back. Physiol. Rev. 2019, 99, 21–78. [Google Scholar] [CrossRef]
- Liebner, S.; Dijkhuizen, R.M.; Reiss, Y.; Plate, K.H.; Agalliu, D.; Constantin, G. Functional morphology of the blood–brain barrier in health and disease. Acta Neuropathol. 2018, 135, 311–336. [Google Scholar] [CrossRef] [PubMed]
- Nortley, R.; Attwell, D. Control of brain energy supply by astrocytes. Curr. Opin. Neurobiol. 2017, 47, 80–85. [Google Scholar] [CrossRef]
- Pellerin, L.; Magistretti, P.J. Sweet Sixteen for ANLS. Br. J. Pharm. 2011, 32, 1152–1166. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A Paravascular Pathway Facilitates CSF Flow Through the Brain Parenchyma and the Clearance of Interstitial Solutes, Inluding Amyloid β. Sci. Transl. Med. 2012, 15, 147ra111. [Google Scholar] [CrossRef]
- Nedergaard, M. Garbage Truck of the Brain. Science 2013, 340, 1529–1530. [Google Scholar] [CrossRef] [PubMed]
- Oberheim, N.A.; Wang, X.; Goldman, S.; Nedergaard, M. Astrocytic Complexity Distinguishes the Human Brain. Trends Neurosci. 2006, 29, 547–553. [Google Scholar] [CrossRef]
- Bushong, E.A.; Martone, M.E.; Jones, Y.Z.; Ellisman, M.H. Protoplasmic Astrocytes in CA1 Stratum Radiatum Occupy Separate Anatomical Domains. J. Neurosci. 2002, 22, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Oberheim, N.A.; Takano, T.; Han, X.; He, W.; Lin, J.H.C.; Wang, F.; Xu, Q.; Wyatt, J.D.; Pilcher, W.; Ojemann, J.G.; et al. Uniquely Hominid Features of Adult Human Astrocytes. J. Neurosci. 2009, 29, 3276–3287. [Google Scholar] [CrossRef]
- Falcone, C.; Penna, E.; Hong, T.; Tarantal, A.F.; Hof, P.R.; Hopkins, W.D.; Sherwood, C.C.; Noctor, S.C.; Martínez-Cerdeño, V. Cortical Interlaminar Astrocytes Are Generated Prenatally, Mature Postnatally, and Express Unique Markers in Human and Nonhuman Primates. Cereb. Cortex 2021, 31, 379–395. [Google Scholar] [CrossRef] [PubMed]
- Colombo, J.; Quinn, B.; Puissant, V. Disruption of astroglial interlaminar processes in Alzheimer’s disease. Brain Res. Bull. 2002, 58, 235–242. [Google Scholar] [CrossRef]
- Sosunov, A.A.; Wu, X.; Tsankova, N.M.; Guilfoyle, E.; McKhann, G.M.; Goldman, J.E. Phenotypic Heterogeneity and Plasticity of Isocortical and Hippocampal Astrocytes in the Human Brain. J. Neurosci. 2014, 34, 2285–2298. [Google Scholar] [CrossRef]
- Kettenmann, H.; Verkhratsky, A. Glial cells. In Neuroscience in the 21st Century: From Basic to Clinical; Springer: New York, NY, USA, 2013; pp. 475–506. [Google Scholar] [CrossRef]
- Escartin, C.; Guillemaud, O.; Sauvage, M.C. Questions and (some) answers on reactive astrocytes. Glia 2019, 67, 2221–2247. [Google Scholar] [CrossRef] [PubMed]
- Jahn, H.M.; Kasakow, C.V.; Helfer, A.; Michely, J.; Verkhratsky, A.; Maurer, H.H.; Scheller, A.; Kirchhoff, F. Refined protocols of tamoxifen injection for inducible DNA recombination in mouse astroglia. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef]
- Yu, X.; Nagai, J.; Khakh, B.S. Improved tools to study astrocytes. Nat. Rev. Neurosci. 2020, 21, 121–138. [Google Scholar] [CrossRef]
- Chai, H.; Diaz-Castro, B.; Shigetomi, E.; Monte, E.; Octeau, J.C.; Yu, X.; Cohn, W.; Rajendran, P.S.; Vondriska, T.M.; Whitelegge, J.P.; et al. Neural Circuit-Specialized Astrocytes: Transcriptomic, Proteomic, Morphological, and Functional Evidence. Neuron 2017, 95, 531–549. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.C.C.; Yu, K.; Hatcher, A.; Huang, T.W.; Lee, H.K.; Carlson, J.; Weston, M.C.; Chen, F.; Zhang, Y.; Zhu, W.; et al. Identification of Diverse Astrocyte Populations and Their Malignant Analogs. Nat. Neurosci. 2017, 20, 396–405. [Google Scholar] [CrossRef]
- Morel, L.; Chiang, M.S.R.; Higashimori, H.; Shoneye, T.; Iyer, L.K.; Yelick, J.; Tai, A.; Yang, Y. Molecular and Functional Properties of Regional Astrocytes in the Adult Brain. J. Neurosci. 2017, 37, 8706–8717. [Google Scholar] [CrossRef]
- Lanjakornsiripan, D.; Pior, B.J.; Kawaguchi, D.; Furutachi, S.; Tahara, T.; Katsuyama, Y.; Suzuki, Y.; Fukazawa, Y.; Gotoh, Y. Layer-Specific Morphological and Molecular Differences in Neo-cortical Astrocytes and Their Dependence on Neuronal Layers. Nat. Commun. 2015, 9, 1623. [Google Scholar] [CrossRef]
- Morel, L.; Men, Y.; Chiang, M.S.R.; Tian, Y.; Jin, S.; Yelick, J.; Higashimori, H.; Yang, Y. Intracortical astrocyte subpopulations defined by astrocyte reporter Mice in the adult brain. Glia 2019, 67, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Bayraktar, O.A.; Bartels, T.; Holmqvist, S.; Kleshchevnikov, V.; Martirosyan, A.; Polioudakis, D.; Ben Haim, L.; Young, A.M.H.; Batiuk, M.Y.; Prakash, K.; et al. Astrocyte Layers in the Mammalian Cerebral Cortex Revealed by a Single-Cell in Situ Tran-scriptomic Map. Nat. Neurosci. 2020, 23, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Batiuk, M.Y.; Martirosyan, A.; Wahis, J.; de Vin, F.; Marneffe, C.; Kusserow, C.; Koeppen, J.; Viana, J.F.; Oliveira, J.F.; Voet, T.; et al. Identification of Region-Specific Astrocyte Subtypes at Single Cell Resolution. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. Embo Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- De Strooper, B.; Karran, E. The Cellular Phase of Alzheimer’s Disease. Cell 2016, 164, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Dubois, B.; Hampel, H.; Feldman, H.H.; Scheltens, P.; Aisen, P.; Andrieu, S.; Bakardjian, H.; Benali, H.; Bertram, L.; Blennow, K.; et al. Preclinical Alzheimer’s Disease: Definition, Natural History, and Diagnostic Criteria. Alzheimer’s Dement. 2016, 12, 292–323. [Google Scholar] [CrossRef] [PubMed]
- Frere, S.; Slutsky, I. Alzheimer’s Disease: From Firing Instability to Homeostasis Network Collapse. Neuron 2018, 97, 32–58. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Zorec, R.; Rodríguez, J.J.; Parpura, V. Astroglia dynamics in ageing and Alzheimer’s disease. Curr. Opin. Pharm. 2016, 26, 74–79. [Google Scholar] [CrossRef]
- Arranz, A.M.; De Strooper, B. The Role of Astroglia in Alzheimer’s Disease: Pathophysi-ology and Clinical Implications. Lancet Neurol. 2019, 18, 406–414. [Google Scholar] [CrossRef]
- Yu, J.-T.; Tan, L.; Hardy, J. Apolipoprotein E in Alzheimer’s Disease: An Update. Annu. Rev. Neurosci. 2014, 37, 79–100. [Google Scholar] [CrossRef] [PubMed]
- Holtzman, D.M.; Bales, K.R.; Tenkova, T.; Fagan, A.M.; Parsadanian, M.; Sartorius, L.J.; Mackey, B.; Olney, J.; McKeel, D.; Wozniak, D.; et al. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2000, 97, 2892–2897. [Google Scholar] [CrossRef]
- Verghese, P.B.; Castellano, J.M.; Garai, K.; Wang, Y.; Jiang, H.; Shah, A.; Bu, G.; Frieden, C.; Holtzman, D.M. ApoE Influences Amyloid-β (Aβ) Clearance despite Minimal ApoE/Aβ As-sociation in Physiological Conditions. Proc. Natl. Acad. Sci. USA 2013, 110, 1807–1816. [Google Scholar] [CrossRef]
- Carter, S.F.; Scholl, M.; Almkvist, O.; Wall, A.; Engler, H.; Langstrom, B.; Nordberg, A. Evidence for Astrocytosis in Prodromal Alzheimer Disease Provided by 11C-Deuterium-L-Deprenyl: A Multitracer PET Paradigm Com-bining 11C-Pittsburgh Compound B and 18F-FDG. J. Nucl. Med. 2012, 53, 37–46. [Google Scholar] [CrossRef]
- Rodriguez-Vieitez, E.; Saint-Aubert, L.; Carter, S.F.; Almkvist, O.; Farid, K.; Schöll, M.; Chiotis, K.; Thordardottir, S.; Graff, C.; Wall, A.; et al. Diverging longitudinal changes in astrocytosis and amyloid PET in autosomal dominant Alzheimer’s disease. Brain 2016, 139, 922–936. [Google Scholar] [CrossRef]
- Scholl, M.; Carter, S.F.; Westman, E.; Rodriguez-Vieitez, E.; Almkvist, O.; Thordardottir, S.; Wall, A.; Graff, C.; Långstrom, B.; Nordberg, A. Early Astrocytosis in Autosomal Dominant Alzheimer’s Disease Measured in Vivo by MultiTracer Positron Emission Tomography. Sci. Rep. 2015, 5, 1–14. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Muzikansky, A.; Gómez-Isla, T.; Growdon, J.H.; Betensky, R.A.; Frosch, M.P.; Hyman, B.T. Differential Relationships of Reactive Astrocytes and Microglia to Fibrillar Amyloid Deposits in Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2013, 72, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Escartin, C.; Galea, E.; Lakatos, A.; O’Callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhäuser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 2021, 1–14. [Google Scholar] [CrossRef]
- Beach, T.; McGeer, E. Lamina-specific arrangement of astrocytic gliosis and senile plaques in Alzheimer’s disease visual cortex. Brain Res. 1988, 463, 357–361. [Google Scholar] [CrossRef]
- Rodríguez, J.J.; Olabarria, M.; Chvatal, A.; Verkhratsky, A.; Rodr, J.J. Astroglia in dementia and Alzheimer’s disease. Cell Death Differ. 2008, 16, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Zorec, R.; Parpura, V. Stratification of Astrocytes in Healthy and Dis-eased Brain. Brain Pathol. 2017, 27, 629–644. [Google Scholar] [CrossRef] [PubMed]
- Alberdi, E.; Wyssenbach, A.; Alberdi, M.; Sanchez-Gomez, M.V.; Cavaliere, F.; Rodriguez, J.J.; Verkhratsky, A.; Matute, C. Ca(2+)—dependent endoplasmic reticulum stress correlates with astrogliosis in oligomeric amyloid beta-treated astrocytes and in a model of Alzheimer’s disease. Aging Cell 2013, 12, 292–302. [Google Scholar] [CrossRef]
- Kraft, A.W.; Hu, X.; Yoon, H.; Yan, P.; Xiao, O.; Wang, Y.; Gil, S.H.; Brown, J.; Wilhelmsson, U.; Restivo, J.L.; et al. Attenuating Astrocyte Activation Accelerates Plaque Pathogenesis in APP/PS1 Mice. FASEB J. 2013, 27, 187–198. [Google Scholar] [CrossRef]
- Agulhon, C.; Sun, M.-Y.; Murphy, T.; Myers, T.; Lauderdale, K.; Fiacco, T. Calcium Signal-ing and Glio-transmission in Normal vs. Reactive Astrocytes. Front. Pharmacol. 2012, 3, 139. [Google Scholar] [CrossRef]
- Kuchibhotla, K.V.; Lattarulo, C.R.; Hyman, B.T.; Bacskai, B.J. Synchronous hyperactivi-ty and intercellular calcium waves in astrocytes in Alzheimer mice. Science 2009, 323, 1211–1215. [Google Scholar] [CrossRef]
- Frost, G.R.; Li, Y.-M. The Role of Astrocytes in Amyloid Production and Alzheimer’s Disease. Open Biol. 2017, 7, 170228. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Rodríguez-Arellano, J.; Parpura, V.; Zorec, R. Astroglial calcium signalling in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2017, 483, 1005–1012. [Google Scholar] [CrossRef]
- Hsu, E.T.; Gangolli, M.; Su, S.; Holleran, L.; Stein, T.D.; Alvarez, V.E.; McKee, A.C.; Schmidt, R.E.; Brody, D.L. Astrocytic degeneration in chronic traumatic encephalopathy. Acta Neuropathol. 2018, 136, 955–972. [Google Scholar] [CrossRef]
- Yeh, C.-Y.; Vadhwana, B.; Verkhratsky, A.; Rodriguez, J.J. Early Astrocytic Atrophy in the Entorhinal Cortex of a Triple Transgenic Animal Model of Alzheimer’s Disease. ASN Neuro 2011, 3, AN20110025. [Google Scholar] [CrossRef] [PubMed]
- Olabarria, M.; Noristani, H.N.; Verkhratsky, A.; Rodríguez, J.J. Age-Dependent Decrease in Glutamine Synthetase Expression in the Hippocampal Astroglia of the Triple Transgenic Alzheimer’s Disease Mouse Model: Mechanism for Deficient Glutamatergic Transmission? Mol. Neurodegener. 2011, 6, 55. [Google Scholar] [CrossRef]
- Beauquis, J.; Vinuesa, A.; Pomilio, C.; Pavía, P.; Galván, V.; Saravia, F. Neuronal and Glial Alterations, Increased Anxiety, and Cognitive Impairment before Hippocampal Amyloid Deposition in PDAPP Mice, Model of Alzheimer’s Disease. Hippocampus 2014, 24, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Diniz, L.P.; Tortelli, V.; Matias, I.; Morgado, J.; Araujo, A.P.B.; Melo, H.M.; Da Silva, G.S.S.; Alves-Leon, S.V.; De Souza, J.M.; Ferreira, S.T.; et al. Astrocyte Transforming Growth Factor Beta 1 Protects Synapses against Aβ Oligomers in Alzheimer’s Disease Model. J. Neurosci. 2017, 37, 6797–6809. [Google Scholar] [CrossRef]
- Iram, T.; Trudler, D.; Kain, D.; Kanner, S.; Galron, R.; Vassar, R.; Barzilai, A.; Blinder, P.; Fishelson, Z.; Frenkel, D. Astrocytes from old Alzheimer’s disease mice are impaired in Aβ uptake and in neuroprotection. Neurobiol. Dis. 2016, 96, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Polis, B.; Srikanth, K.D.; Elliott, E.; Gil-Henn, H.; Samson, A.O. L-Norvaline Reverses Cognitive Decline and Synaptic Loss in a Murine Model of Alzheimer’s Disease. Neurotherapeutics 2018, 15, 1036–1054. [Google Scholar] [CrossRef]
- Jones, V.C.; Atkinson-Dell, R.; Verkhratsky, A.; Mohamet, L. Aberrant iPSC-Derived Human Astrocytes in Alzheimer’s Disease. Cell Death Dis. 2017, 8, e2696. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Akinyemi, R.O.; Hase, Y.; Firbank, M.J.; Ndung’U, M.N.; Foster, V.; Craggs, L.J.L.; Washida, K.; Okamoto, Y.; Thomas, A.J.; et al. Frontal white matter hyperintensities, clasmatodendrosis and gliovascular abnormalities in ageing and post-stroke dementia. Brain 2016, 139, 242–258. [Google Scholar] [CrossRef]
- Ries, M.; Sastre, M. Mechanisms of Aβ Clearance and Degradation by Glial Cells. Front. Aging Neurosci. 2016, 8, 160. [Google Scholar] [CrossRef]
- Lin, Y.-T.; Seo, J.; Gao, F.; Feldman, H.M.; Wen, H.-L.; Penney, J.; Cam, H.P.; Gjoneska, E.; Raja, W.K.; Cheng, J.; et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron 2018, 98, 1141–1154. [Google Scholar] [CrossRef]
- Simonovitch, S.; Schmukler, E.; Bespalko, A.; Iram, T.; Frenkel, D.; Holtzman, D.M.; Masliah, E.; Michaelson, D.M.; Pinkas-Kramarski, R. Impaired Autophagy in APOE4 Astrocytes. J. Alzheimer’s Dis. 2016, 51, 915–927. [Google Scholar] [CrossRef]
- Huynh, T.-P.V.; Liao, F.; Francis, C.M.; Robinson, G.O.; Serrano, J.R.; Jiang, H.; Roh, J.; Finn, M.B.; Sullivan, P.M.; Esparza, T.J.; et al. Age-Dependent Effects of apoE Reduction Using Antisense Oligonucleotides in a Model of β-amyloidosis. Neuron 2017, 96, 1013–1023.e4. [Google Scholar] [CrossRef]
- Liu, C.-C.; Zhao, N.; Fu, Y.; Wang, N.; Linares, C.; Tsai, C.-W.; Bu, G. ApoE4 Accelerates Early Seeding of Amyloid Pathology. Neuron 2017, 96, 1024–1032.e3. [Google Scholar] [CrossRef]
- Montagne, A.; Nation, D.A.; Sagare, A.P.; Barisano, G.; Sweeney, M.D.; Chakhoyan, A.; Pachicano, M.; Joe, E.; Nelson, A.R.; D’Orazio, L.M.; et al. APOE4 leads to blood–brain barrier dysfunction predicting cognitive decline. Nat. Cell Biol. 2020, 581, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Lian, H.; Zheng, H. Signaling Pathways Regulating Neuron-Glia Interaction and Their Implications in Alzheimer’s Disease. J. Neurochem. 2016, 136, 475–491. [Google Scholar] [CrossRef] [PubMed]
- Lian, H.; Yang, L.; Cole, A.; Sun, L.; Chiang, A.C.A.; Fowler, S.W.; Shim, D.J.; Rodriguez-Rivera, J.; Taglialatela, G.; Jankowsky, J.L.; et al. NFkB-Activated Astroglial Re-lease of Complement C3 Compromises Neuronal Morphology and Function Associated with Alzheimer’s Disease. Neuron 2015, 85, 101–115. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Jo, S.; Yarishkin, O.; Hwang, Y.J.; Chun, Y.E.; Park, M.; Woo, D.H.; Bae, J.Y.; Kim, T.; Lee, J.; Chun, H.; et al. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat. Med. 2014, 20, 886–896. [Google Scholar] [CrossRef]
- Ghatak, S.; Dolatabadi, N.; Trudler, D.; Zhang, X.; Wu, Y.; Mohata, M.; Ambasudhan, R.; Talantova, M.; Lipton, S.A. Mechanisms of hyperexcitability in Alzheimer’s disease hiPSC-derived neurons and cerebral organoids vs isogenic controls. Elife 2019, 8, e50333. [Google Scholar] [CrossRef] [PubMed]
- Garaschuk, O.; Verkhratsky, A. GABAergic astrocytes in Alzheimer’s disease. Aging 2019, 11, 1602–1604. [Google Scholar] [CrossRef]
- Chun, H.; Im, H.; Kang, Y.J.; Kim, Y.; Shin, J.H.; Won, W.; Lim, J.; Ju, Y.; Park, Y.M.; Kim, S.; et al. Severe reactive astrocytes precipitate pathological hallmarks of Alzheimer’s disease via H2O2− production. Nat. Neurosci. 2020, 23, 1555–1566. [Google Scholar] [CrossRef] [PubMed]
- Kapogiannis, D.; Mattson, M.P. Disrupted energy metabolism and neuronal circuit dys-function in cognitive impairment and Alzheimer’s disease. Lancet Neurol. 2011, 10, 187–198. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Molecular Neurodegeneration 2020, 15, 30. [Google Scholar] [CrossRef]
- Bubber, P.; Haroutunian, V.; Fisch, G.; Blass, J.P.; Gibson, G.E. Mitochondrial abnor-malities in Alzheimer brain: Mechanistic implications. Ann. Neurol. 2005, 57, 695–703. [Google Scholar] [CrossRef]
- Brooks, W.M.; Lynch, P.J.; Ingle, C.C.; Hatton, A.; Emson, P.C.; Faull, R.L.; Starkey, M.P. Gene expression profiles of metabolic enzyme transcripts in Alzheimer’s disease. Brain Res. 2007, 1127, 127–135. [Google Scholar] [CrossRef]
- Adav, S.S.; Park, J.E.; Sze, S.K. Quantitative profiling brain proteomes revealed mito-chondrial dysfunction in Alzheimer’s disease. Mol. Brain 2019, 12, 8. [Google Scholar] [CrossRef] [PubMed]
- Sekar, S.; McDonald, J.; Cuyugan, L.; Aldrich, J.; Kurdoglu, A.; Adkins, J.; Serrano, G.; Beach, T.G.; Craig, D.W.; Valla, J.; et al. Alzheimer’s Disease Is Associated with Altered Expression of Genes Involved in Immune Response and Mitochondrial Processes in As-trocytes. Neurobiol. Aging 2015, 36, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Grubman, A.; Chew, G.; Ouyang, J.F.; Sun, G.; Choo, X.Y.; McLean, C.; Simmons, R.K.; Buckberry, S.; Vargas-Landin, D.B.; Poppe, D.; et al. A Single-Cell Atlas of Entorhinal Cortex from Individuals with Alzheimer’s Disease Reveals Cell-Type-Specific Gene Expression Regulation. Nat. Neurosci. 2019, 22, 2087–2097. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, P.; Zaja, I.; Bienengraeber, M.; Rarick, K.R.; Terashvili, M.; Canfield, S.; Falck, J.R.; Harder, D.R. Epoxyeicosatrienoic acids pretreatment improves amyloid beta-induced mitochondrial dysfunction in cultured rat hippocampal astrocytes. Am. J. Physiol.-Heart Circ. Physiol. 2014, 306, 475–484. [Google Scholar] [CrossRef]
- Kaminsky, Y.G.; Kosenko, E.A. Effects of amyloid-beta peptides on hydrogen peroxide-metabolizing enzymes in rat brain in vivo. Free Radic. Res. 2008, 42, 564–573. [Google Scholar] [CrossRef]
- Lee, H.P.; Pancholi, N.; Esposito, L.; Previll, L.A.; Wang, X.; Zhu, X.; Smith, M.A.; Lee, H.G. Early induction of oxidative stress in mouse model of Alzheimer disease with reduced mito-chondrial superoxide dismutase activity. PLoS ONE 2012, 7, e28033. [Google Scholar] [CrossRef]
- Abramov, A.Y.; Canevari, L.; Duchen, M.R. β-amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J. Neurosci. 2004, 24, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Genda, E.N.; Jackson, J.G.; Sheldon, A.L.; Locke, S.F.; Greco, T.M.; O’Donnell, J.C.; Spruce, L.A.; Xiao, R.; Guo, W.; Putt, M.; et al. Co-compartmentalization of the astroglial glutamate transporter, GLT-1, with glycolytic enzymes and mitochondria. J. Neurosci. 2011, 31, 18275–18288. [Google Scholar] [CrossRef]
- Jackson, J.G.; O’Donnell, J.C.; Takano, H.; Coulter, D.A.; Robinson, M.B. Neuronal ac-tivity and glutamate uptake decrease mitochondrial mobility in astrocytes and position mitochondria near glutamate transporters. J. Neurosci. 2014, 34, 1613–1624. [Google Scholar] [CrossRef] [PubMed]
- Schmukler, E.; Solomon, S.; Simonovitch, S.; Goldshmit, Y.; Wolfson, E.; Michaelson, D.M.; Pinkas-Kramarski, R. Altered mitochondrial dynamics and function in APOE4-expressing astrocytes. Cell Death Dis. 2020, 11, 578. [Google Scholar] [CrossRef]
- Davis, C.-H.O.; Kim, K.-Y.; Bushong, E.A.; Mills, E.A.; Boassa, D.; Shih, T.; Kinebuchi, M.; Phan, S.; Zhou, Y.; Bihlmeyer, N.A.; et al. Transcellular degradation of axonal mitochondria. Proc. Natl. Acad. Sci. USA 2014, 111, 9633–9638. [Google Scholar] [CrossRef]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after stroke. Nat. Cell Biol. 2016, 535, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Morales, I.; Sanchez, A.; Puertas-Avendano, R.; Rodriguez-Sabate, C.; Perez-Barreto, A.; Rodriguez, M. Neuroglial transmitophagy and Parkinson’s disease. Glia 2020, 68, 2277–2299. [Google Scholar] [CrossRef]
- Gu, X.-L.; Long, C.-X.; Sun, L.; Xie, C.; Lin, X.; Cai, H. Astrocytic expression of Parkinson’s disease-related A53T α-synuclein causes neurodegeneration in mice. Mol. Brain 2010, 3, 12. [Google Scholar] [CrossRef] [PubMed]
- Solano, R.M.; Casarejos, M.J.; Menéndez-Cuervo, J.; Rodriguez-Navarro, J.A.; De Yébenes, J.G.; Mena, M.A. Glial Dysfunction in Parkin Null Mice: Effects of Aging. J. Neurosci. 2008, 28, 598–611. [Google Scholar] [CrossRef]
- Yun, S.P.; Kam, T.-I.; Panicker, N.; Kim, S.; Oh, Y.; Park, J.-S.; Kwon, S.-H.; Park, Y.J.; Karuppagounder, S.S.; Park, H.; et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat. Med. 2018, 24, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Castro, B.; Gangwani, M.R.; Yu, X.; Coppola, G.; Khakh, B.S. Astrocyte molecular signatures in Huntington’s disease. Sci. Transl. Med. 2019, 11, eaaw8546. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; Ao, Y.; Faas, G.C.; Nwaobi, S.E.; Xu, J.; Haustein, M.D.; Anderson, M.A.; Mody, I.; Olsen, M.L.; Sofro-niew, M.V.; et al. Astrocyte Kir4.1 Ion Channel Deficits Con-tribute to Neuronal Dysfunction in Huntington’s Disease Model Mice. Nat. Neurosci. 2014, 17, 694–703. [Google Scholar] [CrossRef] [PubMed]
- Alami, N.O.; Schurr, C.; Heuvel, F.O.; Tang, L.; Li, Q.; Tasdogan, A.; Kimbara, A.; Nettekoven, M.; Ottaviani, G.; Raposo, C.; et al. NF-κB Activation in Astrocytes Drives a Stage-specific Beneficial Neuroimmunological Response in ALS. EMBO J. 2018, 37, e98697. [Google Scholar] [CrossRef]
- Wheeler, M.A.; Quintana, F.J. Regulation of Astrocyte Functions in Multiple Sclerosis. Cold Spring Harbor Perspect. Med. 2019, 9, a029009. [Google Scholar] [CrossRef]
- Di Giorgio, F.P.; Carrasco, M.A.; Siao, M.S.; Maniatis, T.; Eggan, K. Non-Cell Autono-mous Effect of Glia on Motor Neurons in an Embryonic Stem Cell-Based ALS Model. Nat. Neurosci. 2007, 10, 608–614. [Google Scholar] [CrossRef]
- Gomez-Arboledas, A.; Davila, J.C.; Sanchez-Mejias, E.; Navarro, V.; Nuñez-Diaz, C.; Sanchez-Varo, R.; Sanchez-Mico, M.V.; Trujillo-Estrada, L.; Fernandez-Valenzuela, J.J.; Vizuete, M.; et al. Phagocytic clearance of presynaptic dystrophies by reactive astrocytes in Alzheimer’s disease. Glia 2018, 66, 637–653. [Google Scholar] [CrossRef]
- Orre, M.; Kamphuis, W.; Osborn, L.M.; Jansen, A.H.; Kooijman, L.; Bossers, K.; Hol, E.M. Isolation of glia from Alzheimer’s mice reveals inflammation and dysfunction. Neurobiol. Aging 2014, 35, 2746–2760. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Ma, N.; Yu, B.; Zhang, W.; Wan, J. Transcriptomic profiling of microglia and astrocytes throughout aging. J. Neuroinflammation 2020, 17, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, K.; Friedman, B.A.; Larson, J.L.; Lauffer, B.E.; Goldstein, L.D.; Appling, L.L.; Borneo, J.; Poon, C.; Ho, T.; Cai, F.; et al. Untangling the Brain’s Neuroinflammatory and Neurodegenerative Transcrip-tional Responses. Nat. Commun. 2016, 7, 11295. [Google Scholar] [CrossRef]
- Simpson, J.E.; Ince, P.G.; Shaw, P.J.; Heath, P.R.; Raman, R.; Garwood, C.J.; Gelsthorpe, C.; Baxter, L.; Forster, G.; Matthews, F.E.; et al. Microarray analysis of the astrocyte transcriptome in the aging brain: Relationship to Alzheimer’s pathology and APOE genotype. Neurobiol. Aging 2011, 32, 1795–1807. [Google Scholar] [CrossRef] [PubMed]
- Mills, J.D.; Nalpathamkalam, T.; Jacobs, H.I.; Janitz, C.; Merico, D.; Hu, P.; Janitz, M. RNA-Seq analysis of the parietal cortex in Alzheimer’s disease reveals alternatively spliced isoforms related to lipid metabolism. Neurosci. Lett. 2013, 536, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Habib, N.; McCabe, C.; Medina, S.; Varshavsky, M.; Kitsberg, D.; Dvir-Szternfeld, R.; Green, G.; Dionne, D.; Nguyen, L.; Marshall, J.L.; et al. Disease-associated astrocytes in Alzheimer’s disease and aging. Nat. Neurosci. 2020, 23, 701–706. [Google Scholar] [CrossRef]
- Zhou, Y.; Song, W.M.; Andhey, P.S.; Swain, A.; Levy, T.; Miller, K.R.; Poliani, P.L.; Cominelli, M.; Grover, S.; Gilfillan, S.; et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat. Med. 2020, 26, 131–142. [Google Scholar] [CrossRef]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef]
- Leng, K.; Li, E.; Eser, R.; Piergies, A.; Sit, R.; Tan, M.; Neff, N.; Li, S.H.; Rodriguez, R.D.; Suemoto, C.K.; et al. Molecular characterization of selectively vulnerable neurons in Alzheimer’s disease. Nat. Neurosci. 2021, 24, 276–287. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Shin, Y.J.; Kang, S.W.; Jeong, S.Y.; Shim, Y.J.; Kim, Y.H.; Kim, B.M.; Kee, S.H.; Park, J.J.; Park, I.S.; Min, B.H. Clusterin Enhances Proliferation of Primary Astrocytes through Extracellular Signal-Regulated Kinase Activation. NeuroReport 2006, 17, 1871–1875. [Google Scholar] [CrossRef]
- Kajiwara, Y.; Wang, E.; Wang, M.; Sin, W.C.; Brennand, K.J.; Schadt, E.; Naus, C.C.; Buxbaum, J.; Zhang, B. GJA1 (connexin43) is a key regulator of Alzheimer’s disease pathogenesis. Acta Neuropathol. Commun. 2018, 6, 1–20. [Google Scholar] [CrossRef]
- Chen, W.-T.; Lu, A.; Craessaerts, K.; Pavie, B.; Frigerio, C.S.; Corthout, N.; Qian, X.; Laláková, J.; Kühnemund, M.; Voytyuk, I.; et al. Spatial Transcriptomics and In Situ Sequencing to Study Alzheimer’s Disease. Cell 2020, 182, 976–991.e19. [Google Scholar] [CrossRef] [PubMed]
- Prokop, S.; Miller, K.R.; Labra, S.R.; Pitkin, R.M.; Hoxha, K.; Narasimhan, S.; Changolkar, L.; Rosenbloom, A.; Lee, V.M.Y.; Trojanowski, J.Q. Impact of TREM2 Risk Variants on Brain Region-Specific Immune Activation and Plaque Microenvironment in Alzheimer’s Disease Pa-tient Brain Samples. Acta Neuropathol. 2019, 138, 613–630. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.C.B.; Dammer, E.B.; Duong, D.M.; Ping, L.; Zhou, M.; Yin, L.; Higginbotham, L.A.; Guajardo, A.; White, B.; Troncoso, J.C.; et al. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat. Med. 2020, 26, 769–780. [Google Scholar] [CrossRef]
- Klein, H.U.; Schäfer, M.; Bennett, D.A.; Schwender, H.; de Jager, P.L. Bayesian Integra-tive Analysis of Epigenomic and Transcriptomic Data Identifies Alzheimer’s Disease Candidate Genes and Networks. PLoS Comput. Biol. 2020, 16, e10007771. [Google Scholar] [CrossRef] [PubMed]
- Swarup, V.; Chang, T.S.; Duong, D.M.; Dammer, E.B.; Dai, J.; Lah, J.J.; Johnson, E.C.; Seyfried, N.T.; Levey, A.I.; Geschwind, D.H. Identification of Conserved Proteomic Networks in Neurodegenerative Dementia. Cell Rep. 2020, 31, 107807. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Species | Brain Region | RNA-Seq Technique | Isolation Method | DEGs | Pathways | Refs |
|---|---|---|---|---|---|---|
| Mouse APP/PS1 | Cortex | Bulk RNA-seq | GLT-1 | Cst7 Ccl4 Il1b Clec7a Tyrob Hes5 Tm7sf2 Cyp51 Mvd | Inflammatory response; Neuronal support; Cholesterol biosynthesis | [133] |
| Mouse PS2APP | Cortex | Bulk RNA-seq | GFAP | Gfap Bcl3 Serpina3n C4a C4b | [135] | |
| Mouse APP/PS1 | Whole brain | Bulk RNA-seq | ACSA2 | Cyb5r2 Chil4 Bdkrb2 Rnase4 C4b | [134] | |
| Human | Lateral temporal cortex | Microarray | GFAP | MYO6 KIF21A ACTNB IGF1R PIK3R1 MAP3K12 GJC1 ZO1 TJAP1 | Cytoskeleton; Cell signalling; Cell junctions | [136] |
| Human | Parietal cortex | Bulk RNA-seq | unbiased | ACOT1 ACOT2 | Lipid metabolism | [137] |
| Human | Posterior cingulate cortex | Bulk RNA-seq | ALDH1L1 | TRMT61B FASTKD2 NDUFA4L2 CLU C3 CD74 | Mitochondria; Immune response | [112] |
| Mouse 5XFAD | Hippocampus | Single-nuclei | unbiased | Gfap Serpina3n Apoe Clu Ctsb Ctsd Ctsl | Disease associated astrocytes (DAA) cluster | [138] |
| Mouse 5XFAD | Cortex | Single-nuclei | unbiased | Gfap C4b | [139] | |
| Human | Prefrontal cortex | Single-nuclei | unbiased | NCAN COL5A3 FABP5 HILPDA SOD2 | Extracellular matrix; Lipid and oxidative metabolism | [139] |
| Human | Entorhinal cortex | Single-nuclei | unbiased | C3 ADAMTS18 KCNN3 BIN1 TFEB RGS20 FRMD4A APOE CLDN1 POLN STK32B EDIL3 AKAP12 HECW1 WDR5 LEMD2 DLC1 | Ribosomal function; Mitochondrial function; Neurone differentiation; Heat shock responses; TGFβ signalling; Immune response | [113] |
| Human | Prefrontal cortex | Single-nuclei | unbiased | GLUL CLU APOE | [140] | |
| Human | Entorhinal cortex; Superior frontal gyrus | Single-nuclei | unbiased | CD44 TNC HSPB1 HSP90AA1 SLC1A2 SLC1A3 GLUL SLC6A11 NRXN1 CADM2 PTN GPC5 | Extracellular matrix interactions; Proteostasis; Glutamate/GABA homeostasis; Synaptic adhesion/maintenance | [141] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Preman, P.; Alfonso-Triguero, M.; Alberdi, E.; Verkhratsky, A.; Arranz, A.M. Astrocytes in Alzheimer’s Disease: Pathological Significance and Molecular Pathways. Cells 2021, 10, 540. https://doi.org/10.3390/cells10030540
Preman P, Alfonso-Triguero M, Alberdi E, Verkhratsky A, Arranz AM. Astrocytes in Alzheimer’s Disease: Pathological Significance and Molecular Pathways. Cells. 2021; 10(3):540. https://doi.org/10.3390/cells10030540
Chicago/Turabian StylePreman, Pranav, Maria Alfonso-Triguero, Elena Alberdi, Alexei Verkhratsky, and Amaia M. Arranz. 2021. "Astrocytes in Alzheimer’s Disease: Pathological Significance and Molecular Pathways" Cells 10, no. 3: 540. https://doi.org/10.3390/cells10030540
APA StylePreman, P., Alfonso-Triguero, M., Alberdi, E., Verkhratsky, A., & Arranz, A. M. (2021). Astrocytes in Alzheimer’s Disease: Pathological Significance and Molecular Pathways. Cells, 10(3), 540. https://doi.org/10.3390/cells10030540