Unraveling Cell Death Pathways during Malaria Infection: What Do We Know So Far?

 , and
, and 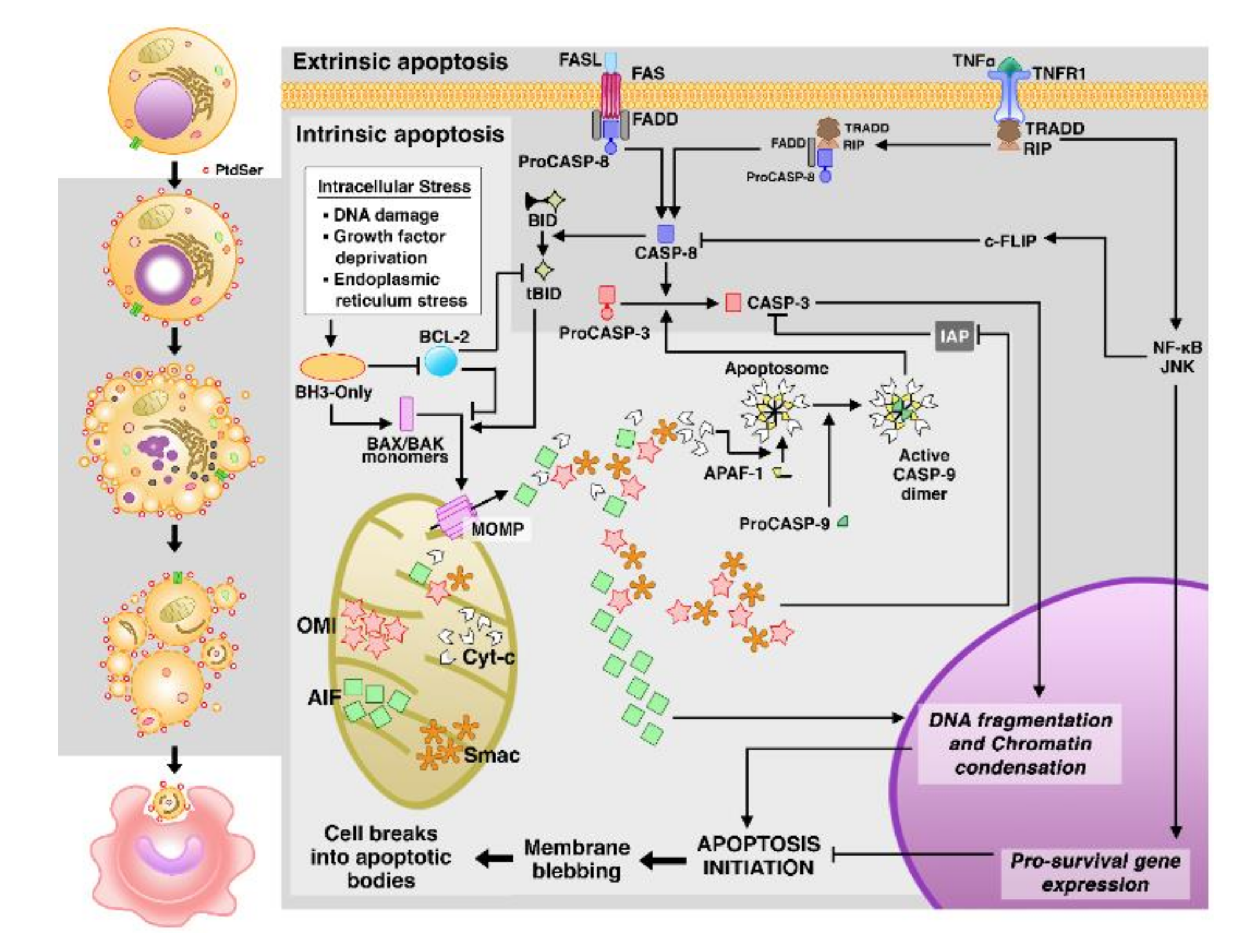{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Malaria
Malaria Immune Response and Cell Death
3. Cell Death
3.1. Apoptosis
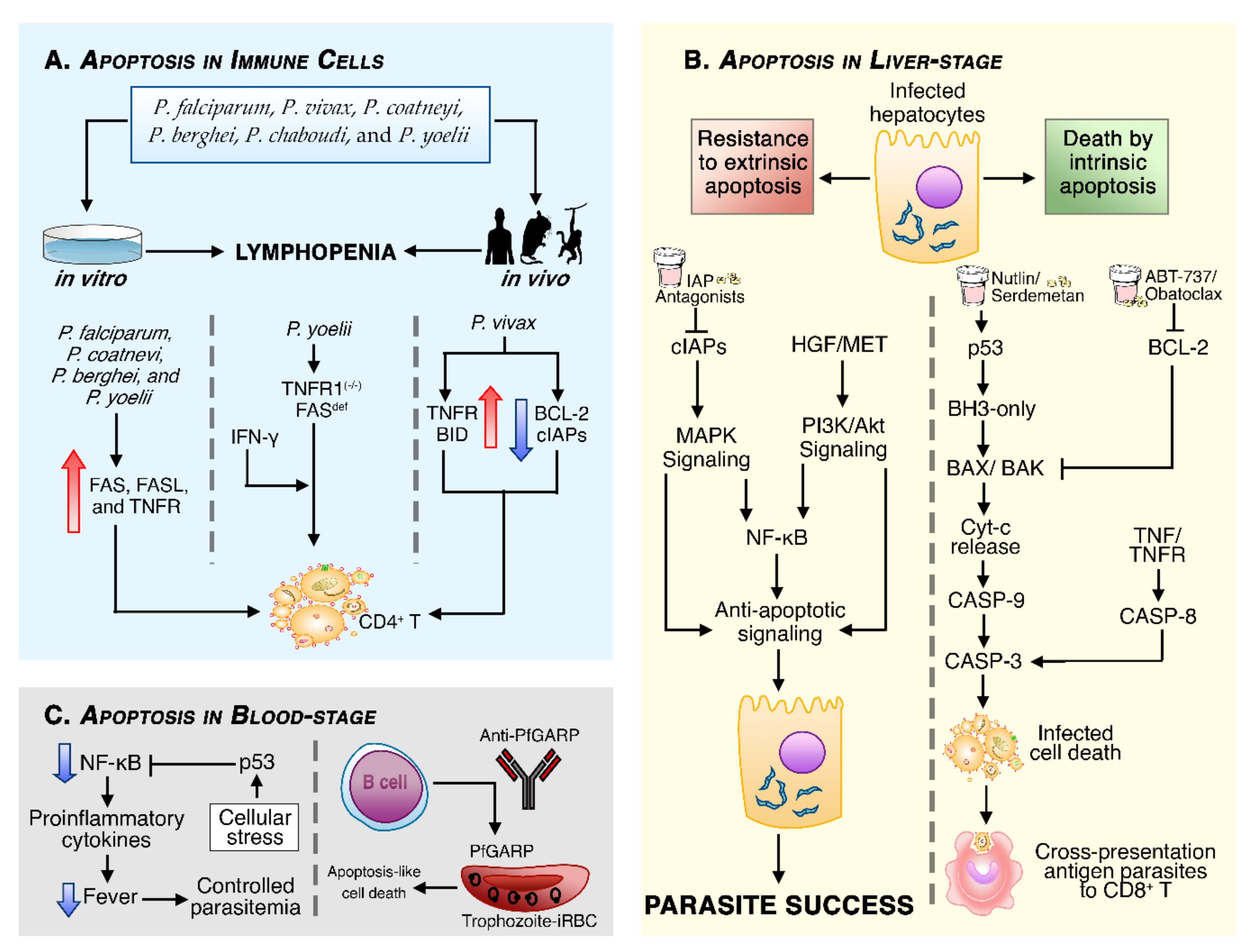
Apoptosis in Malaria
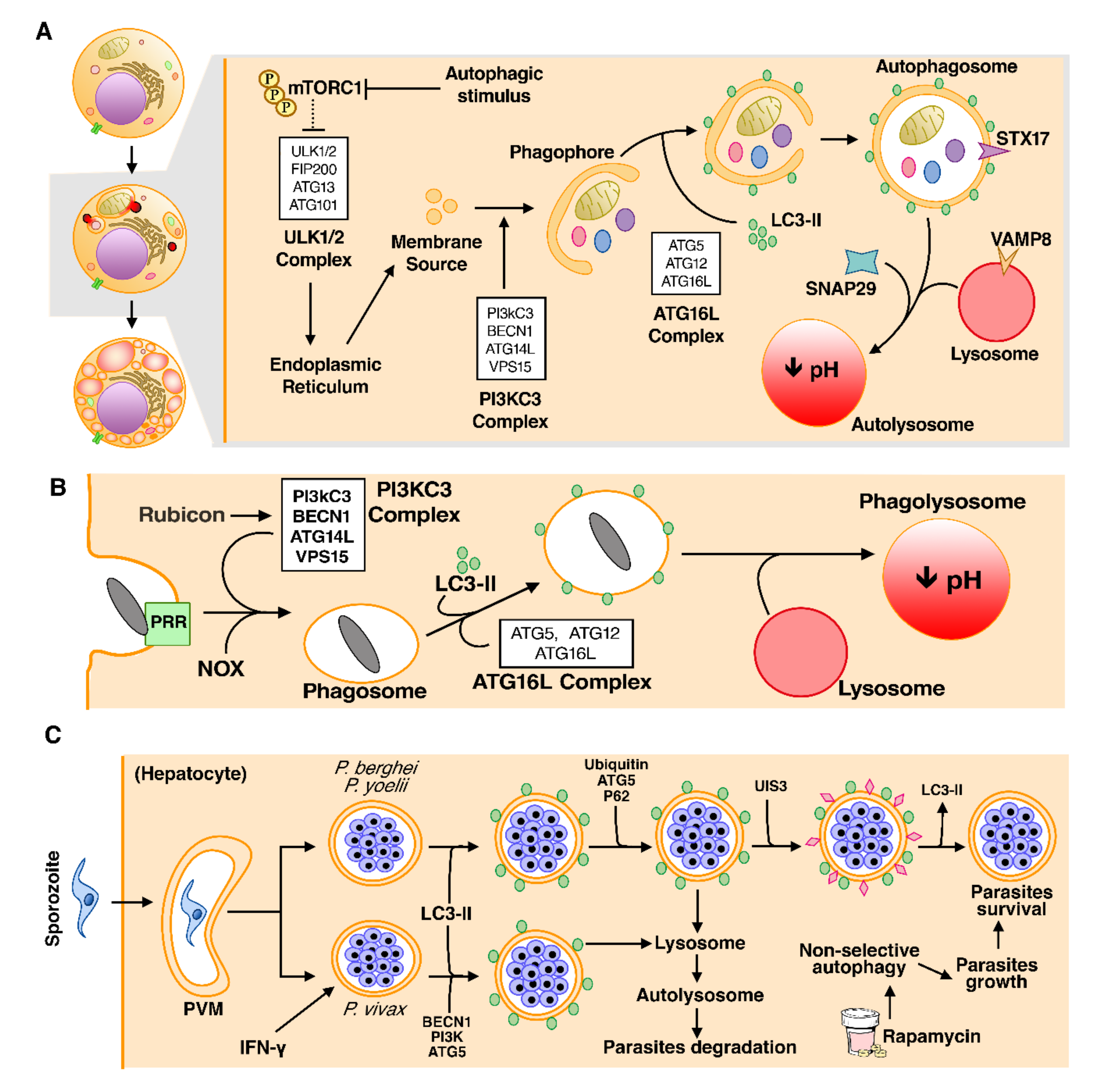
3.2. Autophagy
Autophagy in Malaria
3.3. Necrosis
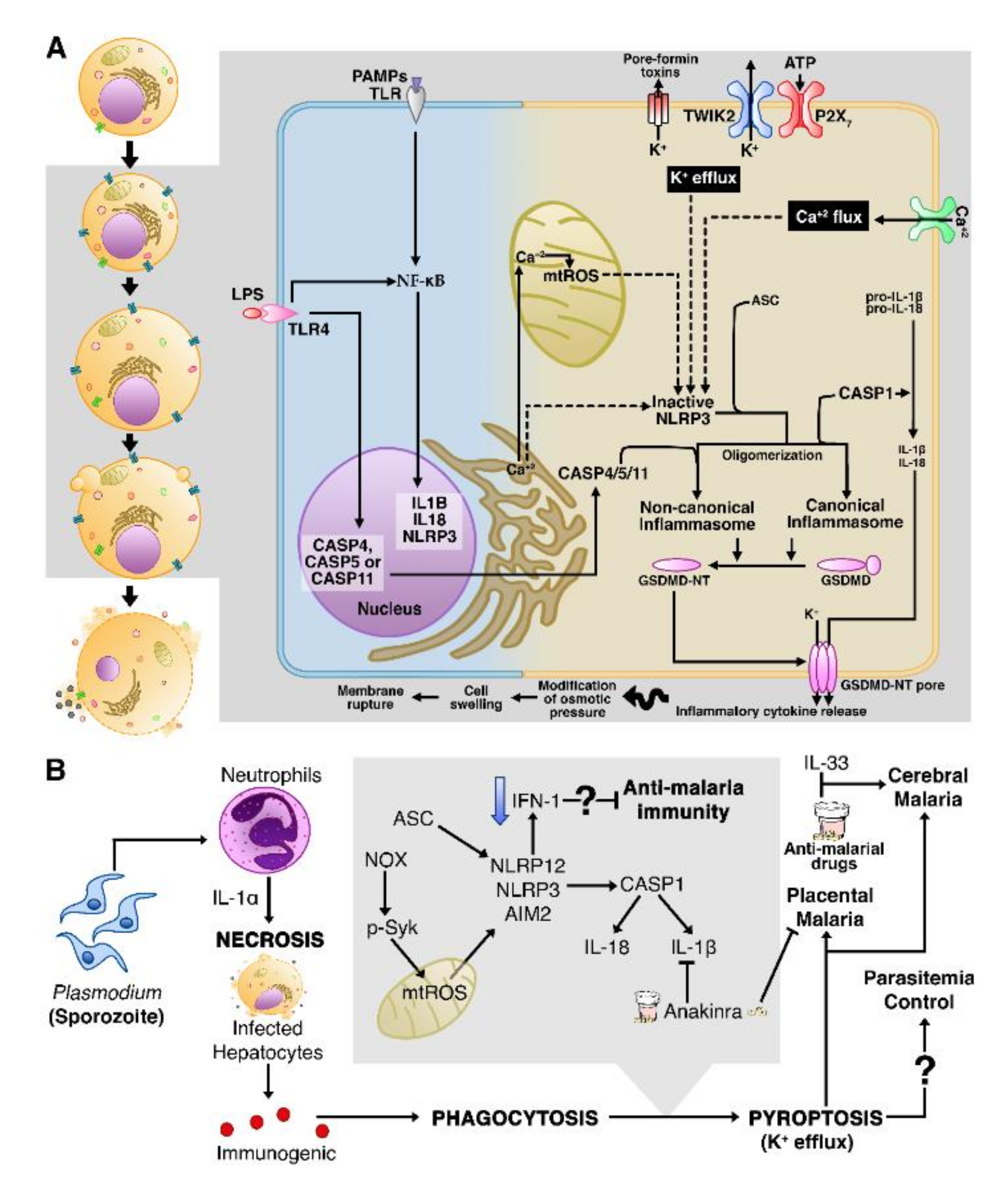
3.4. Pyroptosis
Necrosis in Infected Cells Can Trigger Pyroptotic Cell Death
3.5. NETosis
NETosis Leads to Severe Malaria
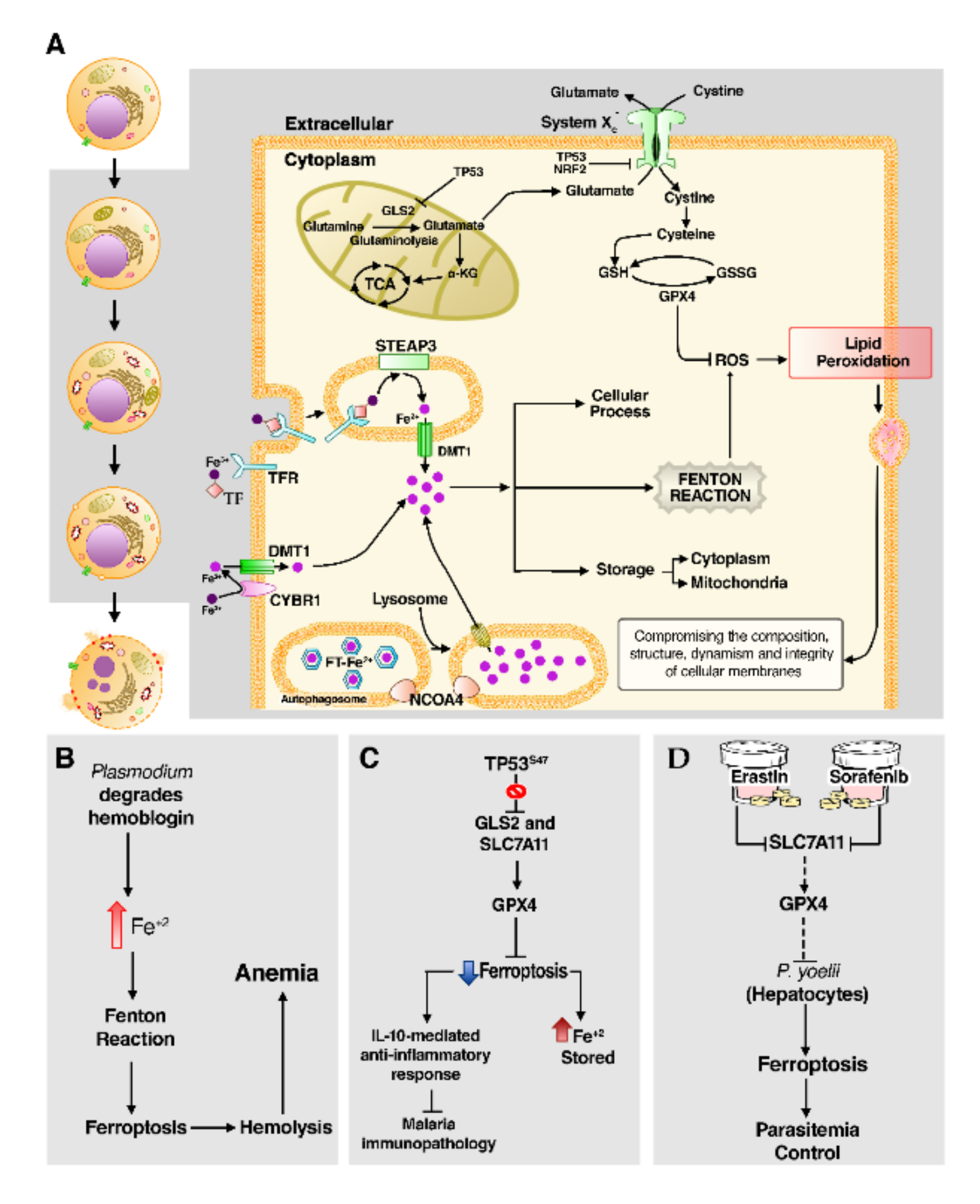
3.6. Ferroptosis
Ferroptosis Leads to Lipid Peroxidation in Malaria
4. Future Perspectives
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- World Malaria Report. Available online: https://www.who.int/publications-detail-redirect/9789241565721 (accessed on 19 August 2020).
- Phillips, M.A.; Burrows, J.N.; Manyando, C.; van Huijsduijnen, R.H.; Van Voorhis, W.C.; Wells, T.N.C. Malaria. Nat. Rev. Dis. Primers 2017, 3, 17050. [Google Scholar] [CrossRef] [PubMed]
- Coelho, C.H.; Doritchamou, J.Y.A.; Zaidi, I.; Duffy, P.E. Advances in malaria vaccine development: Report from the 2017 malaria vaccine symposium. NPJ Vaccines 2017, 2, 34. [Google Scholar] [CrossRef] [PubMed]
- Cockburn, I.A.; Seder, R.A. Malaria prevention: From immunological concepts to effective vaccines and protective antibodies. Nat. Immunol. 2018, 19, 1199–1211. [Google Scholar] [CrossRef] [PubMed]
- Duffy, P.E.; Patrick Gorres, J. Malaria vaccines since 2000: Progress, priorities, products. NPJ Vaccines 2020, 5, 48. [Google Scholar] [CrossRef]
- McQueen, P.G.; Williamson, K.C.; McKenzie, F.E. Host immune constraints on malaria transmission: Insights from population biology of within-host parasites. Malar. J. 2013, 12, 206. [Google Scholar] [CrossRef]
- Nagata, S.; Tanaka, M. Programmed cell death and the immune system. Nat. Rev. Immunol. 2017, 17, 333–340. [Google Scholar] [CrossRef]
- Labbé, K.; Saleh, M. Cell death in the host response to infection. Cell Death Differ. 2008, 15, 1339–1349. [Google Scholar] [CrossRef]
- Legrand, A.J.; Konstantinou, M.; Goode, E.F.; Meier, P. The Diversification of Cell Death and Immunity: Memento Mori. Mol. Cell 2019, 76, 232–242. [Google Scholar] [CrossRef]
- Collins, W.E.; Jeffery, G.M. Plasmodium ovale: Parasite and Disease. Clin. Microbiol. Rev. 2005, 18, 570–581. [Google Scholar] [CrossRef]
- White, N.J. Plasmodium knowlesi: The Fifth Human Malaria Parasite. Clin. Infect. Dis. 2008, 46, 172–173. [Google Scholar] [CrossRef]
- Sutherland, C.J.; Tanomsing, N.; Nolder, D.; Oguike, M.; Jennison, C.; Pukrittayakamee, S.; Dolecek, C.; Hien, T.T.; do Rosário, V.E.; Arez, A.P.; et al. Two Nonrecombining Sympatric Forms of the Human Malaria Parasite Plasmodium ovale Occur Globally. J. Infect. Dis. 2010, 201, 1544–1550. [Google Scholar] [CrossRef] [PubMed]
- Keeling, P.J.; Rayner, J.C. The origins of malaria: There are more things in heaven and earth …. Parasitology 2015, 142, S16–S25. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wassmer, S.C.; Taylor, T.E.; Rathod, P.K.; Mishra, S.K.; Mohanty, S.; Arevalo-Herrera, M.; Duraisingh, M.T.; Smith, J.D. Investigating the Pathogenesis of Severe Malaria: A Multidisciplinary and Cross-Geographical Approach. Am. J. Trop Med. Hyg. 2015, 93, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Wassmer, S.C.; Grau, G.E.R. Severe malaria: What’s new on the pathogenesis front? Int. J. Parasitol. 2017, 47, 145–152. [Google Scholar] [CrossRef]
- Mueller, I.; Zimmerman, P.A.; Reeder, J.C. Plasmodium malariae and Plasmodium ovale—The ‘bashful’ malaria parasites. Trends Parasitol. 2007, 23, 278–283. [Google Scholar] [CrossRef]
- Guerra, C.A.; Howes, R.E.; Patil, A.P.; Gething, P.W.; Van Boeckel, T.P.; Temperley, W.H.; Kabaria, C.W.; Tatem, A.J.; Manh, B.H.; Elyazar, I.R.F.; et al. The International Limits and Population at Risk of Plasmodium vivax Transmission in 2009. PLoS Negl. Trop Dis. 2010, 4, e774. [Google Scholar] [CrossRef]
- Autino, B.; Corbett, Y.; Castelli, F.; Taramelli, D. Pathogenesis of Malaria in Tissues and Blood. Mediterr. J. Hematol. Infect. Dis. 2012, 4. [Google Scholar] [CrossRef]
- Gething, P.W.; Elyazar, I.R.F.; Moyes, C.L.; Smith, D.L.; Battle, K.E.; Guerra, C.A.; Patil, A.P.; Tatem, A.J.; Howes, R.E.; Myers, M.F.; et al. A Long Neglected World Malaria Map: Plasmodium vivax Endemicity in 2010. PLoS Negl. Trop. Dis. 2012, 6, e1814. [Google Scholar] [CrossRef]
- Cox-Singh, J.; Davis, T.M.E.; Lee, K.-S.; Shamsul, S.S.G.; Matusop, A.; Ratnam, S.; Rahman, H.A.; Conway, D.J.; Singh, B. Plasmodium knowlesi malaria in humans is widely distributed and potentially life-threatening. Clin. Infect. Dis. 2008, 46, 165–171. [Google Scholar] [CrossRef]
- Anstey, N.M.; Russell, B.; Yeo, T.W.; Price, R.N. The pathophysiology of vivax malaria. Trends Parasitol. 2009, 25, 220–227. [Google Scholar] [CrossRef]
- Otto, T.D.; Böhme, U.; Jackson, A.P.; Hunt, M.; Franke-Fayard, B.; Hoeijmakers, W.A.M.; Religa, A.A.; Robertson, L.; Sanders, M.; Ogun, S.A.; et al. A comprehensive evaluation of rodent malaria parasite genomes and gene expression. BMC Biol. 2014, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, A.D.; Chiodini, P.L. Malaria and blood transfusion. Vox Sang. 2006, 90, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Poespoprodjo, J.R.; Fobia, W.; Kenangalem, E.; Hasanuddin, A.; Sugiarto, P.; Tjitra, E.; Anstey, N.M.; Price, R.N. Highly Effective Therapy for Maternal Malaria Associated With a Lower Risk of Vertical Transmission. J. Infect. Dis. 2011, 204, 1613–1619. [Google Scholar] [CrossRef]
- Prudêncio, M.; Rodriguez, A.; Mota, M.M. The silent path to thousands of merozoites: The Plasmodium liver stage. Nat. Rev. Microbiol. 2006, 4, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Amino, R.; Thiberge, S.; Martin, B.; Celli, S.; Shorte, S.; Frischknecht, F.; Ménard, R. Quantitative imaging of Plasmodium transmission from mosquito to mammal. Nat. Med. 2006, 12, 220–224. [Google Scholar] [CrossRef]
- Mota, M.M. Migration of Plasmodium Sporozoites Through Cells Before Infection. Science 2001, 291, 141–144. [Google Scholar] [CrossRef]
- Prudêncio, M.; Mota, M.M.; Mendes, A.M. A toolbox to study liver stage malaria. Trends Parasitol. 2011, 27, 565–574. [Google Scholar] [CrossRef]
- Miller, L.H.; Baruch, D.I.; Marsh, K.; Doumbo, O.K. The pathogenic basis of malaria. Nature 2002, 415, 673–679. [Google Scholar] [CrossRef]
- Cowman, A.F.; Crabb, B.S. Invasion of Red Blood Cells by Malaria Parasites. Cell 2006, 124, 755–766. [Google Scholar] [CrossRef]
- Cowman, A.F.; Healer, J.; Marapana, D.; Marsh, K. Malaria: Biology and Disease. Cell 2016, 167, 610–624. [Google Scholar] [CrossRef]
- Wells, T.N.C.; Burrows, J.N.; Baird, J.K. Targeting the hypnozoite reservoir of Plasmodium vivax: The hidden obstacle to malaria elimination. Trends Parasitol. 2010, 26, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Gazzinelli, R.T.; Kalantari, P.; Fitzgerald, K.A.; Golenbock, D.T. Innate sensing of malaria parasites. Nat. Rev. Immunol. 2014, 14, 744–757. [Google Scholar] [CrossRef] [PubMed]
- Gowda, D.C.; Wu, X. Parasite Recognition and Signaling Mechanisms in Innate Immune Responses to Malaria. Front. Immunol. 2018, 9, 3006. [Google Scholar] [CrossRef] [PubMed]
- Liehl, P.; Zuzarte-Luís, V.; Chan, J.; Zillinger, T.; Baptista, F.; Carapau, D.; Konert, M.; Hanson, K.K.; Carret, C.; Lassnig, C.; et al. Host-cell sensors for Plasmodium activate innate immunity against liver-stage infection. Nat. Med. 2014, 20, 47–53. [Google Scholar] [CrossRef]
- Miller, J.L.; Sack, B.K.; Baldwin, M.; Vaughan, A.M.; Kappe, S.H.I. Interferon-Mediated Innate Immune Responses against Malaria Parasite Liver Stages. Cell Rep. 2014, 7, 436–447. [Google Scholar] [CrossRef]
- Arese, P.; Schwarzer, E. Malarial pigment (haemozoin): A very active “inert” substance. Ann. Trop. Med. Parasitol. 1997, 91, 501–516. [Google Scholar] [CrossRef]
- Francis, S.E.; Sullivan, D.J.; Goldberg, D.E. Hemoglobin metabolism in the malaria parasite Plasmodium falciparum. Annu. Rev. Microbiol. 1997, 51, 97–123. [Google Scholar] [CrossRef]
- Krishnegowda, G.; Hajjar, A.M.; Zhu, J.; Douglass, E.J.; Uematsu, S.; Akira, S.; Woods, A.S.; Gowda, D.C. Induction of Proinflammatory Responses in Macrophages by the Glycosylphosphatidylinositols of Plasmodium falciparum: Cell Signaling Receptors, Glycosylphosphatidylinositol (Gpi) Structural Requirement, and Regulation of Gpi Activity. J. Biol. Chem. 2005, 280, 8606–8616. [Google Scholar] [CrossRef]
- Zhu, J.; Krishnegowda, G.; Gowda, D.C. Induction of Proinflammatory Responses in Macrophages by the Glycosylphosphatidylinositols (GPIs) of Plasmodium falciparum: The requirement of ERK, p38, JNK and NF-κB pathways for the expression of proinflammatory cytokines and nitric oxide. J. Biol. Chem. 2005, 280, 8617–8627. [Google Scholar] [CrossRef]
- Parroche, P.; Lauw, F.N.; Goutagny, N.; Latz, E.; Monks, B.G.; Visintin, A.; Halmen, K.A.; Lamphier, M.; Olivier, M.; Bartholomeu, D.C.; et al. Malaria hemozoin is immunologically inert but radically enhances innate responses by presenting malaria DNA to Toll-like receptor 9. Proc. Natl. Acad. Sci. USA 2007, 104, 1919–1924. [Google Scholar] [CrossRef]
- Schofield, L.; Hackett, F. Signal transduction in host cells by a glycosylphosphatidylinositol toxin of malaria parasites. J. Exp. Med. 1993, 177, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Seguin, M.C.; Klotz, F.W.; Schneider, I.; Weir, J.P.; Goodbary, M.; Slayter, M.; Raney, J.J.; Aniagolu, J.U.; Green, S.J. Induction of nitric oxide synthase protects against malaria in mice exposed to irradiated Plasmodium berghei infected mosquitoes: Involvement of interferon gamma and CD8+ T cells. J. Exp. Med. 1994, 180, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Doolan, D.L.; Sedegah, M.; Hedstrom, R.C.; Hobart, P.; Charoenvit, Y.; Hoffman, S.L. Circumventing genetic restriction of protection against malaria with multigene DNA immunization: CD8+ cell-, interferon gamma-, and nitric oxide-dependent immunity. J. Exp. Med. 1996, 183, 1739–1746. [Google Scholar] [CrossRef] [PubMed]
- Coban, C.; Ishii, K.J.; Kawai, T.; Hemmi, H.; Sato, S.; Uematsu, S.; Yamamoto, M.; Takeuchi, O.; Itagaki, S.; Kumar, N.; et al. Toll-like receptor 9 mediates innate immune activation by the malaria pigment hemozoin. J. Exp. Med. 2005, 201, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Giribaldi, G.; Prato, M.; Ulliers, D.; Gallo, V.; Schwarzer, E.; Akide-Ndunge, O.B.; Valente, E.; Saviozzi, S.; Calogero, R.A.; Arese, P. Involvement of Inflammatory Chemokines in Survival of Human Monocytes Fed with Malarial Pigment. IAI 2010, 78, 4912–4921. [Google Scholar] [CrossRef] [PubMed]
- Kalantari, P.; DeOliveira, R.B.; Chan, J.; Corbett, Y.; Rathinam, V.; Stutz, A.; Latz, E.; Gazzinelli, R.T.; Golenbock, D.T.; Fitzgerald, K.A. Dual engagement of the NLRP3 and AIM2 inflammasomes by plasmodial-derived hemozoin and DNA during malaria. Cell Rep. 2014, 6, 196–210. [Google Scholar] [CrossRef]
- Ataide, M.A.; Andrade, W.A.; Zamboni, D.S.; Wang, D.; Souza, M.D.C.; Franklin, B.S.; Elian, S.; Martins, F.S.; Pereira, D.; Reed, G.; et al. Malaria-Induced NLRP12/NLRP3-Dependent Caspase-1 Activation Mediates Inflammation and Hypersensitivity to Bacterial Superinfection. PLoS Pathog. 2014, 10, e1003885. [Google Scholar] [CrossRef]
- Amarante-Mendes, G.P.; Adjemian, S.; Branco, L.M.; Zanetti, L.C.; Weinlich, R.; Bortoluci, K.R. Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Alnemri, E.S.; Altucci, L.; Andrews, D.; Annicchiarico-Petruzzelli, M.; et al. Essential versus accessory aspects of cell death: Recommendations of the NCCD 2015. Cell Death Differ. 2015, 22, 58–73. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [PubMed]
- Deter, R.L.; Baudhuin, P.; de Duve, C. Participation of lysosomes in cellular autophagy induced in rat liver by glucagon. J. Cell Biol. 1967, 35, C11–C16. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.F.R.; Wyllie, A.H.; Currie, A.R. Apoptosis: A Basic Biological Phenomenon with Wide-ranging Implications in Tissue Kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Yatim, N.; Cullen, S.; Albert, M.L. Dying cells actively regulate adaptive immune responses. Nat. Rev. Immunol. 2017, 17, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Schulze-Osthoff, K.; Ferrari, D.; Los, M.; Wesselborg, S.; Peter, M.E. Apoptosis signaling by death receptors. Eur. J. Biochem. 1998, 254, 439–459. [Google Scholar] [CrossRef] [PubMed]
- Chinnaiyan, A.M.; O’Rourke, K.; Tewari, M.; Dixit, V.M. FADD, a novel death domain-containing protein, interacts with the death domain of fas and initiates apoptosis. Cell 1995, 81, 505–512. [Google Scholar] [CrossRef]
- Ashkenazi, A. Death Receptors: Signaling and Modulation. Science 1998, 281, 1305–1308. [Google Scholar] [CrossRef]
- Kischkel, F.C.; Hellbardt, S.; Behrmann, I.; Germer, M.; Pawlita, M.; Krammer, P.H.; Peter, M.E. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J. 1995, 14, 5579–5588. [Google Scholar] [CrossRef]
- Strasser, A.; Jost, P.J.; Nagata, S. The many roles of FAS receptor signaling in the immune system. Immunity 2009, 30, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Vince, J.E.; Pantaki, D.; Feltham, R.; Mace, P.D.; Cordier, S.M.; Schmukle, A.C.; Davidson, A.J.; Callus, B.A.; Wong, W.W.-L.; Gentle, I.E.; et al. TRAF2 Must Bind to Cellular Inhibitors of Apoptosis for Tumor Necrosis Factor (TNF) to Efficiently Activate NF-κB and to Prevent TNF-induced Apoptosis. J. Biol. Chem. 2009, 284, 35906–35915. [Google Scholar] [CrossRef] [PubMed]
- Kelliher, M.A.; Grimm, S.; Ishida, Y.; Kuo, F.; Stanger, B.Z.; Leder, P. The Death Domain Kinase RIP Mediates the TNF-Induced NF-κB Signal. Immunity 1998, 8, 297–303. [Google Scholar] [CrossRef]
- Kucharczak, J.; Simmons, M.J.; Fan, Y.; Gélinas, C. To be, or not to be: NF-κB is the answer—Role of Rel/NF-κB in the regulation of apoptosis. Oncogene 2003, 22, 8961–8982. [Google Scholar] [CrossRef] [PubMed]
- Petros, A.M.; Olejniczak, E.T.; Fesik, S.W. Structural biology of the Bcl-2 family of proteins. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2004, 1644, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Giam, M.; Huang, D.C.S.; Bouillet, P. BH3-only proteins and their roles in programmed cell death. Oncogene 2008, 27, S128–S136. [Google Scholar] [CrossRef]
- Tait, S.W.G.; Green, D.R. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef]
- Panduri, V.; Weitzman, S.A.; Chandel, N.S.; Kamp, D.W. Mitochondrial-derived free radicals mediate asbestos-induced alveolar epithelial cell apoptosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 286, L1220–L1227. [Google Scholar] [CrossRef]
- Corazzari, M.; Gagliardi, M.; Fimia, G.M.; Piacentini, M. Endoplasmic Reticulum Stress, Unfolded Protein Response, and Cancer Cell Fate. Front. Oncol. 2017, 7, 78. [Google Scholar] [CrossRef]
- Toshiyuki, M.; Reed, J.C. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 1995, 80, 293–299. [Google Scholar] [CrossRef]
- Thornborrow, E.C.; Patel, S.; Mastropietro, A.E.; Schwartzfarb, E.M.; Manfredi, J.J. A conserved intronic response element mediates direct p53-dependent transcriptional activation of both the human and murine bax genes. Oncogene 2002, 21, 990–999. [Google Scholar] [CrossRef] [PubMed]
- Haupt, S. Apoptosis—The p53 network. J. Cell Sci. 2003, 116, 4077–4085. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Pinedo, C.; Guio-Carrion, A.; Goldstein, J.C.; Fitzgerald, P.; Newmeyer, D.D.; Green, D.R. Different mitochondrial intermembrane space proteins are released during apoptosis in a manner that is coordinately initiated but can vary in duration. Proc. Natl. Acad. Sci. USA 2006, 103, 11573–11578. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-E.; Du, F.; Fang, M.; Wang, X. Formation of apoptosome is initiated by cytochrome c-induced dATP hydrolysis and subsequent nucleotide exchange on Apaf-1. Proc. Natl. Acad. Sci. USA 2005, 102, 17545–17550. [Google Scholar] [CrossRef]
- Parrish, A.B.; Freel, C.D.; Kornbluth, S. Cellular Mechanisms Controlling Caspase Activation and Function. Cold Spring Harb. Perspect. Biol. 2013, 5, a008672. [Google Scholar] [CrossRef]
- Enari, M.; Sakahira, H.; Yokoyama, H.; Okawa, K.; Iwamatsu, A.; Nagata, S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature 1998, 391, 43–50. [Google Scholar] [CrossRef]
- Sakahira, H.; Enari, M.; Nagata, S. Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature 1998, 391, 96–99. [Google Scholar] [CrossRef]
- Brunner, T. Fas (CD95/Apo-1) ligand regulation in T cell homeostasis, cell-mediated cytotoxicity and immune pathology. Semin. Immunol. 2003, 15, 167–176. [Google Scholar] [CrossRef]
- Voskoboinik, I.; Whisstock, J.C.; Trapani, J.A. Perforin and granzymes: Function, dysfunction and human pathology. Nat. Rev. Immunol. 2015, 15, 388–400. [Google Scholar] [CrossRef]
- Osińska, I.; Popko, K.; Demkow, U. Perforin: An important player in immune response. Cejoi 2014, 1, 109–115. [Google Scholar] [CrossRef]
- Atkinson, E.A.; Barry, M.; Darmon, A.J.; Shostak, I.; Turner, P.C.; Moyer, R.W.; Bleackley, R.C. Cytotoxic T Lymphocyte-assisted Suicide: Caspase 3 Activation is Primarily the Result of the Direct Action of Granzyme B. J. Biol. Chem. 1998, 273, 21261–21266. [Google Scholar] [CrossRef] [PubMed]
- Barry, M.; Heibein, J.A.; Pinkoski, M.J.; Lee, S.-F.; Moyer, R.W.; Green, D.R.; Bleackley, R.C. Granzyme B Short-Circuits the Need for Caspase 8 Activity during Granule-Mediated Cytotoxic T-Lymphocyte Killing by Directly Cleaving Bid. Mol. Cell. Biol. 2000, 20, 3781–3794. [Google Scholar] [CrossRef] [PubMed]
- Jacquemin, G.; Margiotta, D.; Kasahara, A.; Bassoy, E.Y.; Walch, M.; Thiery, J.; Lieberman, J.; Martinvalet, D. Granzyme B-induced mitochondrial ROS are required for apoptosis. Cell Death Differ. 2015, 22, 862–874. [Google Scholar] [CrossRef] [PubMed]
- Martinvalet, D.; Zhu, P.; Lieberman, J. Granzyme A Induces Caspase-Independent Mitochondrial Damage, a Required First Step for Apoptosis. Immunity 2005, 22, 355–370. [Google Scholar] [CrossRef] [PubMed]
- Baldé, A.T.; Sarthou, J.-L.; Roussilhon, C. Acute Plasmodium falciparum infection is associated with increased percentages of apoptotic cells. Immunol. Lett. 1995, 46, 59–62. [Google Scholar] [CrossRef]
- Toure-Balde, A.; Sarthou, J.L.; Aribot, G.; Michel, P.; Trape, J.F.; Rogier, C.; Roussilhon, C. Plasmodium falciparum induces apoptosis in human mononuclear cells. Infect. Immun. 1996, 64, 744–750. [Google Scholar] [CrossRef]
- Kern, P.; Dietrich, M.; Hemmer, C.; Wellinghausen, N. Increased Levels of Soluble Fas Ligand in Serum in Plasmodium falciparum Malaria. Infect. Immun. 2000, 68, 3061–3063. [Google Scholar] [CrossRef]
- Hojo-Souza, N.S.; Pereira, D.B.; Mendes, T.A.; Passos, L.S.; Gazzinelli-Guimarães, A.C.; Gazzinelli-Guimarães, P.H.; Tada, M.S.; Zanini, G.M.; Bartholomeu, D.C.; Fujiwara, R.T.; et al. CD4+ T cells apoptosis in Plasmodium vivax infection is mediated by activation of both intrinsic and extrinsic pathways. Malar. J. 2015, 14. [Google Scholar] [CrossRef]
- Riccio, E.K.P.; Júnior, I.N.; Riccio, L.R.P.; das Graças Alecrim, M.; Corte-Real, S.; Morgado, M.; Daniel-Ribeiro, C.T.; de Fátima Ferreira-da-Cruz, M. Malaria associated apoptosis is not significantly correlated with either parasitemia or the number of previous malaria attacks. Parasitol. Res. 2003, 90, 9–18. [Google Scholar] [CrossRef]
- Matsumoto, J.; Kawai, S.; Terao, K.; Kirinoki, M.; Yasutomi, Y.; Aikawa, M.; Matsuda, H. Malaria Infection Induces Rapid Elevation of the Soluble Fas Ligand Level in Serum and Subsequent T Lymphocytopenia: Possible Factors Responsible for the Differences in Susceptibility of Two Species of Macaca Monkeys to Plasmodium coatneyi Infection. Infect. Immun. 2000, 68, 1183–1188. [Google Scholar] [CrossRef]
- Hirunpetcharat, C.; Good, M.F. Deletion of Plasmodium berghei-specific CD4+ T cells adoptively transferred into recipient mice after challenge with homologous parasite. Proc. Natl. Acad. Sci. USA 1998, 95, 1715–1720. [Google Scholar] [CrossRef] [PubMed]
- Helmby, H.; Jönsson, G.; Troye-Blomberg, M. Cellular Changes and Apoptosis in the Spleens and Peripheral Blood of Mice Infected with Blood-Stage Plasmodium chabaudi chabaudi AS. Infect. Immun. 2000, 68, 1485–1490. [Google Scholar] [CrossRef] [PubMed]
- Wipasa, J.; Xu, H.; Stowers, A.; Good, M.F. Apoptotic Deletion of Th Cells Specific for the 19-kDa Carboxyl-Terminal Fragment of Merozoite Surface Protein 1 During Malaria Infection. J. Immunol. 2001, 167, 3903–3909. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Wipasa, J.; Yan, H.; Zeng, M.; Makobongo, M.O.; Finkelman, F.D.; Kelso, A.; Good, M.F. The Mechanism and Significance of Deletion of Parasite-specific CD4+ T Cells in Malaria Infection. J. Exp. Med. 2002, 195, 881–892. [Google Scholar] [CrossRef] [PubMed]
- Lacerda-Queiroz, N.; Riteau, N.; Eastman, R.T.; Bock, K.W.; Orandle, M.S.; Moore, I.N.; Sher, A.; Long, C.A.; Jankovic, D.; Su, X. Mechanism of splenic cell death and host mortality in a Plasmodium yoelii malaria model. Sci. Rep. 2017, 7, 10438. [Google Scholar] [CrossRef] [PubMed]
- Villegas-Mendez, A.; de Souza, J.B.; Murungi, L.; Hafalla, J.C.R.; Shaw, T.N.; Greig, R.; Riley, E.M.; Couper, K.N. Heterogeneous and Tissue-Specific Regulation of Effector T Cell Responses by IFN-γ during Plasmodium berghei ANKA Infection. J. Immunol. 2011, 187, 2885–2897. [Google Scholar] [CrossRef]
- Woodberry, T.; Minigo, G.; Piera, K.A.; Amante, F.H.; Pinzon-Charry, A.; Good, M.F.; Lopez, J.A.; Engwerda, C.R.; McCarthy, J.S.; Anstey, N.M. Low-Level Plasmodium falciparum Blood-Stage Infection Causes Dendritic Cell Apoptosis and Dysfunction in Healthy Volunteers. J. Infect. Dis. 2012, 206, 333–340. [Google Scholar] [CrossRef]
- Pinzon-Charry, A.; Woodberry, T.; Kienzle, V.; McPhun, V.; Minigo, G.; Lampah, D.A.; Kenangalem, E.; Engwerda, C.; López, J.A.; Anstey, N.M.; et al. Apoptosis and dysfunction of blood dendritic cells in patients with falciparum and vivax malaria. J. Exp. Med. 2013, 210, 1635–1646. [Google Scholar] [CrossRef]
- Lüder, C.G.K.; Gross, U.; Lopes, M.F. Intracellular protozoan parasites and apoptosis: Diverse strategies to modulate parasite–host interactions. Trends Parasitol. 2001, 17, 480–486. [Google Scholar] [CrossRef]
- van Dijk, M.R.; Douradinha, B.; Franke-Fayard, B.; Heussler, V.; van Dooren, M.W.; van Schaijk, B.; van Gemert, G.-J.; Sauerwein, R.W.; Mota, M.M.; Waters, A.P.; et al. Genetically attenuated, P36p-deficient malarial sporozoites induce protective immunity and apoptosis of infected liver cells. Proc. Natl. Acad. Sci. USA 2005, 102, 12194–12199. [Google Scholar] [CrossRef]
- Leiriao, P.; Mota, M.M.; Rodriguez, A. Apoptotic Plasmodium- Infected Hepatocytes Provide Antigens to Liver Dendritic Cells. J. Infect. Dis. 2005, 191, 1576–1581. [Google Scholar] [CrossRef] [PubMed]
- Kaushansky, A.; Metzger, P.G.; Douglass, A.N.; Mikolajczak, S.A.; Lakshmanan, V.; Kain, H.S.; Kappe, S.H. Malaria parasite liver stages render host hepatocytes susceptible to mitochondria-initiated apoptosis. Cell Death Dis. 2013, 4, e762. [Google Scholar] [CrossRef] [PubMed]
- Ebert, G.; Lopaticki, S.; O’Neill, M.T.; Steel, R.W.J.; Doerflinger, M.; Rajasekaran, P.; Yang, A.S.P.; Erickson, S.; Ioannidis, L.; Arandjelovic, P.; et al. Targeting the Extrinsic Pathway of Hepatocyte Apoptosis Promotes Clearance of Plasmodium Liver Infection. Cell Rep. 2020, 30, 4343–4354. [Google Scholar] [CrossRef] [PubMed]
- Kaushansky, A.; Ye, A.S.; Austin, L.S.; Mikolajczak, S.A.; Vaughan, A.M.; Camargo, N.; Metzger, P.G.; Douglass, A.N.; MacBeath, G.; Kappe, S.H.I. Suppression of Host p53 Is Critical for Plasmodium Liver-Stage Infection. Cell Rep. 2013, 3, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Douglass, A.N.; Kain, H.S.; Abdullahi, M.; Arang, N.; Austin, L.S.; Mikolajczak, S.A.; Billman, Z.P.; Hume, J.C.C.; Murphy, S.C.; Kappe, S.H.I.; et al. Host-based Prophylaxis Successfully Targets Liver Stage Malaria Parasites. Mol. Ther. 2015, 23, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Renggli, J.; Hahne, M.; Matile, H.; Betschart, B.; Tschopp, J.; Corradin, G. Elimination of P. berghei liver stages is independent of Fas (CD95/Apo-I) or perforin-mediated cytotoxicity. Parasite Immunol. 1997, 19, 145–148. [Google Scholar] [CrossRef] [PubMed]
- van de Sand, C.; Horstmann, S.; Schmidt, A.; Sturm, A.; Bolte, S.; Krueger, A.; Lütgehetmann, M.; Pollok, J.-M.; Libert, C.; Heussler, V.T. The liver stage of Plasmodium berghei inhibits host cell apoptosis. Mol. Microbiol. 2005, 58, 731–742. [Google Scholar] [CrossRef]
- Leirião, P.; Albuquerque, S.S.; Corso, S.; Van Gemert, G.-J.; Sauerwein, R.W.; Rodriguez, A.; Giordano, S.; Mota, M.M. HGF/MET signalling protects Plasmodium-infected host cells from apoptosis: Anti-apoptotic HGF/MET signalling in malaria infection. Cell. Microbiol. 2005, 7, 603–609. [Google Scholar] [CrossRef]
- Albuquerque, S.S.; Carret, C.; Grosso, A.; Tarun, A.S.; Peng, X.; Kappe, S.H.; Prudêncio, M.; Mota, M.M. Host cell transcriptional profiling during malaria liver stage infection reveals a coordinated and sequential set of biological events. BMC Genom. 2009, 10, 270. [Google Scholar] [CrossRef]
- Guha, M.; Kumar, S.; Choubey, V.; Maity, P.; Bandyopadhyay, U.; Guha, M.; Kumar, S.; Choubey, V.; Maity, P.; Bandyopadhyay, U. Apoptosis in liver during malaria: Role of oxidative stress and implication of mitochondrial pathway. FASEB J. 2006, 20, 1224–1226. [Google Scholar] [CrossRef]
- Dey, S.; Guha, M.; Alam, A.; Goyal, M.; Bindu, S.; Pal, C.; Maity, P.; Mitra, K.; Bandyopadhyay, U. Malarial infection develops mitochondrial pathology and mitochondrial oxidative stress to promote hepatocyte apoptosis. Free Radic. Biol. Med. 2009, 46, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Ferguson, T.; Zitvogel, L.; Kroemer, G. Immunogenic and tolerogenic cell death. Nat. Rev. Immunol. 2009, 9, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Albert, M.L.; Sauter, B.; Bhardwaj, N. Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature 1998, 392, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.M.; Guha, R.; Portugal, S.; Skinner, J.; Ongoiba, A.; Bhardwaj, J.; Jones, M.; Moebius, J.; Venepally, P.; Doumbo, S.; et al. A Molecular Signature in Blood Reveals a Role for p53 in Regulating Malaria-Induced Inflammation. Immunity 2019, 51, 750–765. [Google Scholar] [CrossRef] [PubMed]
- Raj, D.K.; Das Mohapatra, A.; Jnawali, A.; Zuromski, J.; Jha, A.; Cham-Kpu, G.; Sherman, B.; Rudlaff, R.M.; Nixon, C.E.; Hilton, N.; et al. Anti-PfGARP activates programmed cell death of parasites and reduces severe malaria. Nature 2020, 582, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Totino, P.R.; Magalhães, A.D.; Silva, L.A.; Banic, D.M.; Daniel-Ribeiro, C.T.; Ferreira-da-Cruz, M. Apoptosis of non-parasitized red blood cells in malaria: A putative mechanism involved in the pathogenesis of anaemia. Malar. J. 2010, 9, 350. [Google Scholar] [CrossRef]
- Sharma, L.; Kaur, J.; Shukla, G. Role of Oxidative Stress and Apoptosis in the Placental Pathology of Plasmodium berghei Infected Mice. PLoS ONE 2012, 7, e32694. [Google Scholar] [CrossRef]
- Liu, M.; Hassana, S.; Stiles, J.K. Heme-mediated apoptosis and fusion damage in BeWo trophoblast cells. Sci. Rep. 2016, 6, 36193. [Google Scholar] [CrossRef]
- Lackner, P.; Burger, C.; Pfaller, K.; Heussler, V.; Helbok, R.; Morandell, M.; Broessner, G.; Tannich, E.; Schmutzhard, E.; Beer, R. Apoptosis in experimental cerebral malaria: Spatial profile of cleaved caspase-3 and ultrastructural alterations in different disease stages. Neuropathol. Appl. Neurobiol. 2007. [Google Scholar] [CrossRef]
- Boulet, C.; Doerig, C.D.; Carvalho, T.G. Manipulating Eryptosis of Human Red Blood Cells: A Novel Antimalarial Strategy? Front. Cell. Infect. Microbiol. 2018, 8, 419. [Google Scholar] [CrossRef] [PubMed]
- Harbuzariu, A.; Pitts, S.; Cespedes, J.C.; Harp, K.O.; Nti, A.; Shaw, A.P.; Liu, M.; Stiles, J.K. Modelling heme-mediated brain injury associated with cerebral malaria in human brain cortical organoids. Sci. Rep. 2019, 9, 19162. [Google Scholar] [CrossRef]
- Nishanth, G.; Schlüter, D. Blood–Brain Barrier in Cerebral Malaria: Pathogenesis and Therapeutic Intervention. Trends Parasitol. 2019, 35, 516–528. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Debnath, J. Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell Biol. 2015, 16, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Klionsky, D.J. Autophagy wins the 2016 Nobel Prize in Physiology or Medicine: Breakthroughs in baker’s yeast fuel advances in biomedical research. Proc. Natl. Acad. Sci. USA 2017, 114, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Green, D.R. Autophagy-Independent Functions of the Autophagy Machinery. Cell 2019, 177, 1682–1699. [Google Scholar] [CrossRef]
- Denton, D.; Kumar, S. Autophagy-dependent cell death. Cell Death Differ. 2019, 26, 605–616. [Google Scholar] [CrossRef]
- Mijaljica, D.; Prescott, M.; Devenish, R.J. Microautophagy in mammalian cells: Revisiting a 40-year-old conundrum. Autophagy 2011, 7, 673–682. [Google Scholar] [CrossRef]
- Dong, S.; Aguirre-Hernandez, C.; Scrivo, A.; Eliscovich, C.; Arias, E.; Bravo-Cordero, J.J.; Cuervo, A.M. Monitoring spatiotemporal changes in chaperone-mediated autophagy in vivo. Nat. Commun. 2020, 11, 645. [Google Scholar] [CrossRef]
- Yang, Y.; Klionsky, D.J. Autophagy and disease: Unanswered questions. Cell Death Differ. 2020, 27, 858–871. [Google Scholar] [CrossRef]
- Shang, L.; Wang, X. AMPK and mTOR coordinate the regulation of Ulk1 and mammalian autophagy initiation. Autophagy 2011, 7, 924–926. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.-M.; Puente, C.; Ganley, I.G.; Jiang, X. The ULK1 complex. Autophagy 2013, 9, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, N.; Sasaki, T.; Iemura, S.; Natsume, T.; Hara, T.; Mizushima, N. Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy 2009, 5, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Takamura, A.; Kishi, C.; Iemura, S.; Natsume, T.; Guan, J.-L.; Mizushima, N. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J. Cell Biol. 2008, 181, 497–510. [Google Scholar] [CrossRef]
- Hurley, J.H.; Young, L.N. Mechanisms of Autophagy Initiation. Annu. Rev. Biochem. 2017, 86, 225–244. [Google Scholar] [CrossRef]
- Young, L.N.; Goerdeler, F.; Hurley, J.H. Structural pathway for allosteric activation of the autophagic PI 3-kinase complex I. Proc. Natl. Acad. Sci. USA 2019, 116, 21508–21513. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Shpilka, T.; Elazar, Z. Mechanisms of Autophagosome Biogenesis. Curr. Biol. 2012, 22, R29–R34. [Google Scholar] [CrossRef]
- Kawabata, T.; Yoshimori, T. Autophagosome biogenesis and human health. Cell Discov. 2020, 6, 33. [Google Scholar] [CrossRef]
- Mizushima, N.; Noda, T.; Yoshimori, T.; Tanaka, Y.; Ishii, T.; George, M.D.; Klionsky, D.J.; Ohsumi, M.; Ohsumi, Y. A protein conjugation system essential for autophagy. Nature 1998, 395, 395–398. [Google Scholar] [CrossRef]
- Dancourt, J.; Melia, T.J. Lipidation of the autophagy proteins LC3 and GABARAP is a membrane-curvature dependent process. Autophagy 2014, 10, 1470–1471. [Google Scholar] [CrossRef][Green Version]
- Keller, M.D.; Torres, V.J.; Cadwell, K. Autophagy and microbial pathogenesis. Cell Death Differ. 2020, 27, 872–886. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Bowman, J.W.; Jung, J.U. Autophagy during viral infection—A double-edged sword. Nat. Rev. Microbiol. 2018, 16, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.; Malireddi, R.S.; Lu, Q.; Cunha, L.D.; Pelletier, S.; Gingras, S.; Orchard, R.; Guan, J.-L.; Tan, H.; Peng, J.; et al. Molecular characterization of LC3-associated phagocytosis (LAP) reveals distinct roles for Rubicon, NOX2, and autophagy proteins. Nat. Cell Biol. 2015, 17, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.P.; Chakraborty, P. Malaria: Autophagy as a Potential Therapeutic Target. JPP 2016, 4, 298–306. [Google Scholar] [CrossRef][Green Version]
- Cervantes, S.; Bunnik, E.M.; Saraf, A.; Conner, C.M.; Escalante, A.; Sardiu, M.E.; Ponts, N.; Prudhomme, J.; Florens, L.; Le Roch, K.G. The multifunctional autophagy pathway in the human malaria parasite, Plasmodium falciparum. Autophagy 2014, 10, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Hain, A.U.P.; Bosch, J. Autophagy in Plasmodium, a multifunctional pathway? Comput. Struct. Biotechnol. J. 2013, 8. [Google Scholar] [CrossRef]
- Coppens, I. Metamorphoses of Malaria: The role of autophagy in parasite differentiation. Essays Biochem. 2011, 51, 127–136. [Google Scholar] [CrossRef]
- Tomlins, A.M.; Ben-Rached, F.; Williams, R.A.; Proto, W.R.; Coppens, I.; Ruch, U.; Gilberger, T.W.; Coombs, G.H.; Mottram, J.C.; Müller, S.; et al. Plasmodium falciparum ATG8 implicated in both autophagy and apicoplast formation. Autophagy 2013, 9, 1540–1552. [Google Scholar] [CrossRef]
- Joy, S.; Thirunavukkarasu, L.; Agrawal, P.; Singh, A.; Sagar, B.K.C.; Manjithaya, R.; Surolia, N. Basal and starvation-induced autophagy mediates parasite survival during intraerythrocytic stages of Plasmodium falciparum. Cell Death Discov. 2018, 4, 43. [Google Scholar] [CrossRef]
- Brito, T.; Barone, A.A.; Faria, R.M. Human liver biopsy inP. falciparum andP. vivax malaria: A light and electron microscopy study. Virchows Arch. Abt. A Path. Anat. 1969, 348, 220–229. [Google Scholar] [CrossRef]
- Evans, R.J.; Sundaramurthy, V.; Frickel, E.-M. The Interplay of Host Autophagy and Eukaryotic Pathogens. Front. Cell Dev. Biol. 2018, 6, 118. [Google Scholar] [CrossRef] [PubMed]
- Agop-Nersesian, C.; Niklaus, L.; Wacker, R.; Theo Heussler, V. Host cell cytosolic immune response during Plasmodium liver stage development. FEMS Microbiol. Rev. 2018, 42, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Schmuckli-Maurer, J.; Reber, V.; Wacker, R.; Bindschedler, A.; Zakher, A.; Heussler, V.T. Inverted recruitment of autophagy proteins to the Plasmodium berghei parasitophorous vacuole membrane. PLoS ONE 2017, 12, e0183797. [Google Scholar] [CrossRef] [PubMed]
- Prado, M.; Eickel, N.; De Niz, M.; Heitmann, A.; Agop-Nersesian, C.; Wacker, R.; Schmuckli-Maurer, J.; Caldelari, R.; Janse, C.J.; Khan, S.M.; et al. Long-term live imaging reveals cytosolic immune responses of host hepatocytes against Plasmodium infection and parasite escape mechanisms. Autophagy 2015, 11, 1561–1579. [Google Scholar] [CrossRef]
- Wacker, R.; Eickel, N.; Schmuckli-Maurer, J.; Annoura, T.; Niklaus, L.; Khan, S.M.; Guan, J.-L.; Heussler, V.T. LC3-association with the parasitophorous vacuole membrane of Plasmodium berghei liver stages follows a noncanonical autophagy pathway. Cell. Microbiol. 2017, 19, e12754. [Google Scholar] [CrossRef]
- Boonhok, R.; Rachaphaew, N.; Duangmanee, A.; Chobson, P.; Pattaradilokrat, S.; Utaisincharoen, P.; Sattabongkot, J.; Ponpuak, M. LAP-like process as an immune mechanism downstream of IFN-γ in control of the human malaria Plasmodium vivax liver stage. Proc. Natl. Acad. Sci. USA 2016, 113, E3519–E3528. [Google Scholar] [CrossRef]
- Ghartey-Kwansah, G.; Adu-Nti, F.; Aboagye, B.; Ankobil, A.; Essuman, E.E.; Opoku, Y.K.; Abokyi, S.; Abu, E.K.; Boampong, J.N. Autophagy in the control and pathogenesis of parasitic infections. Cell Biosci. 2020, 10, 101. [Google Scholar] [CrossRef]
- Thieleke-Matos, C.; Lopes da Silva, M.; Cabrita-Santos, L.; Portal, M.D.; Rodrigues, I.P.; Zuzarte-Luis, V.; Ramalho, J.S.; Futter, C.E.; Mota, M.M.; Barral, D.C.; et al. Host cell autophagy contributes to Plasmodium liver development. Cell. Microbiol. 2016, 18, 437–450. [Google Scholar] [CrossRef]
- Zhao, C.; Liu, T.; Zhou, T.; Fu, Y.; Zheng, H.; Ding, Y.; Zhang, K.; Xu, W. The rodent malaria liver stage survives in the rapamycin-induced autophagosome of infected Hepa1–6 cells. Sci. Rep. 2016, 6, 38170. [Google Scholar] [CrossRef]
- Real, E.; Rodrigues, L.; Cabal, G.G.; Enguita, F.J.; Mancio-Silva, L.; Mello-Vieira, J.; Beatty, W.; Vera, I.M.; Zuzarte-Luís, V.; Figueira, T.N.; et al. Plasmodium UIS3 sequesters host LC3 to avoid elimination by autophagy in hepatocytes. Nat. Microbiol. 2018, 3, 17–25. [Google Scholar] [CrossRef]
- Longley, R.J.; Halbroth, B.R.; Salman, A.M.; Ewer, K.J.; Hodgson, S.H.; Janse, C.J.; Khan, S.M.; Hill, A.V.S.; Spencer, A.J. Assessment of the Plasmodium falciparum Preerythrocytic Antigen UIS3 as a Potential Candidate for a Malaria Vaccine. Infect. Immun. 2017, 85, e00641-16. [Google Scholar] [CrossRef] [PubMed]
- Lima, F.A.; Barateiro, A.; Dombrowski, J.G.; de Souza, R.M.; Costa, D.D.S.; Murillo, O.; Epiphanio, S.; Gonçalves, L.A.; Marinho, C.R.F. Plasmodium falciparum infection dysregulates placental autophagy. PLoS ONE 2019, 14, e0226117. [Google Scholar] [CrossRef] [PubMed]
- Dimasuay, K.G.; Aitken, E.H.; Rosario, F.; Njie, M.; Glazier, J.; Rogerson, S.J.; Fowkes, F.J.I.; Beeson, J.G.; Powell, T.; Jansson, T.; et al. Inhibition of placental mTOR signaling provides a link between placental malaria and reduced birthweight. BMC Med. 2017, 15, 1. [Google Scholar] [CrossRef] [PubMed]
- Dimasuay, K.G.; Gong, L.; Rosario, F.; McBryde, E.; Spelman, T.; Glazier, J.; Rogerson, S.J.; Beeson, J.G.; Jansson, T.; Devenish, R.J.; et al. Impaired placental autophagy in placental malaria. PLoS ONE 2017, 12, e0187291. [Google Scholar] [CrossRef]
- Kain, H.S.; Glennon, E.K.K.; Vijayan, K.; Arang, N.; Douglass, A.N.; Fortin, C.L.; Zuck, M.; Lewis, A.J.; Whiteside, S.L.; Dudgeon, D.R.; et al. Liver stage malaria infection is controlled by host regulators of lipid peroxidation. Cell Death Differ. 2020, 27, 44–54. [Google Scholar] [CrossRef]
- Kho, S.; Minigo, G.; Andries, B.; Leonardo, L.; Prayoga, P.; Poespoprodjo, J.R.; Kenangalem, E.; Price, R.N.; Woodberry, T.; Anstey, N.M.; et al. Circulating Neutrophil Extracellular Traps and Neutrophil Activation Are Increased in Proportion to Disease Severity in Human Malaria. J. Infect. Dis. 2019, 219, 1994–2004. [Google Scholar] [CrossRef]
- Zychlinsky, A.; Prevost, M.C.; Sansonetti, P.J. Shigella flexneri induces apoptosis in infected macrophages. Nature 1992, 358, 167–169. [Google Scholar] [CrossRef]
- Cookson, B.T.; Brennan, M.A. Pro-inflammatory programmed cell death. Trends Microbiol. 2001, 9, 113–114. [Google Scholar] [CrossRef]
- Aachoui, Y.; Sagulenko, V.; Miao, E.A.; Stacey, K.J. Inflammasome-mediated pyroptotic and apoptotic cell death, and defense against infection. Curr. Opin. Microbiol. 2013, 16, 319–326. [Google Scholar] [CrossRef]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting Edge: NF-κB Activating Pattern Recognition and Cytokine Receptors License NLRP3 Inflammasome Activation by Regulating NLRP3 Expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Enosi Tuipulotu, D.; Tan, W.H.; Kay, C.; Man, S.M. Emerging Activators and Regulators of Inflammasomes and Pyroptosis. Trends Immunol. 2019, 40, 1035–1052. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The Inflammasome. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Planillo, R.; Kuffa, P.; Martínez-Colón, G.; Smith, B.L.; Rajendiran, T.M.; Núñez, G. K+ Efflux Is the Common Trigger of NLRP3 Inflammasome Activation by Bacterial Toxins and Particulate Matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.; Vince, J.E. Pyroptosis versus necroptosis: Similarities, differences, and crosstalk. Cell Death Differ. 2019, 26, 99–114. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; Hu, L.; Shao, F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014, 514, 187–192. [Google Scholar] [CrossRef]
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Walle, L.V.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.; Heldens, S.; et al. Non-canonical inflammasome activation targets caspase-11. Nature 2011, 479, 117–121. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, W.; Shi, X.; Ding, J.; Liu, W.; He, H.; Wang, K.; Shao, F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 2017, 547, 99–103. [Google Scholar] [CrossRef]
- Sarhan, J.; Liu, B.C.; Muendlein, H.I.; Li, P.; Nilson, R.; Tang, A.Y.; Rongvaux, A.; Bunnell, S.C.; Shao, F.; Green, D.R.; et al. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc. Natl. Acad. Sci. USA 2018, 115, E10888–E10897. [Google Scholar] [CrossRef]
- Taabazuing, C.Y.; Okondo, M.C.; Bachovchin, D.A. Pyroptosis and apoptosis pathways engage in bidirectional crosstalk in monocytes and macrophages. Cell Chem. Biol. 2017, 24, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.W.; Monteleone, M.; Boucher, D.; Sollberger, G.; Ramnath, D.; Condon, N.D.; von Pein, J.B.; Broz, P.; Sweet, M.J.; Schroder, K. Noncanonical inflammasome signaling elicits gasdermin D–dependent neutrophil extracellular traps. Sci. Immunol. 2018, 3, eaar6676. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhao, Y.; Lai, D.; Zhang, P.; Yang, Y.; Li, Y.; Fei, K.; Jiang, G.; Fan, J. Neutrophil extracellular traps promote macrophage pyroptosis in sepsis. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Dey, S.; Bindu, S.; Goyal, M.; Pal, C.; Alam, A.; Iqbal, M.S.; Kumar, R.; Sarkar, S.; Bandyopadhyay, U. Impact of Intravascular Hemolysis in Malaria on Liver Dysfunction: Involvement of Hepatic Free Heme Overload, Nf-Κb Activation, And Neutrophil Infiltration. J. Biol. Chem. 2012, 287, 26630–26646. [Google Scholar] [CrossRef]
- Nair, R.; Rao, K.; Mukherjee, D.; Datt, B.; Sharma, S.; Prakash, S. Acute kidney injury due to acute cortical necrosis following vivax malaria. Saudi J. Kidney Dis. Transp. 2019, 30, 960. [Google Scholar] [CrossRef]
- Rupani, A.B.; Amarapurkar, A.D. Hepatic changes in fatal malaria: An emerging problem. Ann. Trop. Med. Parasitol. 2009, 103, 119–127. [Google Scholar] [CrossRef]
- de Menezes, M.N.; Salles, É.M.; Vieira, F.; Amaral, E.P.; Zuzarte-Luís, V.; Cassado, A.; Epiphanio, S.; Alvarez, J.M.; Alves-Filho, J.C.; Mota, M.M.; et al. IL-1α promotes liver inflammation and necrosis during blood-stage Plasmodium chabaudi malaria. Sci. Rep. 2019, 9, 7575. [Google Scholar] [CrossRef]
- Dunstan, S.J.; Rockett, K.A.; Ngoc Quyen, N.T.; Teo, Y.Y.; Thai, C.Q.; Hang, N.T.; Jeffreys, A.; Clark, T.G.; Small, K.S.; Simmons, C.P.; et al. Variation in Human Genes Encoding Adhesion and Pro-inflammatory Molecules are Associated with Severe Malaria in the Vietnamese. Genes Immun. 2012, 13, 503–508. [Google Scholar] [CrossRef]
- Shio, M.T.; Eisenbarth, S.C.; Savaria, M.; Vinet, A.F.; Harder, K.W.; Sutterwala, F.S.; Bohle, D.S.; Descoteaux, A.; Olivier, M. Malarial Hemozoin Activates the NLRP3 Inflammasome through Lyn and Syk Kinases. PLoS Pathog. 2009, 5, 14. [Google Scholar] [CrossRef]
- Dostert, C.; Guarda, G.; Romero, J.F.; Menu, P.; Gross, O.; Tardivel, A.; Suva, M.-L.; Stehle, J.-C.; Kopf, M.; Stamenkovic, I.; et al. Malarial Hemozoin Is a Nalp3 Inflammasome Activating Danger Signal. PLoS ONE 2009, 4, e6510. [Google Scholar] [CrossRef]
- Griffith, J.W.; Sun, T.; McIntosh, M.T.; Bucala, R. Pure Hemozoin Is Inflammatory In Vivo and Activates the NALP3 Inflammasome via Release of Uric Acid. J. Immunol. 2009, 183, 5208–5220. [Google Scholar] [CrossRef] [PubMed]
- Gallego-Delgado, J.; Ty, M.; Orengo, J.M.; van de Hoef, D.; Rodriguez, A. A Surprising Role for Uric Acid: The Inflammatory Malaria Response. Curr. Rheumatol. Rep. 2014, 16, 401. [Google Scholar] [CrossRef] [PubMed]
- Dutra, F.F.; Alves, L.S.; Rodrigues, D.; Fernandez, P.L.; de Oliveira, R.B.; Golenbock, D.T.; Zamboni, D.S.; Bozza, M.T. Hemolysis-induced lethality involves inflammasome activation by heme. Proc. Natl. Acad. Sci. USA 2014, 111, E4110–E4118. [Google Scholar] [CrossRef] [PubMed]
- Man, S.M.; Karki, R.; Kanneganti, T.-D. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol. Rev. 2017, 277, 61–75. [Google Scholar] [CrossRef] [PubMed]
- Kordes, M.; Matuschewski, K.; Hafalla, J.C.R. Caspase-1 Activation of Interleukin-1β (IL-1β) and IL-18 Is Dispensable for Induction of Experimental Cerebral Malaria. Infect. Immun. 2011, 79, 3633–3641. [Google Scholar] [CrossRef]
- Pereira, L.M.N.; Assis, P.A.; de Araújo, N.M.; Durso, D.F.; Junqueira, C.; Ataíde, M.A.; Pereira, D.B.; Lien, E.; Fitzgerald, K.A.; Zamboni, D.S.; et al. Caspase-8 mediates inflammation and disease in rodent malaria. Nat. Commun. 2020, 11, 4596. [Google Scholar] [CrossRef]
- Reis, A.S.; Barboza, R.; Murillo, O.; Barateiro, A.; Peixoto, E.P.M.; Lima, F.A.; Gomes, V.M.; Dombrowski, J.G.; Leal, V.N.C.; Araujo, F.; et al. Inflammasome activation and IL-1 signaling during placental malaria induce poor pregnancy outcomes. Sci. Adv. 2020, 6, eaax6346. [Google Scholar] [CrossRef]
- Yu, X.; Du, Y.; Cai, C.; Cai, B.; Zhu, M.; Xing, C.; Tan, P.; Lin, M.; Wu, J.; Li, J.; et al. Inflammasome activation negatively regulates MyD88-IRF7 type I IFN signaling and anti-malaria immunity. Nat. Commun. 2018, 9, 4964. [Google Scholar] [CrossRef]
- Santos, M.L.S.; Reis, E.C.; Bricher, P.N.; Sousa, T.N.; Brito, C.F.A.; Lacerda, M.V.G.; Fontes, C.J.F.; Carvalho, L.H.; Pontillo, A. Contribution of inflammasome genetics in Plasmodium vivax malaria. Infect. Genet. Evol. 2016, 40, 162–166. [Google Scholar] [CrossRef]
- Strangward, P.; Haley, M.J.; Albornoz, M.G.; Barrington, J.; Shaw, T.; Dookie, R.; Zeef, L.; Baker, S.M.; Winter, E.; Tzeng, T.-C.; et al. Targeting the IL33–NLRP3 axis improves therapy for experimental cerebral malaria. Proc. Natl. Acad. Sci. USA 2018, 115, 7404–7409. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V. Neutrophil Extracellular Traps Kill Bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, S.; Stojkov, D.; Germic, N.; Simon, D.; Wang, X.; Benarafa, C.; Simon, H. Untangling “NETosis” from NETs. Eur. J. Immunol. 2019, 49, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, M.J.; Radic, M. Neutrophil extracellular traps (NETs): Double-edged swords of innate immunity. J. Immunol. 2012, 189, 2689–2695. [Google Scholar] [CrossRef]
- Metzler, K.D.; Goosmann, C.; Lubojemska, A.; Zychlinsky, A.; Papayannopoulos, V. A Myeloperoxidase-Containing Complex Regulates Neutrophil Elastase Release and Actin Dynamics during NETosis. Cell Rep. 2014, 8, 883–896. [Google Scholar] [CrossRef]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef]
- Li, P.; Li, M.; Lindberg, M.R.; Kennett, M.J.; Xiong, N.; Wang, Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 2010, 207, 1853–1862. [Google Scholar] [CrossRef]
- Wang, Y.; Li, M.; Stadler, S.; Correll, S.; Li, P.; Wang, D.; Hayama, R.; Leonelli, L.; Han, H.; Grigoryev, S.A.; et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J. Cell Biol. 2009, 184, 205–213. [Google Scholar] [CrossRef]
- Neubert, E.; Meyer, D.; Rocca, F.; Günay, G.; Kwaczala-Tessmann, A.; Grandke, J.; Senger-Sander, S.; Geisler, C.; Egner, A.; Schön, M.P.; et al. Chromatin swelling drives neutrophil extracellular trap release. Nat. Commun. 2018, 9, 3767. [Google Scholar] [CrossRef]
- Sollberger, G.; Choidas, A.; Burn, G.L.; Habenberger, P.; Di Lucrezia, R.; Kordes, S.; Menninger, S.; Eickhoff, J.; Nussbaumer, P.; Klebl, B.; et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci. Immunol. 2018, 3, eaar6689. [Google Scholar] [CrossRef]
- Wartha, F.; Henriques-Normark, B. ETosis: A Novel Cell Death Pathway. Sci. Signal. 2008, 1, pe25. [Google Scholar] [CrossRef]
- Radic, M. NETosis and ETosis: Incompletely Understood Types of Granulocyte Death and their Proposed Adaptive Benefits and Costs. In Apoptosis and Beyond; Radosevich, J., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2018; pp. 511–534. ISBN 978-1-119-43246-3. [Google Scholar]
- Aitken, E.H.; Alemu, A.; Rogerson, S.J. Neutrophils and Malaria. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Amulic, B.; Moxon, C.A.; Cunnington, A.J. A More Granular View of Neutrophils in Malaria. Trends Parasitol. 2020, 36, 501–503. [Google Scholar] [CrossRef]
- Chen, L.; Sendo, F. Cytokine and chemokine mRNA expression in neutrophils from CBA/NSlc mice infected with Plasmodium berghei ANKA that induces experimental cerebral malaria. Parasitol. Int. 2001, 50, 139–143. [Google Scholar] [CrossRef]
- Feintuch, C.M.; Saidi, A.; Seydel, K.; Chen, G.; Goldman-Yassen, A.; Mita-Mendoza, N.K.; Kim, R.S.; Frenette, P.S.; Taylor, T.; Daily, J.P. Activated Neutrophils Are Associated with Pediatric Cerebral Malaria Vasculopathy in Malawian Children. mBio 2016, 7, e01300-15. [Google Scholar] [CrossRef]
- Rocha, B.C.; Marques, P.E.; Leoratti, F.M.D.S.; Junqueira, C.; Pereira, D.B.; Antonelli, L.R.D.V.; Menezes, G.B.; Golenbock, D.T.; Gazzinelli, R.T. Type I Interferon Transcriptional Signature in Neutrophils and Low-Density Granulocytes Are Associated with Tissue Damage in Malaria. Cell Rep. 2015, 13, 2829–2841. [Google Scholar] [CrossRef]
- Sercundes, M.K.; Ortolan, L.S.; Debone, D.; Soeiro-Pereira, P.V.; Gomes, E.; Aitken, E.H.; Neto, A.C.; Russo, M.; D’ Império Lima, M.R.; Alvarez, J.M.; et al. Targeting Neutrophils to Prevent Malaria-Associated Acute Lung Injury/Acute Respiratory Distress Syndrome in Mice. PLoS Pathog. 2016, 12, e1006054. [Google Scholar] [CrossRef]
- Boström, S.; Schmiegelow, C.; Abu Abed, U.; Minja, D.T.R.; Lusingu, J.; Brinkmann, V.; Honkpehedji, Y.J.; Loembe, M.M.; Adegnika, A.A.; Mordmüller, B.; et al. Neutrophil alterations in pregnancy-associated malaria and induction of neutrophil chemotaxis by Plasmodium falciparum. Parasite Immunol. 2017, 39, e12433. [Google Scholar] [CrossRef]
- Baker, V.S.; Imade, G.E.; Molta, N.B.; Tawde, P.; Pam, S.D.; Obadofin, M.O.; Sagay, S.A.; Egah, D.Z.; Iya, D.; Afolabi, B.B.; et al. Cytokine-associated neutrophil extracellular traps and antinuclear antibodies in Plasmodium falciparum infected children under six years of age. Malar. J. 2008, 7, 41. [Google Scholar] [CrossRef]
- Boeltz, S.; Muñoz, L.E.; Fuchs, T.A.; Herrmann, M. Neutrophil Extracellular Traps Open the Pandora’s Box in Severe Malaria. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef]
- Knackstedt, S.L.; Georgiadou, A.; Apel, F.; Abu-Abed, U.; Moxon, C.A.; Cunnington, A.J.; Raupach, B.; Cunningham, D.; Langhorne, J.; Krüger, R.; et al. Neutrophil extracellular traps drive inflammatory pathogenesis in malaria. Sci. Immunol. 2019, 4, eaaw0336. [Google Scholar] [CrossRef]
- Rodrigues, D.A.S.; Prestes, E.B.; Gama, A.M.S.; Silva, L.D.S.; Pinheiro, A.A.S.; Ribeiro, J.M.C.; Campos, R.M.P.; Pimentel-Coelho, P.M.; De Souza, H.S.; Dicko, A.; et al. CXCR4 and MIF are required for neutrophil extracellular trap release triggered by Plasmodium-infected erythrocytes. PLoS Pathog. 2020, 16, e1008230. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Angeli, J.P.F.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.; et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Cassat, J.E.; Skaar, E.P. Iron in Infection and Immunity. Cell Host Microbe 2013, 13, 509–519. [Google Scholar] [CrossRef]
- Frazer, D.M.; Anderson, G.J. The regulation of iron transport: The Regulation of Iron Transport. BioFactors 2014, 40, 206–214. [Google Scholar] [CrossRef]
- Lane, D.J.R.; Merlot, A.M.; Huang, M.L.-H.; Bae, D.-H.; Jansson, P.J.; Sahni, S.; Kalinowski, D.S.; Richardson, D.R. Cellular iron uptake, trafficking and metabolism: Key molecules and mechanisms and their roles in disease. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2015, 1853, 1130–1144. [Google Scholar] [CrossRef]
- Mancias, J.D.; Vaites, L.P.; Nissim, S.; Biancur, D.E.; Kim, A.J.; Wang, X.; Liu, Y.; Goessling, W.; Kimmelman, A.C.; Harper, J.W. Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2-mediated proteolysis. eLife 2015, 4, e10308. [Google Scholar] [CrossRef]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef]
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem. Biophys. Res. Commun. 2016, 478, 1338–1343. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Tamba, M.; Ishii, T.; Bannai, S. Cloning and Expression of a Plasma Membrane Cystine/Glutamate Exchange Transporter Composed of Two Distinct Proteins. J. Biol. Chem. 1999, 274, 11455–11458. [Google Scholar] [CrossRef] [PubMed]
- Conrad, M.; Sato, H. The oxidative stress-inducible cystine/glutamate antiporter, system xc−: Cystine supplier and beyond. Amino Acids 2012, 42, 231–246. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Lewerenz, J.; Hewett, S.J.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The Cystine/Glutamate Antiporter System xc− in Health and Disease: From Molecular Mechanisms to Novel Therapeutic Opportunities. Antioxid. Redox Signal. 2013, 18, 522–555. [Google Scholar] [CrossRef]
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cell 2015, 59, 298–308. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Jiang, X.; Gu, W. Emerging Mechanisms and Disease Relevance of Ferroptosis. Trends Cell Biol. 2020, 30, 478–490. [Google Scholar] [CrossRef]
- Hu, W.; Zhang, C.; Wu, R.; Sun, Y.; Levine, A.; Feng, Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc. Natl. Acad. Sci. USA 2010, 107, 7455–7460. [Google Scholar] [CrossRef]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.-J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells: Hepatobiliary Malignancies. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Fearnhead, H.O.; Vandenabeele, P.; Vanden Berghe, T. How do we fit ferroptosis in the family of regulated cell death? Cell Death Differ. 2017, 24, 1991–1998. [Google Scholar] [CrossRef] [PubMed]
- Das, B.S.; Nanda, N.K. Evidence for erythrocyte lipid peroxidation in acute falciparum malaria. Trans. R. Soc. Trop. Med. Hyg. 1999, 93, 58–62. [Google Scholar] [CrossRef]
- Drakesmith, H.; Prentice, A.M. Hepcidin and the Iron-Infection Axis. Science 2012, 338, 768–772. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.-L.; Wu, J.; Shah, B.N.; Greutélaers, K.C.; Ghosh, M.C.; Ollivierre, H.; Su, X.; Thuma, P.E.; Bedu-Addo, G.; Mockenhaupt, F.P.; et al. Erythrocytic ferroportin reduces intracellular iron accumulation, hemolysis, and malaria risk. Science 2018, 359, 1520–1523. [Google Scholar] [CrossRef] [PubMed]
- Fortes, G.B.; Alves, L.S.; de Oliveira, R.; Dutra, F.F.; Rodrigues, D.; Fernandez, P.L.; Souto-Padron, T.; De Rosa, M.J.; Kelliher, M.; Golenbock, D.; et al. Heme induces programmed necrosis on macrophages through autocrine TNF and ROS production. Blood 2012, 119, 2368–2375. [Google Scholar] [CrossRef]
- Percário, S.; Moreira, D.R.; Gomes, B.A.Q.; Ferreira, M.E.S.; Gonçalves, A.C.M.; Laurindo, P.S.O.C.; Vilhena, T.C.; Dolabela, M.F.; Green, M.D. Oxidative Stress in Malaria. Int. J. Mol. Sci 2012, 13, 16346–16372. [Google Scholar] [CrossRef]
- Fabbri, C.; de Cássia Mascarenhas-Netto, R.; Lalwani, P.; Melo, G.C.; Magalhães, B.M.; Alexandre, M.A.; Lacerda, M.V.; Lima, E.S. Lipid peroxidation and antioxidant enzymes activity in Plasmodium vivax malaria patients evolving with cholestatic jaundice. Malar. J. 2013, 12, 315. [Google Scholar] [CrossRef]
- Prasannachandra; D’Souza, V.; D’Souza, B. Comparative study on lipid peroxidation and antioxidant vitamins E and C inFalciparum andVivax malaria. Indian J. Clin. Biochem. 2006, 21, 103–106. [Google Scholar] [CrossRef]
- Jennis, M.; Kung, C.-P.; Basu, S.; Budina-Kolomets, A.; Leu, J.I.-J.; Khaku, S.; Scott, J.P.; Cai, K.Q.; Campbell, M.R.; Porter, D.K.; et al. An African-specific polymorphism in the TP53 gene impairs p53 tumor suppressor function in a mouse model. Genes Dev. 2016, 30, 918–930. [Google Scholar] [CrossRef]
- Singh, K.S.; Leu, J.I.-J.; Barnoud, T.; Vonteddu, P.; Gnanapradeepan, K.; Lin, C.; Liu, Q.; Barton, J.C.; Kossenkov, A.V.; George, D.L.; et al. African-centric TP53 variant increases iron accumulation and bacterial pathogenesis but improves response to malaria toxin. Nat. Commun. 2020, 11, 473. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.A.; Goheen, M.M.; Fulford, A.; Prentice, A.M.; Elnagheeb, M.A.; Patel, J.; Fisher, N.; Taylor, S.M.; Kasthuri, R.S.; Cerami, C. Host iron status and iron supplementation mediate susceptibility to erythrocytic stage Plasmodium falciparum. Nat. Commun. 2014, 5, 4446. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sena-dos-Santos, C.; Braga-da-Silva, C.; Marques, D.; Azevedo dos Santos Pinheiro, J.; Ribeiro-dos-Santos, Â.; Cavalcante, G.C. Unraveling Cell Death Pathways during Malaria Infection: What Do We Know So Far? Cells 2021, 10, 479. https://doi.org/10.3390/cells10020479
Sena-dos-Santos C, Braga-da-Silva C, Marques D, Azevedo dos Santos Pinheiro J, Ribeiro-dos-Santos Â, Cavalcante GC. Unraveling Cell Death Pathways during Malaria Infection: What Do We Know So Far? Cells. 2021; 10(2):479. https://doi.org/10.3390/cells10020479
Chicago/Turabian StyleSena-dos-Santos, Camille, Cíntia Braga-da-Silva, Diego Marques, Jhully Azevedo dos Santos Pinheiro, Ândrea Ribeiro-dos-Santos, and Giovanna C. Cavalcante. 2021. "Unraveling Cell Death Pathways during Malaria Infection: What Do We Know So Far?" Cells 10, no. 2: 479. https://doi.org/10.3390/cells10020479
APA StyleSena-dos-Santos, C., Braga-da-Silva, C., Marques, D., Azevedo dos Santos Pinheiro, J., Ribeiro-dos-Santos, Â., & Cavalcante, G. C. (2021). Unraveling Cell Death Pathways during Malaria Infection: What Do We Know So Far? Cells, 10(2), 479. https://doi.org/10.3390/cells10020479