
Estrogen Receptors in Polycystic Ovary Syndrome



Abstract
1. Introduction
2. The Characteristics of Estrogen Receptors
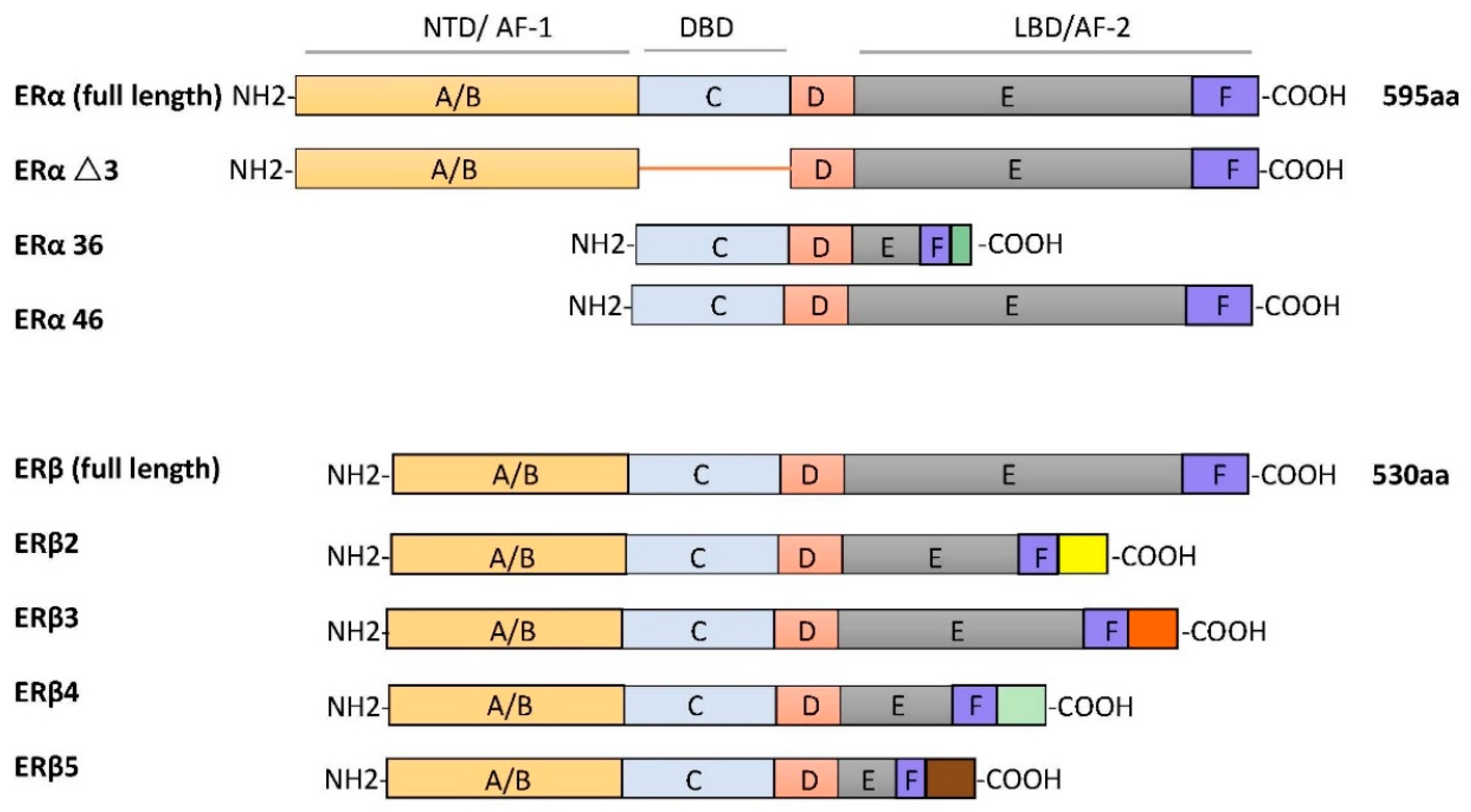
2.1. Estrogen Receptors: Expression, Structure
2.2. Estrogen Receptor Ligands
3. Physiological and Pathological Function of ERs
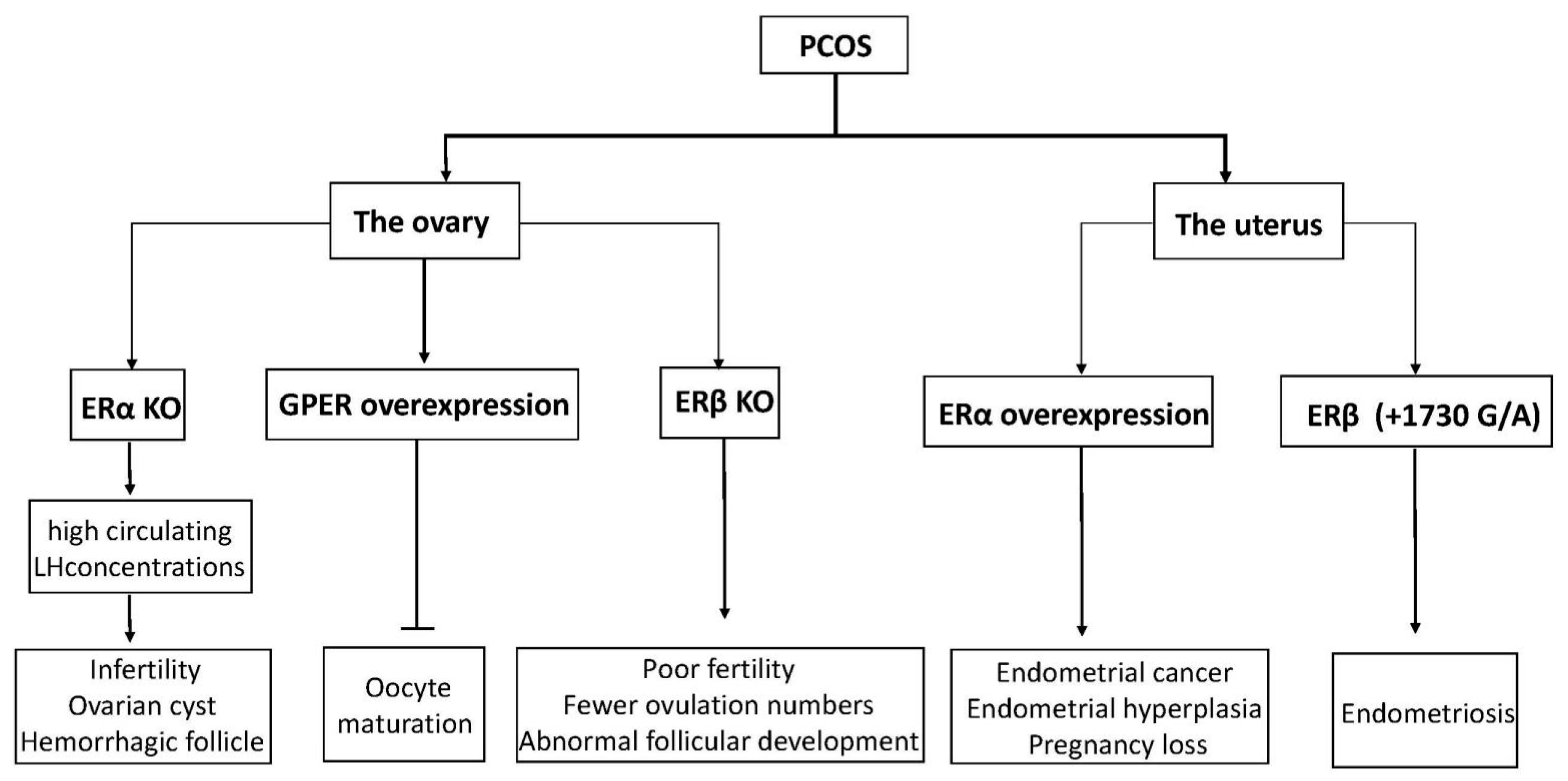
4. Functions of ERs in PCOS
4.1. ERs with Follicular Formation/Ovulation
4.2. ER Changes Associated with Endometrium
5. Selective Estrogen Receptor Modulators
6. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rosenfield, R.L.; Ehrmann, D.A. The Pathogenesis of Polycystic Ovary Syndrome (PCOS): The Hypothesis of PCOS as Functional Ovarian Hyperandrogenism Revisited. Endocr. Rev. 2016, 37, 467–520. [Google Scholar] [CrossRef] [PubMed]
- Dumesic, D.A.; Oberfield, S.E.; Stener-Victorin, E.; Marshall, J.C.; Laven, J.S.; Legro, R.S. Scientific Statement on the Diagnostic Criteria, Epidemiology, Pathophysiology, and Molecular Genetics of Polycystic Ovary Syndrome. Endocr. Rev. 2015, 36, 487–525. [Google Scholar] [CrossRef]
- Khan, M.J.; Ullah, A.; Basit, S. Genetic Basis of Polycystic Ovary Syndrome (PCOS): Current Perspectives. Appl. Clin. Genet. 2019, 12, 249–260. [Google Scholar] [CrossRef] [PubMed]
- March, W.A.; Moore, V.M.; Willson, K.J.; Phillips, D.I.W.; Norman, R.J.; Davies, M.J. The prevalence of polycystic ovary syndrome in a community sample assessed under contrasting diagnostic criteria. Hum. Reprod. 2010, 25, 544–551. [Google Scholar] [CrossRef]
- Azziz, R.; Carmina, E.; Chen, Z.; Dunaif, A.; Laven, J.S.E.; Legro, R.S.; Lizneva, D.; Natterson-Horowtiz, B.; Teede, H.J.; Yildiz, B.O. Polycystic ovary syndrome. Nat. Rev. Dis. Primers. 2016, 2, 16057. [Google Scholar] [CrossRef] [PubMed]
- Boomsma, C.M.; Eijkemans, M.J.C.; Hughes, E.G.; Visser, G.H.A.; Fauser, B.C.J.M.; Macklon, N.S. A meta-analysis of pregnancy outcomes in women with polycystic ovary syndrome. Hum. Reprod. Update 2006, 12, 673–683. [Google Scholar] [CrossRef]
- Dumesic, D.A.; Lobo, R.A. Cancer risk and PCOS. Steroids 2013, 78, 782–785. [Google Scholar] [CrossRef]
- Cobin, R.H. Cardiovascular and metabolic risks associated with PCOS. Intern. Emerg. Med. 2013, 8 (Suppl. 1), S61–S64. [Google Scholar] [CrossRef]
- Azziz, R. Polycystic Ovary Syndrome. Obstet. Gynecol. 2018, 132, 321–336. [Google Scholar] [CrossRef]
- Richards, J.S.; Pangas, S.A. The ovary: Basic biology and clinical implications. J. Clin. Investig. 2010, 120, 963–972. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, Y.; Li, Y.; Liu, X.; Wang, X.; Zhang, C.; Hao, C.; Deng, S. Regulation of follicular development and differentiation by intra-ovarian factors and endocrine hormones. Front. Biosci. (Landmark Ed.) 2019, 24, 983–993. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Sait, S.F. Luteinizing hormone and its dilemma in ovulation induction. J. Hum. Reprod. Sci. 2011, 4, 2–7. [Google Scholar] [CrossRef]
- Tang, Z.; Xu, X.; Deng, S.; Lian, Z.; Yu, K. Oestrogenic Endocrine Disruptors in the Placenta and the Fetus. Int. J. Mol. Sci. 2020, 21, 1519. [Google Scholar] [CrossRef]
- Li, Y.; Guo, J.; Deng, S.; Gao, Z.; Liu, Y.; Gu, Q. Fibrin Facilitates Mesenchymal Stem Cells to Ameliorate Rats with Polycystic Ovary Syndrome. Appl. Sci. 2020, 10, 3598. [Google Scholar] [CrossRef]
- Gustafsson, J. What pharmacologists can learn from recent advances in estrogen signalling. Trends Pharmacol. Sci. 2003, 24, 479–485. [Google Scholar] [CrossRef]
- Jensen, E.V.; Desombre, E.R.; Kawashima, T.; Suzuki, T.; Kyser, K.; Jungblut, P.W. Estrogen-binding substances of target tissues. Science 1967, 158, 529–530. [Google Scholar] [CrossRef]
- Jensen, E.V.; Suzuki, T.; Kawashima, T.; Stumpf, W.E.; Jungblut, P.W.; DeSombre, E.R. A two-step mechanism for the interaction of estradiol with rat uterus. Proc. Natl. Acad. Sci. USA 1968, 59, 632–638. [Google Scholar] [CrossRef]
- Kuiper, G.G.; Enmark, E.; Pelto-Huikko, M.; Nilsson, S.; Gustafsson, J.A. Cloning of a novel receptor expressed in rat prostate and ovary. Proc. Natl. Acad. Sci. USA 1996, 93, 5925–5930. [Google Scholar] [CrossRef]
- Brandenberger, A.W.; Tee, M.K.; Lee, J.Y.; Chao, V.; Jaffe, R.B. Tissue distribution of estrogen receptors alpha (ER-alpha) and beta (ER-beta) mRNA in the midgestational human fetus. J. Clin. Endocrinol. Metab. 1997, 82, 3509–3512. [Google Scholar]
- Shi, L.; Dong, B.; Li, Z.; Lu, Y.; Ouyang, T.; Li, J.; Wang, T.; Fan, Z.; Fan, T.; Lin, B.; et al. Expression of ER-{alpha}36, a novel variant of estrogen receptor {alpha}, and resistance to tamoxifen treatment in breast cancer. J. Clin. Oncol. 2009, 27, 3423–3429. [Google Scholar] [CrossRef] [PubMed]
- Denger, S.; Reid, G.; Kos, M.; Flouriot, G.; Parsch, D.; Brand, H.; Korach, K.S.; Sonntag-Buck, V.; Gannon, F. ERalpha gene expression in human primary osteoblasts: Evidence for the expression of two receptor proteins. Mol. Endocrinol. 2001, 15, 2064–2077. [Google Scholar]
- Moore, J.T.; McKee, D.D.; Slentz-Kesler, K.; Moore, L.B.; Jones, S.A.; Horne, E.L.; Su, J.L.; Kliewer, S.A.; Lehmann, J.M.; Willson, T.M. Cloning and characterization of human estrogen receptor beta isoforms. Biochem. Biophys. Res. Commun. 1998, 247, 75–78. [Google Scholar] [CrossRef]
- Jia, M.; Dahlman-Wright, K.; Gustafsson, J. Estrogen receptor alpha and beta in health and disease. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 557–568. [Google Scholar] [CrossRef]
- Kumar, R.; Zakharov, M.N.; Khan, S.H.; Miki, R.; Jang, H.; Toraldo, G.; Singh, R.; Bhasin, S.; Jasuja, R. The dynamic structure of the estrogen receptor. J. Amino Acids 2011, 2011, 812540. [Google Scholar] [CrossRef]
- Feng, Y.; Gregor, P. Cloning of a novel member of the G protein-coupled receptor family related to peptide receptors. Biochem. Biophys. Res. Commun. 1997, 231, 651–654. [Google Scholar] [CrossRef]
- O’Dowd, B.F.; Nguyen, T.; Marchese, A.; Cheng, R.; Lynch, K.R.; Heng, H.H.; Kolakowski, L.F.J.; George, S.R. Discovery of three novel G-protein-coupled receptor genes. Genomics 1998, 47, 310–313. [Google Scholar] [CrossRef]
- Revankar, C.M.; Cimino, D.F.; Sklar, L.A.; Arterburn, J.B.; Prossnitz, E.R. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science 2005, 307, 1625–1630. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.; Pang, Y.; Filardo, E.J.; Dong, J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology 2005, 146, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Prossnitz, E.R.; Barton, M. Estrogen biology: New insights into GPER function and clinical opportunities. Mol. Cell Endocrinol. 2014, 389, 71–83. [Google Scholar] [CrossRef]
- Barton, M.; Filardo, E.J.; Lolait, S.J.; Thomas, P.; Maggiolini, M.; Prossnitz, E.R. Twenty years of the G protein-coupled estrogen receptor GPER: Historical and personal perspectives. J. Steroid Biochem. Mol. Biol. 2018, 176, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Filardo, E.J.; Quinn, J.A.; Bland, K.I.; Frackelton, A.R.J. Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol. Endocrinol. 2000, 14, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.; Xuan, J.; Liu, Y.; Shi, G. Function of G-Protein-Coupled Estrogen Receptor-1 in Reproductive System Tumors. J. Immunol. Res. 2016, 2016, 7128702. [Google Scholar] [CrossRef] [PubMed]
- Ososki, A.L.; Kennelly, E.J. Phytoestrogens: A review of the present state of research. Phytother. Res. 2003, 17, 845–869. [Google Scholar] [CrossRef] [PubMed]
- Patisaul, H.B.; Jefferson, W. The pros and cons of phytoestrogens. Front. Neuroendocrinol. 2010, 31, 400–419. [Google Scholar] [CrossRef] [PubMed]
- Lóránd, T.; Vigh, E.; Garai, J. Hormonal action of plant derived and anthropogenic non-steroidal estrogenic compounds: Phytoestrogens and xenoestrogens. Curr. Med. Chem. 2010, 17, 3542–3574. [Google Scholar] [CrossRef]
- Casals-Casas, C.; Desvergne, B. Endocrine disruptors: From endocrine to metabolic disruption. Annu. Rev. Physiol. 2011, 73, 135–162. [Google Scholar] [CrossRef] [PubMed]
- Singleton, D.W.; Khan, S.A. Xenoestrogen exposure and mechanisms of endocrine disruption. Front. Biosci. 2003, 8, s110–s118. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.; Dong, J. Binding and activation of the seven-transmembrane estrogen receptor GPR30 by environmental estrogens: A potential novel mechanism of endocrine disruption. J. Steroid Biochem. Mol. Biol. 2006, 102, 175–179. [Google Scholar] [CrossRef]
- Jordan, V.C. SERMs: Meeting the promise of multifunctional medicines. J. Natl. Cancer Inst. 2007, 99, 350–356. [Google Scholar] [CrossRef]
- Morello, K.C.; Wurz, G.T.; DeGregorio, M.W. SERMs: Current status and future trends. Crit. Rev. Oncol. Hematol. 2002, 43, 63–76. [Google Scholar] [CrossRef]
- Huang, D.; Yang, F.; Wang, Y.; Guan, X. Mechanisms of resistance to selective estrogen receptor down-regulator in metastatic breast cancer. Biochim. Biophys. Acta. Rev. Cancer 2017, 1868, 148–156. [Google Scholar] [CrossRef]
- Dai, X.; Xiang, L.; Li, T.; Bai, Z. Cancer Hallmarks, Biomarkers and Breast Cancer Molecular Subtypes. J. Cancer 2016, 7, 1281–1294. [Google Scholar] [CrossRef]
- Albanito, L.; Madeo, A.; Lappano, R.; Vivacqua, A.; Rago, V.; Carpino, A.; Oprea, T.I.; Prossnitz, E.R.; Musti, A.M.; Andò, S.; et al. G protein-coupled receptor 30 (GPR30) mediates gene expression changes and growth response to 17beta-estradiol and selective GPR30 ligand G-1 in ovarian cancer cells. Cancer Res. 2007, 67, 1859–1866. [Google Scholar] [CrossRef]
- Gambrell, R.D.J.; Bagnell, C.A.; Greenblatt, R.B. Role of estrogens and progesterone in the etiology and prevention of endometrial cancer: Review. Am. J. Obstet. Gynecol. 1983, 146, 696–707. [Google Scholar] [CrossRef]
- Shang, K.; Jia, X.; Qiao, J.; Kang, J.; Guan, Y. Endometrial abnormality in women with polycystic ovary syndrome. Reprod. Sci. 2012, 19, 674–683. [Google Scholar] [CrossRef]
- Tang, Z.; Deng, S.; Lian, Z.; Yu, K. Terazosin reduces steroidogenic factor 1 and upregulates heat shock protein 90 expression in LH-induced bovine ovarian theca cells. Free Radic. Biol. Med. 2021, 163, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Ryu, Y.; Kim, S.W.; Kim, Y.Y.; Ku, S. Animal Models for Human Polycystic Ovary Syndrome (PCOS) Focused on the Use of Indirect Hormonal Perturbations: A Review of the Literature. Int. J. Mol. Sci. 2019, 20, 2720. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kang, D.; Hudgins-Spivey, S.; Krust, A.; Lee, E.; Koo, Y.; Cheon, Y.; Gye, M.C.; Chambon, P.; Ko, C. Theca-specific estrogen receptor-alpha knockout mice lose fertility prematurely. Endocrinology 2009, 150, 3855–3862. [Google Scholar] [CrossRef] [PubMed]
- Brawer, J.R.; Munoz, M.; Farookhi, R. Development of the polycystic ovarian condition (PCO) in the estradiol valerate-treated rat. Biol. Reprod. 1986, 35, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Couse, J.F.; Yates, M.M.; Sanford, R.; Nyska, A.; Nilson, J.H.; Korach, K.S. Formation of cystic ovarian follicles associated with elevated luteinizing hormone requires estrogen receptor-beta. Endocrinology 2004, 145, 4693–4702. [Google Scholar] [CrossRef]
- Arifin, E.; Shively, C.A.; Register, T.C.; Cline, J.M. Polycystic ovary syndrome with endometrial hyperplasia in a cynomolgus monkey (Macaca fascicularis). Vet. Pathol. 2008, 45, 512–515. [Google Scholar] [CrossRef] [PubMed]
- Eisner, J.R.; Barnett, M.A.; Dumesic, D.A.; Abbott, D.H. Ovarian hyperandrogenism in adult female rhesus monkeys exposed to prenatal androgen excess. Fertil. Steril. 2002, 77, 167–172. [Google Scholar] [CrossRef]
- West, C.; Foster, D.L.; Evans, N.P.; Robinson, J.; Padmanabhan, V. Intra-follicular activin availability is altered in prenatally-androgenized lambs. Mol. Cell Endocrinol. 2001, 185, 51–59. [Google Scholar] [CrossRef]
- Isobe, N.; Yoshimura, Y. Deficient proliferation and apoptosis in the granulosa and theca interna cells of the bovine cystic follicle. J. Reprod. Dev. 2007, 53, 1119–1124. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Zhang, R.; Lian, Z.; Deng, S.; Yu, K. Estrogen-Receptor Expression and Function in Female Reproductive Disease. Cells 2019, 8, 1123. [Google Scholar] [CrossRef]
- Majumder, S.; Das, S.; Moulik, S.R.; Mallick, B.; Pal, P.; Mukherjee, D. G-protein coupled estrogen receptor (GPER) inhibits final oocyte maturation in common carp, Cyprinus carpio. Gen. Comp. Endocrinol. 2015, 211, 28–38. [Google Scholar] [CrossRef]
- Zang, L.; Zhang, Q.; Zhou, Y.; Zhao, Y.; Lu, L.; Jiang, Z.; Peng, Z.; Zou, S. Expression pattern of G protein-coupled estrogen receptor 1 (GPER) in human cumulus granulosa cells (CGCs) of patients with PCOS. Syst. Biol. Reprod. Med. 2016, 62, 184–191. [Google Scholar] [CrossRef]
- Wang, C.; Prossnitz, E.R.; Roy, S.K. G protein-coupled receptor 30 expression is required for estrogen stimulation of primordial follicle formation in the hamster ovary. Endocrinology 2008, 149, 4452–4461. [Google Scholar] [CrossRef]
- Richards, J.S. Hormonal control of gene expression in the ovary. Endocr. Rev. 1994, 15, 725–751. [Google Scholar] [CrossRef]
- Schomberg, D.W.; Couse, J.F.; Mukherjee, A.; Lubahn, D.B.; Sar, M.; Mayo, K.E.; Korach, K.S. Targeted disruption of the estrogen receptor-alpha gene in female mice: Characterization of ovarian responses and phenotype in the adult. Endocrinology 1999, 140, 2733–2744. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, K.J.; Arao, Y.; Korach, K.S. Estrogen hormone physiology: Reproductive findings from estrogen receptor mutant mice. Reprod. Biol. 2014, 14, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Jakimiuk, A.J.; Weitsman, S.R.; Yen, H.; Bogusiewicz, M.; Magoffin, D.A. Estrogen receptor alpha and beta expression in theca and granulosa cells from women with polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2002, 87, 5532–5538. [Google Scholar] [CrossRef] [PubMed]
- Rumi, M.A.K.; Dhakal, P.; Kubota, K.; Chakraborty, D.; Lei, T.; Larson, M.A.; Wolfe, M.W.; Roby, K.F.; Vivian, J.L.; Soares, M.J. Generation of Esr1-knockout rats using zinc finger nuclease-mediated genome editing. Endocrinology 2014, 155, 1991–1999. [Google Scholar] [CrossRef] [PubMed]
- Quaynor, S.D.; Stradtman, E.W.J.; Kim, H.; Shen, Y.; Chorich, L.P.; Schreihofer, D.A.; Layman, L.C. Delayed puberty and estrogen resistance in a woman with estrogen receptor α variant. N. Engl. J. Med. 2013, 369, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, S.C.; Winuthayanon, W.; Korach, K.S. What’s new in estrogen receptor action in the female reproductive tract. J. Mol. Endocrinol. 2016, 56, R55–R71. [Google Scholar] [CrossRef]
- Krege, J.H.; Hodgin, J.B.; Couse, J.F.; Enmark, E.; Warner, M.; Mahler, J.F.; Sar, M.; Korach, K.S.; Gustafsson, J.A.; Smithies, O. Generation and reproductive phenotypes of mice lacking estrogen receptor beta. Proc. Natl. Acad. Sci. USA 1998, 95, 15677–15682. [Google Scholar] [CrossRef] [PubMed]
- Harris, H.A. Estrogen receptor-beta: Recent lessons from in vivo studies. Mol. Endocrinol. 2007, 21, 1–13. [Google Scholar] [CrossRef]
- Couse, J.F.; Yates, M.M.; Deroo, B.J.; Korach, K.S. Estrogen receptor-beta is critical to granulosa cell differentiation and the ovulatory response to gonadotropins. Endocrinology 2005, 146, 3247–3262. [Google Scholar] [CrossRef]
- Shiozawa, T.; Li, S.F.; Nakayama, K.; Nikaido, T.; Fujii, S. Relationship between the expression of cyclins/cyclin-dependent kinases and sex-steroid receptors/Ki67 in normal human endometrial glands and stroma during the menstrual cycle. Mol. Hum. Reprod. 1996, 2, 745–752. [Google Scholar] [CrossRef]
- Punyadeera, C.; Verbost, P.; Groothuis, P. Oestrogen and progestin responses in human endometrium. J. Steroid Biochem. Mol. Biol. 2003, 84, 393–410. [Google Scholar] [CrossRef]
- Snijders, M.P.; de Goeij, A.F.; Debets-Te Baerts, M.J.; Rousch, M.J.; Koudstaal, J.; Bosman, F.T. Immunocytochemical analysis of oestrogen receptors and progesterone receptors in the human uterus throughout the menstrual cycle and after the menopause. J. Reprod. Fertil. 1992, 94, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, S.; Fukaya, T.; Suzuki, T.; Murakami, T.; Sasano, H.; Yajima, A. Oestrogen receptor alpha and beta mRNA expression in human endometrium throughout the menstrual cycle. Mol. Hum. Reprod. 1999, 5, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Otto, C.; Rohde-Schulz, B.; Schwarz, G.; Fuchs, I.; Klewer, M.; Brittain, D.; Langer, G.; Bader, B.; Prelle, K.; Nubbemeyer, R.; et al. G protein-coupled receptor 30 localizes to the endoplasmic reticulum and is not activated by estradiol. Endocrinology 2008, 149, 4846–4856. [Google Scholar] [CrossRef]
- Funakoshi, T.; Yanai, A.; Shinoda, K.; Kawano, M.M.; Mizukami, Y. G protein-coupled receptor 30 is an estrogen receptor in the plasma membrane. Biochem. Biophys. Res. Commun. 2006, 346, 904–910. [Google Scholar] [CrossRef]
- Prossnitz, E.R.; Barton, M. The G-protein-coupled estrogen receptor GPER in health and disease. Nature reviews. Endocrinology 2011, 7, 715–726. [Google Scholar]
- Winuthayanon, W.; Hewitt, S.C.; Orvis, G.D.; Behringer, R.R.; Korach, K.S. Uterine epithelial estrogen receptor α is dispensable for proliferation but essential for complete biological and biochemical responses. Proc. Natl. Acad. Sci. USA 2010, 107, 19272–19277. [Google Scholar] [CrossRef] [PubMed]
- Margarit, L.; Taylor, A.; Roberts, M.H.; Hopkins, L.; Davies, C.; Brenton, A.G.; Conlan, R.S.; Bunkheila, A.; Joels, L.; White, J.O.; et al. MUC1 as a discriminator between endometrium from fertile and infertile patients with PCOS and endometriosis. J. Clin. Endocrinol. Metab. 2010, 95, 5320–5329. [Google Scholar] [CrossRef]
- Pillay, O.C.; Te Fong, L.F.W.; Crow, J.C.; Benjamin, E.; Mould, T.; Atiomo, W.; Menon, P.A.; Leonard, A.J.; Hardiman, P. The association between polycystic ovaries and endometrial cancer. Hum. Reprod. 2006, 21, 924–929. [Google Scholar] [CrossRef]
- Haoula, Z.; Salman, M.; Atiomo, W. Evaluating the association between endometrial cancer and polycystic ovary syndrome. Hum. Reprod. 2012, 27, 1327–1331. [Google Scholar] [CrossRef]
- Villavicencio, A.; Bacallao, K.; Gabler, F.; Fuentes, A.; Albornoz, J.; Casals, A.; Vega, M. Deregulation of tissue homeostasis in endometria from patients with polycystic ovarian syndrome with and without endometrial hyperplasia. Gynecol. Oncol. 2007, 104, 290–295. [Google Scholar] [CrossRef]
- Fearnley, E.J.; Marquart, L.; Spurdle, A.B.; Weinstein, P.; Webb, P.M. Polycystic ovary syndrome increases the risk of endometrial cancer in women aged less than 50 years: An Australian case-control study. Cancer Causes Control 2010, 21, 2303–2308. [Google Scholar] [CrossRef]
- Maliqueo, M.; Clementi, M.; Gabler, F.; Johnson, M.C.; Palomino, A.; Sir-Petermann, T.; Vega, M. Expression of steroid receptors and proteins related to apoptosis in endometria of women with polycystic ovary syndrome. Fertil. Steril. 2003, 80 (Suppl. 2), 812–819. [Google Scholar] [CrossRef]
- Navaratnarajah, R.; Pillay, O.C.; Hardiman, P. Polycystic ovary syndrome and endometrial cancer. Semin. Reprod. Med. 2008, 26, 62–71. [Google Scholar] [CrossRef]
- Kim, J.J.; Choi, Y.M.; Choung, S.H.; Yoon, S.H.; Lee, G.H.; Moon, S.Y. Estrogen receptor beta gene +1730 G/A polymorphism in women with polycystic ovary syndrome. Fertil. Steril. 2010, 93, 1942–1947. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Roh, J.W.; Kim, J.W. Single nucleotide polymorphism: A new risk factor for endometrial cancer? Future Oncol. 2005, 1, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Quezada, S.; Avellaira, C.; Johnson, M.C.; Gabler, F.; Fuentes, A.; Vega, M. Evaluation of steroid receptors, coregulators, and molecules associated with uterine receptivity in secretory endometria from untreated women with polycystic ovary syndrome. Fertil. Steril. 2006, 85, 1017–1026. [Google Scholar] [CrossRef]
- Gregory, C.W.; Wilson, E.M.; Apparao, K.B.C.; Lininger, R.A.; Meyer, W.R.; Kowalik, A.; Fritz, M.A.; Lessey, B.A. Steroid receptor coactivator expression throughout the menstrual cycle in normal and abnormal endometrium. J. Clin. Endocrinol. Metab. 2002, 87, 2960–2966. [Google Scholar] [CrossRef]
- Nilsson, S.; Koehler, K.F. Oestrogen receptors and selective oestrogen receptor modulators: Molecular and cellular pharmacology. Basic Clin. Pharmacol. Toxicol. 2005, 96, 15–25. [Google Scholar] [CrossRef]
- Adashi, E.Y. Clomiphene citrate: Mechanism(s) and site(s) of action—A hypothesis revisited. Fertil. Steril. 1984, 42, 331–344. [Google Scholar]
- Tanbo, T.; Mellembakken, J.; Bjercke, S.; Ring, E.; Åbyholm, T.; Fedorcsak, P. Ovulation induction in polycystic ovary syndrome. Acta Obstet. Gynecol. Scand. 2018, 97, 1162–1167. [Google Scholar] [CrossRef]
- Jones, T.; Ho, J.R.; Gualtieri, M.; Bruno-Gaston, J.; Chung, K.; Paulson, R.J.; Bendikson, K.A. Clomiphene Stair-Step Protocol for Women with Polycystic Ovary Syndrome. Obstet. Gynecol. 2018, 131, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Chaabane, S.; Sheehy, O.; Monnier, P.; Fraser, W.; Bissonnette, F.; Trasler, J.M.; Muanda, F.T.; Boukhris, T.; Karam, F.; Santos, F.; et al. Ovarian Stimulation, Intrauterine Insemination, Multiple Pregnancy and Major Congenital Malformations: A Systematic Review and Meta- Analysis- The ART_Rev Study. Curr. Drug Saf. 2016, 11, 222–261. [Google Scholar] [CrossRef] [PubMed]
- Coughlan, C.; Fitzgerald, J.; Milne, P.; Wingfield, M. Is it safe to prescribe clomiphene citrate without ultrasound monitoring facilities? J. Obstet. Gynaecol. 2010, 30, 393–396. [Google Scholar] [CrossRef]
- Weiss, N.S.; van Vliet, M.N.; Limpens, J.; Hompes, P.G.A.; Lambalk, C.B.; Mochtar, M.H.; van der Veen, F.; Mol, B.W.J.; van Wely, M. Endometrial thickness in women undergoing IUI with ovarian stimulation. How thick is too thin? A systematic review and meta-analysis. Hum. Reprod. 2017, 32, 1009–1018. [Google Scholar] [CrossRef] [PubMed]
- Perales-Puchalt, A.; Legro, R.S. Ovulation induction in women with polycystic ovary syndrome. Steroids 2013, 78, 767–772. [Google Scholar] [CrossRef] [PubMed]
- Homburg, R. Clomiphene citrate—End of an era? A mini-review. Hum. Reprod. 2005, 20, 2043–2051. [Google Scholar] [CrossRef] [PubMed]
- Gottardis, M.M.; Robinson, S.P.; Satyaswaroop, P.G.; Jordan, V.C. Contrasting actions of tamoxifen on endometrial and breast tumor growth in the athymic mouse. Cancer Res. 1988, 48, 812–815. [Google Scholar] [PubMed]
- Brown, J.; Farquhar, C. Clomiphene and other antioestrogens for ovulation induction in polycystic ovarian syndrome. Cochrane Database Syst. Rev. 2016, 12, D2249. [Google Scholar] [CrossRef]
- Williamson, J.G.; Ellis, J.D. The induction of ovulation by tamoxifen. J. Obstet. Gynaecol. Br. Commonw. 1973, 80, 844–847. [Google Scholar] [CrossRef] [PubMed]
- Martinkovich, S.; Shah, D.; Planey, S.L.; Arnott, J.A. Selective estrogen receptor modulators: Tissue specificity and clinical utility. Clin. Interv. Aging 2014, 9, 1437–1452. [Google Scholar]
- Thiebaud, D.; Secrest, R.J. Selective estrogen receptor modulators: Mechanism of action and clinical experience. Focus on raloxifene. Reprod. Fertil. Dev. 2001, 13, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Tiboni, G.M.; Ponzano, A. Fetal safety profile of aromatase inhibitors: Animal data. Reprod. Toxicol. 2016, 66, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Chumsri, S. Clinical utilities of aromatase inhibitors in breast cancer. Int. J. Womens Dermatol. 2015, 7, 493–499. [Google Scholar] [CrossRef]
- Pritts, E.A. Letrozole for ovulation induction and controlled ovarian hyperstimulation. Curr. Opin. Obstet. Gynecol. 2010, 22, 289–294. [Google Scholar] [CrossRef]
- Usluogullari, B.; Duvan, C.; Usluogullari, C. Use of aromatase inhibitors in practice of gynecology. J. Ovarian Res. 2015, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Tenti, S.; Correale, P.; Cheleschi, S.; Fioravanti, A.; Pirtoli, L. Aromatase Inhibitors-Induced Musculoskeletal Disorders: Current Knowledge on Clinical and Molecular Aspects. Int. J. Mol. Sci. 2020, 21, 5625. [Google Scholar] [CrossRef]
- Yu, K.; Wang, R.; Li, M.; Sun, T.; Zhou, Y.; Li, Y.; Sun, L.; Zhang, B.; Lian, Z.; Xue, S.; et al. Melatonin Reduces Androgen Production and Upregulates Heme Oxygenase-1 Expression in Granulosa Cells from PCOS Patients with Hypoestrogenia and Hyperandrogenia. Oxid. Med. Cell. Longev. 2019, 2019, 8218650. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Classification of Organisms | Year | Species | Type | Estrous Cycle | Ovarian Cyst | Fertility | Notes | Reference |
|---|---|---|---|---|---|---|---|---|
| Rodent models | 2009 | Mouse | ER α KO | Irregular | Yes | Lost (age of 6 months) | The formation of hemorrhagic follicle; androgen level (↑) | [48] |
| 1986 | Rat | Treated with estradiol-valerate | Cease | Yes | NA | Anovulation; ovarian weights declined significantly | [49] | |
| 2004 | Mouse | ERβ KO (elevated LH) | NA | No | NA | Increased steroidogenic enzyme expression; androgen level (↓) | [50] | |
| Nonhuman primate models | 2008 | Cynomolgus monkey | Spontaneous PCOS | A prolonged menstrual cycle length (up to 161 days) | Yes | NA | Endometrial hyperplasia | [51] |
| 2002 | Female Rhesus Monkeys | Exposed prenatally to androgen | Irregular | Yes | NA | Delayed menarche; androgen level (↑) | [52] | |
| Other models | 2001 | Female sheep | Exposed prenatally to androgen | Irregular | Yes | NA | Increased ovarian volume; anovulation | [53] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, X.-L.; Deng, S.-L.; Lian, Z.-X.; Yu, K. Estrogen Receptors in Polycystic Ovary Syndrome. Cells 2021, 10, 459. https://doi.org/10.3390/cells10020459
Xu X-L, Deng S-L, Lian Z-X, Yu K. Estrogen Receptors in Polycystic Ovary Syndrome. Cells. 2021; 10(2):459. https://doi.org/10.3390/cells10020459
Chicago/Turabian StyleXu, Xue-Ling, Shou-Long Deng, Zheng-Xing Lian, and Kun Yu. 2021. "Estrogen Receptors in Polycystic Ovary Syndrome" Cells 10, no. 2: 459. https://doi.org/10.3390/cells10020459
APA StyleXu, X.-L., Deng, S.-L., Lian, Z.-X., & Yu, K. (2021). Estrogen Receptors in Polycystic Ovary Syndrome. Cells, 10(2), 459. https://doi.org/10.3390/cells10020459