Abstract
Adenosine diphosphate ribosylation (ADP-ribosylation; ADPr), the addition of ADP-ribose moieties onto proteins and nucleic acids, is a highly conserved modification involved in a wide range of cellular functions, from viral defence, DNA damage response (DDR), metabolism, carcinogenesis and neurobiology. Here we study MACROD1 and MACROD2 (mono-ADP-ribosylhydrolases 1 and 2), two of the least well-understood ADPr-mono-hydrolases. MACROD1 has been reported to be largely localized to the mitochondria, while the MACROD2 genomic locus has been associated with various neurological conditions such as autism, attention deficit hyperactivity disorder (ADHD) and schizophrenia; yet the potential significance of disrupting these proteins in the context of mammalian behaviour is unknown. Therefore, here we analysed both Macrod1 and Macrod2 gene knockout (KO) mouse models in a battery of well-defined, spontaneous behavioural testing paradigms. Loss of Macrod1 resulted in a female-specific motor-coordination defect, whereas Macrod2 disruption was associated with hyperactivity that became more pronounced with age, in combination with a bradykinesia-like gait. These data reveal new insights into the importance of ADPr-mono-hydrolases in aspects of behaviour associated with both mitochondrial and neuropsychiatric disorders.
1. Introduction
ADP-ribosylation (ADPr) is a modification of proteins and nucleic acids that controls multiple processes common to all kingdoms of life [1,2,3,4]. ADPr is the addition of one (mono) or more (poly) ADP-ribose units using nicotinamide adenine dinucleotide (NAD+) as a substrate onto molecular targets, known as MAR- or PARylation and their products as e (mono-ADP-ribose) or PAR (poly-ADP-ribose), respectively. Several families of enzymes can synthesise ADPr, the best understood of which are the poly(ADP-ribose) polymerases (PARPs). PARPs have 17 known family members in humans and are considered the major ADPr contributor in cells; PARP1 accounting for some 85–90% of NAD+ consumption [5,6]. ADPr is involved many essential cellular processes such as DNA repair, chromatin remodelling, antiviral responses and WNT signalling [5,7,8,9,10].
Similar to other modifications, ADPr is fully reversible [11]. Removal of ADPr is performed by two main types of ADPr hydrolase families: the macrodomains and ARHs (ADP-ribosylhydrolases) [12]. There many different specificities for hydrolases, dependent upon specific macromolecule modification and the nature of the chemical bond on the macromolecular targets [12,13,14,15,16]. Among the macrodomain family are three enzymes, MACROD1, MACROD2 and TARG1 (terminal ADP-ribose protein glycohydrolase); these share very similar biochemical activities and are responsible for removal of the terminal ADP-ribose moiety from acidic residues [17,18,19]. Interestingly, in terms of neurobiology and behaviour, genetic deficiency of ADPr hydrolases often leads to disease, most commonly neurodegeneration [17,20,21,22,23].
Probably the most poorly understood human ADPr hydrolases are MACROD1 and MACROD2; both share a catalytic macrodomain fold [24] but have different regulatory regions, yet their functions and substrate specificities remain unclear. Both MACROD1 and MACROD2 hydrolyse various mono-ADP-ribosylated substrates in vitro: proteins [18,25,26], nucleic acids [27,28,29] and the O-acetylADP-ribose sirtuin byproduct [30].
Localization of MACROD1 seems largely in mitochondria [29,31] and thus it follows that MACROD1 is highly concentrated in skeletal muscles [29]. Since ADPr- mono-hydrolase activity can be highly specific [32] and perhaps dependent on certain cell types and/or stress, Žaja et al. studied the effect of MACROD1 loss in a skeletal muscle cell line (RD cells—rhabdomyosarcoma) [33]. Genetic knockout of Macrod1 in RD cells did not result in any major growth defects but did subtly alter mitochondrial structure [33], which is in-line with our previous work that demonstrated Macrod1 KO mice are healthy and viable [29]. Previous in vitro studies of MACROD1, using different cell types, have reported MACROD1 as an essential cofactor for androgen receptor [34], estrogen receptor [35,36] and NFκB (nuclear factor kappa-light-chain-enhancer of activated B cells) [37,38] transcriptional activity. Meanwhile, the only confirmed in vivo target of MACROD1 is the aryl hydrocarbon receptor (AHR), working opposite PARP7 (also known as TIPARP), in the toxic response to dioxin [39], which links back again to the mitochondria [40]. Dysfunction of the mitochondria and NAD+ metabolism is a common mechanism in neurodegeneration [41,42,43].
On the other hand, MACROD2 resides largely in the cytoplasm [31,33] and is highly expressed in the brain [44]. Indeed, several genome–wide association studies (GWAS) have reported links between MACROD2 and various neurological and psychiatric conditions such as autism [45], schizophrenia [46], ADHD [47] as well as congenital heart defects [48] (28% of which are linked to neurodegeneration [49]). However, there has been some conflicting reports, for example, MACROD2 is considered a hotspot for mutation [50] and subsequent studies (using different populations, i.e., Chinese) showed no association with autism [51]. Another role suggested for MACROD2 is in the DNA damage response [52] with ATM (ATM serine/threonine kinase) DNA repair kinase controlling the shuttling of MACROD2 between the cytoplasm and nucleus. Interestingly, deregulation of both MACROD1 and MACROD2 has also potentially been linked to cancer [53,54,55], although the validity of this has been queried [56]. Another suggested biological target of MACROD2 includes glycogen synthase kinase 3β (GSK3β), which is involved in WNT-signalling [25].
Therefore, based on the above information, which suggest an important functional role for MACROD1 in skeletal muscles in cells and a link between MACROD2 and various neurological conditions, we decided to understand the physiological role of MACROD1 and MACROD2 hydrolases by subjecting knockout mice to a phenotyping battery to assess multiple, spontaneous aspects of mouse behaviour including locomotor activity, anxiety, motor coordination, grip strength, short-term memory and attention.
Genetic KO models for these genes are available on a cost-recovery basis from the International Mouse Phenotyping Consortium (IMPC) and some preliminary phenotyping is already available on their website [57] (www.mousephenotype.org—accessed 7 December 2020). Briefly, the IMPC website shows, Macrod1 KO was mostly non-significant. While Macrod2 KO reported phenotypes included a variety of metabolic defects; smaller size, increased circulating blood glucose etc. as well as an abnormal locomotor behaviour (KO mice move further in a light-dark test, on both sides of the light-dark box) and an abnormal sleep pattern [58]. Whilst informative, the information gathered from the IMPC is limited, in particular with regards to later time-points. Age is an extremely important consideration for neurodegenerative disorders, where motor-function and/or mental cognition often gets worse with age [59,60,61]. Specifically for Macrod1, mitochondrial decline is age-related [43] hence we chose later time points (12 and 18-months). Whereas Macrod2 has been associated with ADHD which presents differently in childhood [62], hence why the most practicable youngest age (around three months, at 8–12 weeks) was chosen for the Macrod2 starting time point, as well as a later time-point (13 months) to follow up.
Our behavioural characterisation revealed interesting and novel phenotypes related to the loss of both Macrod1 and Macrod2. Loss of Macrod1 resulted in a female-specific motor-coordination defect, whereas Macrod2 disruption was associated with hyperactivity that became more pronounced with age, in combination with a bradykinesia-like gait. These data reveal new insights into the importance of ADPr-mono-hydrolases in aspects of behaviour associated with both mitochondrial and neuropsychiatric disorders.
2. Materials and Methods
2.1. Animals and Housing
Macrod1 and Macrod2 KO mouse models were obtained via the IMPC [57,63,64]. The Macrod1 KO strain is a Knockout Mouse Programme (KOMP)-Regeneron (Velocigene) definitive null design (Project ID:VG13617) whereby most of exons 1 through to 3 are deleted and replaced with a promoter-driven Zen_Ub1 cassette [29]. In total 9303 bp were deleted between positions 7131384–7140686 of Chromosome 19 (Genome Build37). This original ‘tm1a’ allele contains a potentially confounding promoter-driven neomycin cassette that was removed by crossing to a Sox2 promoter-driven cre-recombinase line (Sox2Cre, a gift from Elizabeth Robertson, The University of Oxford [65]) to generate the experimental ‘tm1d’ KO allele. The Macrod2 KO strain is also a Velocigene definitive null design, whereby 19.2 kb (including part of exon 2) is deleted and replaced with a promoter-driven Zen_Ub1 cassette (Project ID:VG12650). In total 19,224 bp were deleted between positions 140226712–140245935 of Chromosome 2 (Genome Build37). Experimental animals were rederived directly from an already-made ‘tm1d’ stock. No overt phenotypes were noticeable from cage-side observations and all mice appeared to age normally.
The genotype of individual animals was confirmed by PCR on genomic DNA extracted from ear-notches using the Phire Animal Tissue Direct PCR Kit (Thermo Fisher Scientific, Life Technologies Ltd, Paisley, UK) as follows: Macrod1 primers 5′-AAGCATGGAGGGCATTTTGG, 5′-GGTCCTAAGGTAGCGACTCG and 5′-TGTGGCTTCATTCCAGACAG amplifies products of 516 and 348 bp (wild-type (WT) and KO respectively). Macrod2 primers, 5′-TTCCTGAGCTCCGTGAATG, 5′-GCAGCAGCTTCCTGAAACAT and 5′-GTCTGTCCTAGCTTCCTCACTG amplifies products of 460 and 552 bp (WT and KO respectively)—see Figure 1 for a scheme.
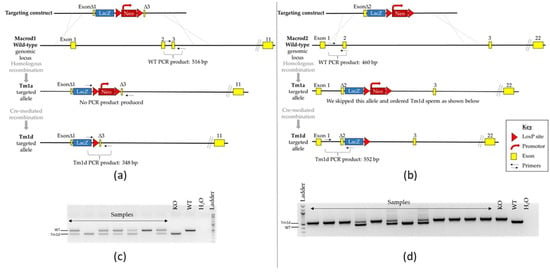
Figure 1.
Confirmation of Macrod1 (left panel) and Macrod2 (right panel) genetic disruption. (a,b) Schematic diagram of the targeting strategy of the WT genomic locus and associated alleles for both genes of interest; note that only mice carrying the tm1d allele were used for behavioural testing. Not to scale. (c,d) Example of PCR genotyping reactions. DNA fragments were separated on a 2% agarose gel by electrophoresis. Expected product sizes are Macrod1: 516 and 348 bp and Macrod2: 460 and 552 bp (WT and KO respectively), presence of a double band indicates a heterozygous animal.
Mice were weaned at three weeks and kept in same sex groups in controlled conditions (12 h light–dark cycle 09:00–21:00, at 21–22 °C with food and water ad libitum). Cohorts of at least 10 age-matched (within three to four weeks), per sex, per genotype (WT and KO) littermates were analysed in the study from heterozygous (HET) intercrosses. Mice from the IMPC were on a C57BL/6N genetic background, however our in-house C57BL6 mice (including the Sox2-Cre) are the C57BL/6J substrain, therefore following at least one backcross our mice were on a mixed C57BL6/J-N genetic background.
All studies were conducted under a valid UK Home Office Animal Project Licence (PPL: 30/3307) which has undergone ethical review by departmental AWERB (Animal Welfare and Ethical Review Body) at the University of Oxford. All work was in accordance with the UK Animals (Scientific Procedures) Act 1986 Amendment Regulations 2012 (ASPA 2012).
2.2. Behavioural Testing
The Macrod1 cohorts were tested at both 12 and 18 months of age (two separate cohorts), while the Macrod2 cohorts were tested at three and 13 months (one cohort, repeated testing, Table 1). We refer to the cohorts by age at the start of testing throughout in order to distinguish each experimental group/round.

Table 1.
Behavioural testing cohort numbers and age at start of testing.
All behavioural testing occurred between 12:00 and 17:00 and allowed the animals to acclimatise to the behavioural testing room for at least 30 min. Mice underwent a battery of six tests, one test per week over the course of seven weeks (see Figure 2). Testing was performed by the same scientist to reduce user and handling bias and the experimenter was blinded to genotype. Mice were excluded from testing if they appeared unwell for any reason, for example dermatitis or cataracts in aged mice.
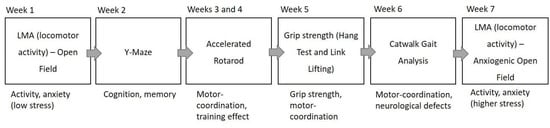
Figure 2.
Scheme of behavioural testing overview. NOTE: Macrod1 KO did not have the second week of accelerated rotarod testing.
2.2.1. Locomotor Activity (LMA)—Open Field
Mice were placed individually in transparent cages (25.4 cm × 47 cm) containing fresh sawdust. The cage was crisscrossed with 4 × 8 evenly spaced infrared beams. The number of beam breaks in the periphery (outermost beams) and centre (innermost 2 × 6 quadrant) were recorded in 10 s intervals using a beam splitter (San Diego Instruments PAS, San Diego, CA, US) for a total of 90 min. Data examined are total beam breaks and central proportion (%).
2.2.2. Y-Maze—Preference Test
The Y-maze consisted of three clear Perspex arms (30 cm × 8 cm × 20 cm) at 120° to one-another with a central zone [66]. The test was performed in two stages. For the habituation stage the mouse was placed at the end of one (start) arm whilst the entry to one arm is blocked and the second (other) arm is open. Activity was recorded for the initial 5 min after the first entry in to the central zone. The mouse was then returned to its home cage for 1 min. Stage two: the mouse was returned to the maze at the end of the same start arm with the third (novel) arm now accessible. Activity was recorded for 2 min after entry into the central zone. Start arm and closed arm were rotated, in a logical ordered fashion, between each mouse (since experimenter was blinded to genotype and mice were kept in cages with mixed genotypes) to reduce room positional bias. Activity data were quantified from an overhead camera using ANY-Maze software (Stoelting, Dublin, IE). The distance travelled, the time spent in each of the arms, the number of arm entries and the number of repeat entries to the same arm were recorded. The preference ratio was calculated as the time in the novel arm/(time in novel arm + other arm).
2.2.3. Accelerating Rotarod
A commercial rotarod device was used (Med Associates, Inc., Fairfax, VT, USA) consisting of a grooved plastic beam 5 cm in diameter (with dividers to stop physical interactions between animals). Mice were placed on the beam (revolving at the default 5 rpm) facing in the opposite orientation to rotation. After 30–60 s to allow mice to become accustomed to walking on the beam (and to load multiple mice, up to five at a time) the speed was gradually accelerated to a maximum of 50 rpm over 5 min by electronic control of the motor. The latency before falling was measured up to a maximum total time of 5 min. Trails were repeated three times in total over three consecutive days. For Macrod1, the average time taken to fall from the three days is presented. For Macrod2 cohort only, because many of the female mice could stay on for the full 5 min, a final test was performed the following week at a faster speed 8–80 rpm, to avoid a ceiling affect with analysis. The Macrod2 final trial was performed on a single day, three runs with one hour’s rest between trails, the mean of which is presented. Data examined are average latency to fall (s).
2.2.4. Grip Strength
Inverted Screen—Four-Limb Hang Test
Mice were placed on the centre of a metal grid (12 mm2 of 1 mm diameter wire, 45 cm2 in size within a 5 cm wooden frame). The grid was then inverted over a padded surface. The time taken to fall from the grid was recorded over three trials in total over three consecutive days. The data are presented as the square root transformed hanging impulse (gs) = hang time (s) × mass (g). There was no maximum time set for Macrod1 (most mice fell off within 2–3 min, with only one or two mice lasting longer than 5 min). However, no maximum time was considered impractical for the three-month-old Macrod2 cohort, where many mice could hang 10+ min. Therefore, we decided instead to screen Macrod2 mice for only 5 min as a benchmark (nearly all mice passed) and then use a different method to assess grip strength (see Link Lifting below) to avoid a ceiling effect in analysis.
Link Lifting
Mice were allowed to grip onto wire balls for kettle descaling that were attached to a varying number of metal chain links of increasing weight [67]. Starting with the lowest weight, mice were allowed to grip the wire ball their front paws only, a stop watch was started and then they were lifted gently by the tail. Mice had three chances to lift the links just clear of the bench, for a criterion time of 3 s (as determined by stopwatch) before trying the next heaviest weight. For mice that failed to lift one set of links, the longest attempt was recorded and used as a score qualifier. A lift score was calculated as: (max link lifted × 3) + (next link × time held in seconds).
2.2.5. Catwalk Gait Analysis
Mice were placed on a narrow, sealed, raised platform (10 cm × ~1 m), and their activity is recorded using a camera placed below the floor (Catwalk XT, Noldus). The Catwalk system uses illuminated footprint technology, in which, briefly, the walking surface is made of glass which internally reflects a green LED light, except where a paw touches the glass, thus footprints or other miscellaneous points of contact glow green. The test is performed in the dark; five compliant runs of ~55 cm in length (six to seven step cycles) were taken from each mouse. Compliant runs were determined as maximum duration 5 s and maximum speed variation of 35%. Compliant runs are classified, and the following data was examined; maximum contact mean intensity (forepaw and hind paw, average of left and right sides used square root transformed), gait velocity (cm/s), gait cadence (steps/minute–, square root transformed), forefoot swing speed (cm/s), forefoot stride length (cm).
2.2.6. LMA Anxiogenic Open Field—Bright Field
Mice were placed in a white circular drum (60 cm × 60 cm lit by four high-fluorescent 7 W OSRAM Deluxe light bulbs) and their activity recorded for 5 min using a camera placed over the centre of the drum. Automated video tracking software (ANY-Maze, Stoelting, Dublin, IE) was used to record the total distance travelled (m), number of entries into the central zone (20 cm diameter) and time (s) in the central zone (s).
2.3. Statistics
The statistical software Minitab 2019 was used to analyse the results. The data were checked for normal distribution by using histogram plots and some data were square root transformed to remove any skewing of data (mentioned in Results, below). Analysis for Macrod1 and Macrod2 was performed separately.
Initial analyses included an all-factors-combined analysis of variance (ANOVA) (described in detail below) to give a highly powered statistical overview. If genotype was statistically significant (p-value of <0.05) we then performed further post hoc analysis to further clarify that genes’ role. Further tests typically included analysis of covariance (ANCOVAs) to calculate significance, as weight tended to highly influence results [68,69,70,71] and Cohen’s d statistic (d-score) to objectively measure effect size (difference in means ÷ population standard deviation).
Since Macrod1 was two independent cohorts, we used the general linear model ANOVA (a factorial-ANCOVA which allows use of covariables such as weight), with genotype, sex and cohort (age) as cofactors and weight as a covariable. The following interactions were also included in the model: genotype*sex, genotype*cohort, weight*sex, weight*sex, weight*genotype, sex*cohort. We continued to model the weight*genotype interaction for Macrod1 because whilst not statistically significant the p-value was borderline (see Results below, Table 2).
Table 2.
Descriptive statistics of weight data. Mean ± SEM is presented. Individual N number is included as well as p-values for all terms and interactions. Macrod1 used a factorial-ANOVA. Macrod2 used a factorial-ANOVA with repeated measures, hence the extra term Mouse Name. x = not an exact F-test.
As the Macrod2 cohort involved repeated testing of the same mice, we used a repeated measures ANOVA, thus Mouse Name (random factor, nested with genotype and sex) was also analysed along with the above cofactors and interactions listed for Macrod1 (excluding weight*genotype which was ruled out after an initial weight analysis—see Table 2). Minitab 2019 automatically corrects for missing values.
Results are presented as the mean ± standard error of the mean (SEM). A p-value of <0.05 was considered significant. Cohen’s d-score was assigned as follows; <0.2 trivial, >0.2 small, 0.5–0.8 medium and >0.8 large effect size. All graphs were generated using Prism 8 and all significant differences are marked with a solid line and an asterisk. A dashed line was used to mark trends (not significant and no asterisk added). The exact p-value can be found in the figure legends as well as various tables.
3. Results
3.1. Confirmation of Genetic Knock-Out
All Macrod1 and Macrod2 KO mice were born at the expected Mendelian ratios. Protein deletion in these genetic models has been reported previously by us for Macrod1 [29] and by two other independent research groups for Macrod2 [53,72].
3.2. Weight Evaluation
Since many aspects of behaviour are affected by an animal’s weight (such as ambulation, dexterity and strength [68,69,70,71]) it was important to first determine if genotype was significantly associated with differences in mass. In both genotypic cohorts, there was no impact of genotype on weight (Macrod1: p = 0.058 and Macrod2: p = 0.701 (Table 2 and Figure 3). Animal weight was, however, significantly affected by sex and age in both groups (p < 0.001) as expected [73,74].
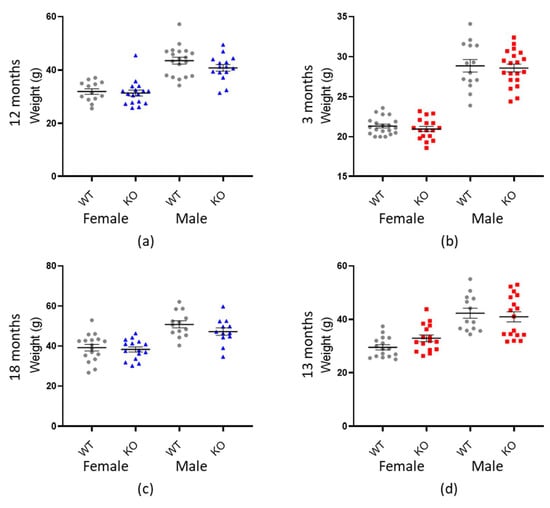
Figure 3.
There is no association between weight and genotype. Graphs show individual values dot plot for female and male mice, WT shown in grey circles, Macrod1 KO in blue triangles and Macrod2 KO in red squares. Error bars are the SEM. Graphs are as follows: (a,c) Macrod1 12 months and 18 months, (b,d) Macrod2 three months and 13 months.
3.3. Macrod1 KO Is Associated with Sex-Specific Reduced Motor-Coordination
We performed a standard test of mouse motor-coordination, the accelerated rotarod, which records one linear variable, the latency to fall (s). Our initial statistical analysis revealed a significant effect of genotype for Macrod1 KO (* p = 0.018) but not Macrod2 KO (p = 0.357) (see Table 3 and Supplementary Table S1 and Figure S1). Main effect in both cohorts was weight (p < 0.001) however the Macrod1 cohort also had a significant effect with sex (* p = 0.006), weight*sex interaction (* p = 0.005) and the weight*genotype interaction (* p = 0.045). Therefore, we postulated there might be a sex-specific difference. Indeed, further investigation and plotting of the data revealed that the effect was most pronounced in 12-month-old female mice, who fell off the rotarod 47.3 s (±15.2 SEM) sooner (* p = 0.005) than their female WT littermates (see Figure 4). Eighteen-month-old female mice followed the same trend and fell 20.62 s (± 9.52 SEM) sooner than their female WT littermates however this was not significant on its own (p = 0.089). A 47.3 s reduction in latency to fall for the 12-month old females is considered a medium to large effect size (d = 0.788), indicating that this is a relevant biological difference. There were no significant differences in latency to fall between the KO and WT males. Taken together our data demonstrate a moderate female-specific motor-coordination defect in the Macrod1 KO mice.
Table 3.
Descriptive statistics of data from Macrod1 accelerated rotarod. Mean ± SEM is presented. Individual N number is included as well as p-values for all terms and interactions. Statistics was performed on all factors combined using a factorial-ANOVA, whereas separated by sex and age cohort comparisons used an ANCOVA (weight as a covariable). The difference between the means is presented alongside a Cohen’s d-score to demonstrate effect size.
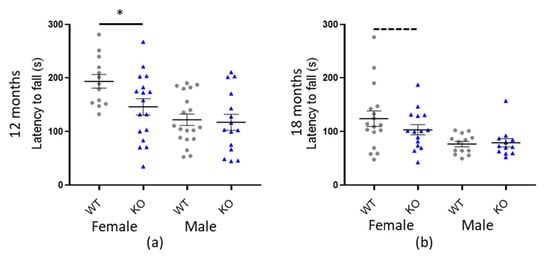
Figure 4.
Macrod1 KO is associated with sex specific reduced fall latency on the rotarod. Graphs show individual values dot plot, separated by sex and cohort, WT shown in grey circles and Macrod1 KO in blue triangles. Error bars are the SEM. Graphs are as follows, (a,b) Macrod1 aged 12 and 18 months. Significant difference for Macrod1 KO 12 months; F(1,27) = 14.53, * p = 0.005 and trending at 18 months; F(1,29) = 3.11, p = 0.089 (ANCOVA).
3.4. Macrod2 KO Increases Total Locomotion Activity
We observed Macrod1 and Macrod2 KO mice in a range of spontaneous behavioural testing paradigms. In nearly all tests where total distance travelled was recorded, the Macrod2 KO mice moved further than their WT littermates, indicating a hyperactivity phenotype. These tests included locomotor activity (LMA), open field (see Table 4), Y-maze preference test (see Table 5) and LMA anxiogenic open field (bright field) (not significant but follows trend—see Supplementary Table S4 and Figure S4).
Table 4.
Descriptive statistics of data from Macrod2 locomotor activity (LMA) open field. Mean ± SEM is presented. Individual N number is included as well as p-values for all terms and interactions. Statistics were performed on all factors combined using a factorial-ANOVA with repeated measures hence the term Mouse name. x = not an exact F-test. Separated by sex and age cohort comparisons used an ANCOVA (weight as a covariable). The difference between the means is presented alongside a Cohen’s d-score to demonstrate effect size.
Table 5.
Descriptive statistics of distance travelled data from Macrod2 distance travelled Y-maze preference test. Mean ± SEM is presented. Individual N number is included as well as p-values for all terms and interactions. Statistics were performed on all factors combined using a factorial-ANOVA with repeated measures hence the term Mouse name. x = not an exact F-test. Separated by sex and age cohort comparisons used an ANCOVA (weight as a covariable). The difference between the means is presented alongside a Cohen’s d-score to demonstrate effect size.
Our main test of locomotor activity is the LMA open field, which records mice for 90 min. Our initial statistical analysis revealed a highly significant effect on total beam breaks with genotype for Macrod2 KO (* p = 0.001), but not Macrod1 KO (p = 0.427) (see Supplementary Table S3 and Figure S3). Additional significant effects were for genotype*cohort (* p = 0.021) and random variable Mouse Name (* p = 0.002). Mouse Name is a term included in the Macrod2 statistical modelling which accounts for repeated testing of individual mice at both time points (see Methods). A significant p-value for Mouse Name indicates that the nature of individual mice is consistent between tests (a mouse that moved further at one time-point was highly likely to move further at the other time-point). An effect for genotype*cohort demonstrates an effect of genotype with age. Indeed, a hyperactivity phenotype was actually more pronounced in the aged Macrod2 KO mice for the LMA open field test (see Figure 5 and Table 4 for a statistical summary). Consistent with this, the three-month-old female and male mice were not significant when examined individually and had a small to medium effect size (d = 0.431 and d = 0.585 respectively). Whereas the 13-month-old Macrod2 KO female (* p = 0.005) and male (* p = 0.015) mice were independently significant and had large effect sizes (d = 0.840 and d = 0.915 respectively). Practically, this translates as the 13-month-old Macrod2 KO mice moved 33–53% more than their WT counterparts in the LMA open field test.
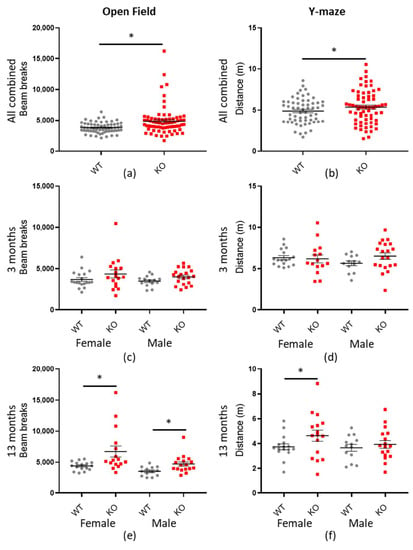
Figure 5.
Macrod2 KO is associated with hyperactivity. Graphs show individual values dot plot for all mice combined or separated by sex and age, WT shown in grey circles and Macrod2 KO in red squares. Error bars are the SEM. Graphs are as follows: (left panel) LMA open field total beam breaks over 90 min; (a) all data combined, (c) three months and (d) 13 months. (Right panel) Y-maze preference test total distance travelled (m) in 2 min (b) all data combined, (d) three months and (f) 13 months. Significant difference for Macrod2 KO LMA open field: (a) F(1,55) = 12.14, * p < 0.001, (e) 13-month-old females F(1,29) = 9.24, * p = 0.005, males F(1,27) = 6.68, * p = 0.015. Significant differences for Macrod2 Y-maze: (b) F(1,51) = 5.74, * p = 0.019, (f) 13-month-old females F(1,29) = 6.09, * p = 0.020.
Similarly, the Y-maze preference test (see Supplementary Tables S6 and S7 and Figures S6 and S7 for preference ratio data—no significant differences) also showed a significant difference in locomotor activity between the genotypes (* p = 0.019—see Table 5 for statistical summary). The effect was most pronounced in the 13-month-old female mice (* p = 0.020) with a medium effect size (d = 0.560), although, the hyperactivity trend was present in all but the three-month-old female mice (see Figure 5). On the other hand, the bright field test did not show any significant differences in total distance travelled (p = 0.072), however, the overall trend is still present (see Supplementary Table S4 and Figure S4). Taken together, our results demonstrate that Macrod2 KO is associated with increased total locomotion activity and that this hyperactivity is more pronounced with age.
3.5. Macrod2 KO Is Associated with Reduced Speed and a Shorter Stride Length
Mouse ambulatory speed was measured by a number of factors, including gait velocity, gait cadence and forepaw swing speed (see Table 6, Table 7 and Table 8 for a statistical summary). In each case, Macrod2 KO mice were consistently and significantly slower than their WT littermates overall; gait velocity −2.922 cm/s (± 0.571 SEM) * p < 0.001: SQRT-gait cadence −0.952 steps/min (±0.272 SEM) * p = 0.026: forepaw swing speed −4.32 cm/s (±1.06 SEM) * p = 0.001. When further analyses were conducted, separated by sex and age, the three- and 13-month-old males and the 13-month-old females had the largest effect size. For gait velocity and forefoot swing speed, three-month-old males had the largest effect size (d = 1.188 and d = 1.053) followed by the 13-month-old males (d = 0.842 and d = 0.912) whereas the three-month-old females had only small differences. Interestingly, the 13-month-old females had a significant, medium effect size for gait velocity but only a negligible and non-significant difference for forefoot swing speed (see Table 6 and Table 8). Another discrepancy was for gait cadence, which showed a small but significant effect size overall (d = 0.420) but not for testing the sexes and ages separately (see Table 7). Nonetheless, the overall trend, for Macrod2 KO mice to be slower, remained present as can be observed in Figure 6.
Table 6.
Descriptive statistics of gait velocity from the Macrod2 catwalk gait analysis. Mean ± SEM is presented. Individual N number is included as well as p-values for all terms and interactions. Statistics were performed on all factors combined using a factorial-ANOVA with repeated measures hence the term Mouse name. x = not an exact F-test. Separated by sex and age cohort comparisons used an ANCOVA (weight as a covariable). The difference between the means is presented alongside a Cohen’s d-score to demonstrate effect size.
Table 7.
Descriptive statistics of SQRT gait cadence from the Macrod2 catwalk gait analysis. SQRT = square root transformed. Mean ± SEM is presented. Individual N number is included as well as p-values for all terms and interactions. Statistics were performed on all factors combined using a factorial-ANOVA with repeated measures hence the term Mouse Name. x = not an exact F-test. Separated by sex and age cohort comparisons used an ANCOVA (weight as a covariable). The difference between the means is presented alongside a Cohen’s d-score to demonstrate effect size.
Table 8.
Descriptive statistics of forefoot swing speed from the Macrod2 catwalk gait analysis. Mean ± SEM is presented. Individual N number is included as well as p-values for all terms and interactions. Statistics were performed on all factors combined using a factorial-ANOVA with repeated measures hence the term Mouse Name. x = not an exact F-test. Separated by sex and age cohort comparisons used an ANCOVA (weight as a covariable). The difference between the means is presented alongside a Cohen’s d-score to demonstrate effect size.
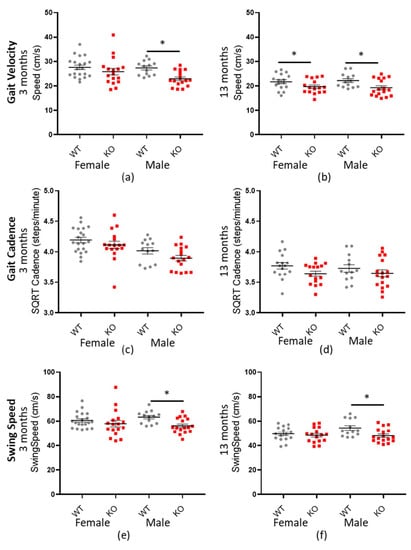
Figure 6.
Macrod2 KO is associated with a slower gait. Graphs show individual values dot plot, separated by sex and age, WT shown in grey circles and Macrod2 KO in red squares. Error bars are the SEM. Graphs are as follows (a,b) gait velocity (average speed) at three and 13 months, (c,d) SQRT gait cadence (steps/minute) at three and 13 months and (e,f) forepaw swing speed (cm/s) at three and 13 months. Significant differences for Macrod2 KO (a) three-month-old males F(1,27) = 15.09, * p = 0.001, (b) 13-month old females F(1,29) = 4.33, * p = 0.046, males F(1,27) = 7.98, * p = 0.009, (e) three-month-old males F(1,27) = 10.82, * p = 0.003 and (f) 13-month-old males F(1,27) = 7.26, * p = 0.012.
Additionally, Macrod2 KO mice have a shorter stride length overall: −0.414 cm (± 0.0734 SEM) * p < 0.001 (all factors and ages combined—see Table 9). The genotype*sex interaction was also significant (* p = 0.033), implying perhaps a different effect in females vs. males, based on genotype, however when we plotted the data (see Figure 7) it appeared as if Macrod2 KO display reduced stride length in both the genders, albeit more for the males. Further analysis showed that the female mice had only a small effect size (d = 0.345 and d = 0.406 for three and 13 months respectively), whilst the male mice had a much larger effect size (d = 1.361 and d = 1.045 for three and 13 months respectively; see Table 9). The male forefoot stride length data was also independently highly statistically significant (* p > 0.001 and * p = 0.003 for three and 13 months, respectively). Reduced stride length does not entirely explain the Macrod2 KO mice decreased average speed, because the gait cadence was also marginally reduced. Only the forepaw data is presented here, however hind paw data followed the same trend. As an additional control, there was no significant differences for these same traits when compared for the Macrod1 cohorts (see Supplementary Tables S10 and S11 and Figure S9).
Table 9.
Descriptive statistics of forefoot stride length from the Macrod2 catwalk gait analysis. Mean ± SEM is presented. Individual N number is included as well as p-values for all terms and interactions. Statistics were performed on all factors combined using a factorial-ANOVA with repeated measures hence the term Mouse Name. x = not an exact F-test. Separated by sex and age cohort comparisons used an ANCOVA (weight as a covariable). The difference between the means is presented alongside a Cohen’s d-score to demonstrate effect size.
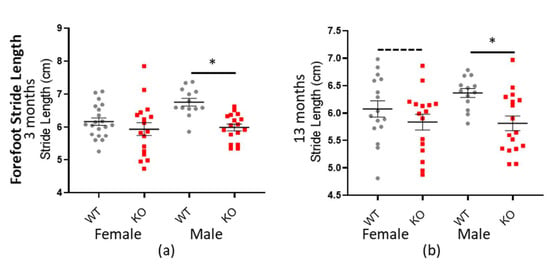
Figure 7.
Macrod2 KO is associated with a shorter stride length. Graphs show individual values’ dot plot, separated by sex and age, WT shown in grey circles and Macrod2 KO in red squares. Error bars are the SEM. Graphs are as follows: (a,b) forefoot stride length (cm), three and 13 months. Significant differences for Macrod2 KO (a) three-month-old males F(1,27) = 23.85, * p > 0.001, (b) 13-month-old males F(1,27) = 7.98, * p = 0.009 and trending for 13-month-old females F(1,29) = 3.63, p = 0.067.
In conclusion, Macrod2 KO mice have an abnormal gait. Peculiarly, since Macrod2 KO mice were determined to be hyperactive (total distance travelled in a variety of testing paradigms) their natural walk, as recorded by the catwalk test (in the dark, least stressful), was actually slower and with shorter steps. This type of gait is known as bradykinesia (a slow shuffling gait as appears in Parkinson’s disease [75]). Males are possibly more affected than the females, however the trend is present in both sexes.
3.6. Further Behavioural Testing Summaries
Key results have been highlighted above however there are some other minor but important points to make as a result of this preliminary behavioural phenotyping. Neither Macrod1 or Macrod2 KO appear to have an effect on short-term working memory or attention (no significance difference in the Y-maze preference test—see Supplementary Tables S6 and S7 and Figures S6 and S7) or grip strength (see Supplementary Tables S13 and S14 and Figures S11 and S12) or anxiety levels (LMA open field and bright field—see Supplementary Tables S2–S5 and Figures S2–S5). Additionally, we initially thought there might be an altered gait in Macrod1 KO mice, since they appeared to place less pressure on their front paws (but not hind paws) when walking (catwalk gait analysis), however further examination determined this phenotype to be due instead to subtle differences in animal weight (see Supplementary Tables S9 and S12 and Figures S8 and S10). Another phenotype we ruled out for Macrod1 KO was hypoactivity for the 12-month old female mice in the Y-maze test only (see Supplementary Table S8). Whilst the effect size seemed moderate (d = 0.740), the p-value was borderline (* p = 0.047) and no other hypoactivity traits could be seen in any of the other testing paradigms.
4. Discussion
Our study shows the first behavioural characterization of the mono-ADP-ribosylation hydrolyses Macrod1 and Macrod2. Since there is nothing known about the physiological targets of these genes and muscular or neurological functional roles are suggested, we decided to investigate them further with some standard spontaneous behavioural testing in KO mouse models.
Genetic loss of Macrod1 resulted in a female-specific motor-coordination defect. The mild to moderate motor-coordination defect observed, whereby Macrod1 KO female mice fell 20–47 s sooner from a rotating rod than their WT littermates, was not likely due to a fundamental difference in grip strength as no differences were measured in the inverted hang test. Additionally, this reduced latency to fall was not obviously due to an abnormal gait, as no differences between genotypes were observed across several parameters during the catwalk test.
Previous studies have demonstrated that reduced latency to fall from the rotarod can be a result of muscle disease [76], mitochondrial disease [77] as well as mitochondrial dysfunction induced by ischemic injury [78]. Therefore, one plausible explanation for Macrod1 KO’s reduced latency to fall from the rotarod, based on the mitochondrial subcellular location of Macrod1 and enrichment in skeletal muscles [29,33] could be, loss of aerobic (mitochondrial) fitness. Indeed, Loss of MACROD1 in RD cells has been shown to cause mitochondrial fragmentation [33] which might become a problem under continued muscle use and increasing energy demands [79], such as the rotarod test. Certainly, altered mitochondrial structure has previously been linked to muscle fatigue [80]. Since Macrod1 KO mice are viable, fertile and healthy it is logical that perhaps MACROD1 is only activated under certain conditions, such as stress or exercise. Further supporting a role in exercise, Macrod1 appeared specifically upregulated as part of evening (but not morning) exercise adaptation in rats [81].
Following on, the intriguing sex-specific differences we see, as well as the smaller difference in the 18-month old females could simply be a matter of shorter exercise duration and thus less mitochondrial energy demands [79]. Males (both ages) and the aged females spend approximately half the total time on the rotarod compared to the 12-month-old WT female mice, it is possible that the phenotype only presents following longer periods of exercise, as in the 12-month-old WT female mice, upon which aerobic mitochondrial respiration takes over from the initial reliance on glycolytic respiration.
Alternatively, sex-specific alterations in Macrod1 regulation in vivo are potentially consistent with Macrod1 being an estrogen and androgen sensitive gene [34,35,36]. There are broad influences of sex hormones within the brain, which can influence many aspects of behaviour [82]. Interestingly, with regards to the mitochondria, non-nuclear estrogen receptors have been documented in close proximity to this organelle [83]. Additionally, there are physiological muscular differences between male and female mice, for example higher mitochondrial content in female mice which likely contributes to their increased rotarod ability and, therefore, lack of said mitochondrial benefit may underpin the reduced rotarod latency to fall observed in Macrod1 deficient females [84].
Could our observed phenotype be due to a subtle maladaptation in mitochondria which only shows up under physical exertion or perhaps other stresses? We did not perform any of the more aversive ‘exhaustion’ type behavioural tests (such as forced swim or treadmill) nor did we conduct any passive activity monitoring (such as voluntary wheel running or home-cage video tracking) which can be an alternative method to detect fatigue in mice [85], as this was outside the scope of our initial screen.
Meanwhile, genetic loss of Macrod2 caused a hyperactive phenotype (as measured in LMA open field (large effect) and Y-maze preference test (medium effect)), which became more pronounced with age. The trend was also present in the bright field test however it wasn’t significant. One explanation for the diminished effect, as compared with the open field test could be that the testing time was much reduced (90 min vs. 2 or 5 min) and that there is more for mice to contend with in a Y-maze and bright field test setting. For example a choice of arms or a very bright light which mice find aversive [86] and can send them to sleep [87], confounding interpretation of results from a purely locomotor activity perspective. Nonetheless, the fact that the same trend of Macrod2 KO mice moving more persists in a multitude of tests where total distance travelled was recorded is promising and further supports the notion that the Macrod2 KO mice are hyperactive.
Interestingly, Macrod2 KO hyperactivity was not correlated with a decrease in anxiety (less anxiety might mean more activity [88]) as determined by LMA open field and bright field. Additionally, since hyperactivity can be associated with decreased attention and increased impulsivity [89] it is interesting to note that short-term working memory and basic cognitive ability appears to be intact in these mice, as measured by Y-maze preference test. In the future, more in-depth cognitive and impulsivity testing, such as the radial arm maze, would provide further insight into the relevance of Macrod2 to ADHD, autism and schizophrenia to which the genomic locus has been linked [45,46,47].
One confounding variable with regards to mouse age and test naivety is that the Macrod2 cohort used repeated testing on the same mice at three months and 13 months. Experience and age can highly impact several aspects of mouse behaviour, including locomotion activity, willingness to explore and anxiety-like behaviours [90]. However, since there was no significant difference in central proportion exploration in either the LMA open field or bright field tests, with regards to the genotype, we hypothesise that the hyperactivity phenotype is genuine.
Most interestingly, the Macrod2 KO hyperactivity phenotype was conversely paired with a bradykinesia type gait (slower, shorter steps as appears in Parkinson’s disease [75]). The unusual gait did not noticeably affect motor-coordination as there was a similar latency to fall in the accelerated rotarod test between genotypes and grip strength measurements were also not altered.
Macrod2 is primarily expressed in neurons [44] (and our unpublished data) and hyperactivity as well as gait disturbances are consistent with a neurological phenotype [91,92,93], although due to the constitutive nature of our KO model we cannot exclude a role of other organs or cell types. Neurological conditions, sleep and metabolism are delicately linked together and commonly disrupted in a plethora of human diseases [94,95,96]. Hence it is also interesting to note that Macrod2 was identified as a novel sleep-related gene as part of a high-throughput screen in mice [58]. Macrod2 KO mice slept significantly more during the dark, active phase versus controls; this corresponds well to the kind of sleep disruptions observed in ADHD (delayed sleep phase disorder, some 73–78% of children and adults with ADHD are affected [97]). Sleep disturbances are also common in autism (50–80% of children) [98]. The role of sleep in ADHD and mental health in general is so integral that some experts are calling for a rethink on how the diseases are perceived and thus treated [95,97].
With regards to the initial phenotyping of the Macrod2 KO mice by the IMPC [57] and our phenotyping, where it overlaps, compares somewhat favourably. We could not confirm a smaller size however both us and the IMPC reported abnormal locomotor behaviour (KO mice moved further in a light-dark test). Since the IMPC do not usually keep mice beyond 16 weeks, it is highly likely they would not detect the increased hyperactivity with old age, or worsening gait. In terms of the IMPC metabolic defects, Lo Re et al. were unable to confirm a role for Macrod2 in metabolism by glucose tolerance test, insulin tolerance test or in high fat diet induced obesity [72].
The lack of stronger phenotypes within these Macrod1 and Macrod2 KO mouse models is perhaps surprising given that genetic deficiency of other ADPr reversal enzymes, such as PARG (poly (ADP-ribose) glycohydrolase) can lead to early embryonic lethality in mice [99] or more severe neurological complications [17,20,22]. One potential explanation is redundancy between these two hydrolyses which might be expected given their similar biochemical activities [18,28,30]. On the other hand, quite distinct cellular localisations and tissue expression specificity (in the characterised cell/tissue models) between MACROD1 and MACROD2 would argue against a redundant/overlapping role in vivo, however the lack of strong phenotypes, indicates there must be sufficient residual activity provided by the remaining MAR and PAR hydrolases to compensate for their loss, at least outside conditions of stress. One benefit of a milder phenotype, is to provide a good proof of concept for the use of small molecule inhibitors against either protein should subsequent studies suggest a use for them (for example, anticancer therapies).
Generally, our newly observed behavioural phenotypes of Macrod1 and Macrod2 KO mice suggest that there is much to learn about these proteins, including roles beyond the ones most commonly studied in the ADPr field such as DNA damage response and genome stability. It will be interesting to extend these studies in the future to determine whether more complex forms of behaviour relevant to neurodevelopmental and neuropsychiatric disease are perturbed in these models. In terms of the underlying molecular mechanism, the physiological, in vivo, ADPr targets for MACROD1 and MACROD2 remain unknown or determined from in vitro studies using model ADPr substrates [18,25,26,27,28,29,30,33]. Experiments showing that KO Macrod1/2 orthologues in fungus Neurospora crassa affect levels of the sirtuin by-product O-acetyADP-ribose [30], suggest that physiological targets of Macrod1/2 enzymes could be beyond protein ADPr. Interestingly, very close homologues of MACROD1/2 are found in some viruses (usually referred to as viral macrodomains or mac1) including coronaviruses [100]. These viral mac1 domains are known to dampen interferon response by opposing activity of antiviral PARPs [9,101] and were shown to act on ADP-ribosylated RNAs in vitro suggestive of a possible role of MACROD1/2 in RNA modification [28]. Whilst the physiologically relevant targets of MACROD1/2 have yet to be determined, whether that be protein, RNA or DNA ADPr adducts or O-acetyADP-ribose, here we show that genetic deficiencies in either gene have physiological consequences relating to neuromuscular function and behaviour in vivo.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4409/10/2/368/s1, Table S1: Descriptive statistics of data from Macrod2 accelerated rotarod. Figure S1: Macrod2 KO is not associated with reduced fall latency on the rotarod. Table S2: Descriptive statistics of central proportion from Macrod2 open field. Figure S2: Macrod2 KO is not associated with anxiety in LMA open field. Table S3: Descriptive statistics of data from Macrod1 LMA open field. Figure S3: Macrod1 KO is not associated with changes in locomotor activity or central proportion in LMA open field. Table S4: Descriptive statistics of data from Macrod2 LMA bright field. Figure S4: Macrod2 KO is not associated with changes in locomotor activity or central proportion in LMA bright field. Table S5: Descriptive statistics of data from Macrod1 LMA bright field. Figure S5: Macrod1 KO is not associated with changes in locomotor activity or central proportion in LMA bright field. Table S6: Descriptive statistics of preference ratio from Macrod2 Y-maze preference test. Figure S6: Macrod2 KO is not associated with changes in short term memory and attention. Table S7: Descriptive statistics of data from Macrod1 LMA bright field. Table S8: Descriptive statistics of distance travelled data from Macrod1 distance travelled Y-maze preference test. Figure S7: Macrod1 KO is not associated with changes in short term memory and attention. Table S9: Descriptive statistics of SQRT-fore and hind paw intensity for Macrod1 from the catwalk gait analysis. Figure S8: Macrod1 KO is not associated with changes fore or hind paw foot pressure. Table S10: Descriptive statistics of gait velocity and SQRT gait cadence for Macrod1 from the catwalk gait analysis. Table S11: Descriptive statistics of forefoot swing speed and forefoot stride length for Macrod1 from the catwalk gait analysis. Figure S9: Macrod1 KO is not associated an altered gait. Table S12: Descriptive statistics of SQRT fore and hind paw intensity for Macrod2 from the catwalk gait analysis. Figure S10: Macrod2 KO is not associated with changes in fore or hind paw foot pressure. Table S13: Descriptive statistics of Macrod1 grip strength. Figure S11: Macrod1 KO is not associated altered grip strength. Table S14: Descriptive statistics of Macrod2 grip strength. Figure S12: Macrod2 KO is not associated altered grip strength.
Author Contributions
Conceptualization, I.A. and P.L.O.; methodology, P.L.O. and B.H.M.H.; formal analysis, K.C.; investigation, K.C. and T.A.; resources, I.A. and P.L.O.; data curation, K.C.; writing—original draft preparation, K.C. and I.A.; writing—review and editing, K.C., P.L.O., T.A., B.H.M.H., and I.A.; visualization, K.C.; supervision, I.A.; project administration, K.C. and I.A.; funding acquisition, I.A. All authors have read and agreed to the published version of the manuscript.
Funding
Work in I.A.’s laboratory is supported by Cancer Research United Kingdom (C35050/A22284), Wellcome Trust (101794, 210634) and Biotechnology and Biological Sciences Research Council (BB/R007195/1).
Institutional Review Board Statement
All animal studies were conducted under a valid UK Home Office Animal Project Licence (PPL: 30/3307) which has undergone ethical review by departmental AWERB (Animal Welfare and Ethical Review Body) at the University of Oxford. All work was in accordance with the UK Animals (Scientific Procedures) Act 1986 Amendment Regulations 2012 (ASPA 2012).
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in the article or Supplementary Materials.
Acknowledgments
Katherine Hewitt, for her endless help in booking and setting up the behavioural testing equipment and facilitating collaboration. Prof Elizabeth Robertson for her Soz2Cre mice and general mouse expertise.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kraus, W.L. Parps and adp-ribosylation: 50 years… And counting. Mol. Cell. 2015, 58, 902–910. [Google Scholar] [CrossRef] [PubMed]
- Jankevicius, G.; Ariza, A.; Ahel, M.; Ahel, I. The toxin-antitoxin system dartg catalyzes reversible adp-ribosylation of DNA. Mol. Cell. 2016, 64, 1109–1116. [Google Scholar] [CrossRef]
- Perina, D.; Mikoc, A.; Ahel, J.; Cetkovic, H.; Zaja, R.; Ahel, I. Distribution of protein poly(adp-ribosyl)ation systems across all domains of life. Dna Repair. (Amst.) 2014, 23, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Palazzo, L.; Mikoc, A.; Ahel, I. Adp-ribosylation: New facets of an ancient modification. Febs J. 2017, 284, 2932–2946. [Google Scholar] [CrossRef]
- Langelier, M.F.; Eisemann, T.; Riccio, A.A.; Pascal, J.M. Parp family enzymes: Regulation and catalysis of the poly(adp-ribose) posttranslational modification. Curr. Opin. Struct. Biol. 2018, 53, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Gupte, R.; Liu, Z.; Kraus, W.L. Parps and adp-ribosylation: Recent advances linking molecular functions to biological outcomes. Genes Dev. 2017, 31, 101–126. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.S.; Chang, P. Insights into the biogenesis, function, and regulation of adp-ribosylation. Nat. Chem. Biol. 2018, 14, 236–243. [Google Scholar] [CrossRef]
- Martin-Hernandez, K.; Rodriguez-Vargas, J.M.; Schreiber, V.; Dantzer, F. Expanding functions of adp-ribosylation in the maintenance of genome integrity. Semin. Cell Dev. Biol. 2017, 63, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Fehr, A.R.; Jankevicius, G.; Ahel, I.; Perlman, S. Viral macrodomains: Unique mediators of viral replication and pathogenesis. Trends Microbiol. 2018, 26, 598–610. [Google Scholar] [CrossRef]
- Suskiewicz, M.J.; Zobel, F.; Ogden, T.E.H.; Fontana, P.; Ariza, A.A.-O.X.; Yang, J.A.-O.; Zhu, K.; Bracken, L.; Hawthorne, W.J.; Ahel, D.; et al. Hpf1 completes the parp active site for DNA damage-induced adp-ribosylation. Nature 2020, 579, 598–602. [Google Scholar] [CrossRef]
- Barkauskaite, E.; Jankevicius, G.; Ladurner, A.G.; Ahel, I.; Timinszky, G. The recognition and removal of cellular poly(adp-ribose) signals. Febs J. 2013, 280, 3491–3507. [Google Scholar] [CrossRef]
- Rack, J.G.M.; Palazzo, L.; Ahel, I. (adp-ribosyl)hydrolases: Structure, function, and biology. Genes Dev. 2020, 34, 263–284. [Google Scholar] [CrossRef]
- Fontana, P.; Bonfiglio, J.J.; Palazzo, L.; Bartlett, E.; Matic, I.; Ahel, I. Serine adp-ribosylation reversal by the hydrolase arh3. Elife 2017, 6, e28533. [Google Scholar] [CrossRef] [PubMed]
- Oka, S.; Kato, J.; Moss, J. Identification and characterization of a mammalian 39-kda poly(adp-ribose) glycohydrolase. J. Biol. Chem. 2006, 281, 705–713. [Google Scholar] [CrossRef]
- Lin, W.; Ame, J.C.; Aboul-Ela, N.; Jacobson, E.L.; Jacobson, M.K. Isolation and characterization of the cdna encoding bovine poly(adp-ribose) glycohydrolase. J. Biol. Chem. 1997, 272, 11895–11901. [Google Scholar] [CrossRef] [PubMed]
- Suskiewicz, M.A.-O.; Palazzo, L.A.-O.; Hughes, R.; Ahel, I.A.-O. Progress and outlook in studying the substrate specificities of parps and related enzymes. Febs J. 2020. [Google Scholar] [CrossRef]
- Sharifi, R.; Morra, R.; Appel, C.D.; Tallis, M.; Chioza, B.; Jankevicius, G.; Simpson, M.A.; Matic, I.; Ozkan, E.; Golia, B.; et al. Deficiency of terminal adp-ribose protein glycohydrolase targ1/c6orf130 in neurodegenerative disease. Embo J. 2013, 32, 1225–1237. [Google Scholar] [CrossRef]
- Jankevicius, G.; Hassler, M.; Golia, B.; Rybin, V.; Zacharias, M.; Timinszky, G.; Ladurner, A.G. A family of macrodomain proteins reverses cellular mono-adp-ribosylation. Nat. Struct Mol. Biol. 2013, 20, 508–514. [Google Scholar] [CrossRef]
- Feijs, K.L.; Forst, A.H.; Verheugd, P.; Luscher, B. Macrodomain-containing proteins: Regulating new intracellular functions of mono(adp-ribosyl)ation. Nat. Rev. Mol. Cell Biol. 2013, 14, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.G.; Becker, K.; Huang, H.; Dixon-Salazar, T.; Chai, G.; Salpietro, V.; Al-Gazali, L.; Waisfisz, Q.; Wang, H.; Vaux, K.K.; et al. Biallelic mutations in adprhl2, encoding adp-ribosylhydrolase 3, lead to a degenerative pediatric stress-induced epileptic ataxia syndrome. Am. J. Hum. Genet. 2018, 103, 431–439. [Google Scholar] [CrossRef]
- Hanai, S.; Kanai, M.; Ohashi, S.; Okamoto, K.; Yamada, M.; Takahashi, H.; Miwa, M. Loss of poly(adp-ribose) glycohydrolase causes progressive neurodegeneration in drosophila melanogaster. Proc. Natl. Acad. Sci. USA 2004, 101, 82–86. [Google Scholar] [CrossRef]
- Danhauser, K.; Alhaddad, B.; Makowski, C.; Piekutowska-Abramczuk, D.; Syrbe, S.; Gomez-Ospina, N.; Manning, M.A.; Kostera-Pruszczyk, A.; Krahn-Peper, C.; Berutti, R.; et al. Bi-allelic adprhl2 mutations cause neurodegeneration with developmental delay, ataxia, and axonal neuropathy. Am. J. Hum. Genet. 2018, 103, 817–825. [Google Scholar] [CrossRef]
- Palazzo, L.; Mikolčević, P.; Mikoč, A.; Ahel, I. Adp-ribosylation signalling and human disease. Open Biol. 2019, 9, 190041. [Google Scholar] [CrossRef]
- Rack, J.G.; Perina, D.; Ahel, I. Macrodomains: Structure, function, evolution, and catalytic activities. Annu. Rev. Biochem. 2016, 85, 431–454. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, F.; Feijs, K.L.; Frugier, E.; Bonalli, M.; Forst, A.H.; Imhof, R.; Winkler, H.C.; Fischer, D.; Caflisch, A.; Hassa, P.O.; et al. Macrodomain-containing proteins are new mono-adp-ribosylhydrolases. Nat. Struct. Mol. Biol. 2013, 20, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Barkauskaite, E.; Brassington, A.; Tan, E.S.; Warwicker, J.; Dunstan, M.S.; Banos, B.; Lafite, P.; Ahel, M.; Mitchison, T.J.; Ahel, I.; et al. Visualization of poly(adp-ribose) bound to parg reveals inherent balance between exo- and endo-glycohydrolase activities. Nat. Commun 2013, 4, 2164. [Google Scholar] [CrossRef] [PubMed]
- Munnur, D.; Ahel, I. Reversible mono-adp-ribosylation of DNA breaks. Febs J. 2017, 284, 4002–4016. [Google Scholar] [CrossRef]
- Munnur, D.; Bartlett, E.; Mikolčević, P.; Kirby, I.T.; Rack, J.G.M.; Mikoč, A.; Cohen, M.S.; Ahel, I. Reversible adp-ribosylation of rna. Nucleic Acids Res. 2019, 47, 5658–5669. [Google Scholar] [CrossRef]
- Agnew, T.; Munnur, D.; Crawford, K.; Palazzo, L.; Mikoč, A.; Ahel, I. Macrod1 is a promiscuous adp-ribosyl hydrolase localized to mitochondria. Front. Microbiol 2018, 9, 20. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Vollmar, M.; Rossi, M.N.; Phillips, C.; Kraehenbuehl, R.; Slade, D.; Mehrotra, P.V.; von Delft, F.; Crosthwaite, S.K.; Gileadi, O.; et al. Identification of macrodomain proteins as novel o-acetyl-adp-ribose deacetylases. J. Biol. Chem. 2011, 286, 13261–13271. [Google Scholar] [CrossRef]
- Neuvonen, M.; Ahola, T. Differential activities of cellular and viral macro domain proteins in binding of adp-ribose metabolites. J. Mol. Biol. 2009, 385, 212–225. [Google Scholar] [CrossRef]
- Crawford, K.; Bonfiglio, J.J.; Mikoč, A.; Matic, I.; Ahel, I. Specificity of reversible adp-ribosylation and regulation of cellular processes. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 64–82. [Google Scholar] [CrossRef] [PubMed]
- Žaja, R.; Aydin, G.; Lippok, B.E.; Feederle, R.; Lüscher, B.; Feijs, K.L.H. Comparative analysis of macrod1, macrod2 and targ1 expression, localisation and interactome. Sci. Rep. 2020, 10, 8286. [Google Scholar]
- Yang, J.; Zhao, Y.L.; Wu, Z.Q.; Si, Y.L.; Meng, Y.G.; Fu, X.B.; Mu, Y.M.; Han, W.D. The single-macro domain protein lrp16 is an essential cofactor of androgen receptor. Endocr. Relat. Cancer 2009, 16, 139–153. [Google Scholar] [CrossRef] [PubMed]
- Han, W.D.; Mu, Y.M.; Lu, X.C.; Xu, Z.M.; Li, X.J.; Yu, L.; Song, H.J.; Li, M.; Lu, J.M.; Zhao, Y.L.; et al. Up-regulation of lrp16 mrna by 17beta-estradiol through activation of estrogen receptor alpha (eralpha), but not erbeta, and promotion of human breast cancer mcf-7 cell proliferation: A preliminary report. Endocr. Relat. Cancer 2003, 10, 217–224. [Google Scholar] [CrossRef]
- Han, W.D.; Zhao, Y.L.; Meng, Y.G.; Zang, L.; Wu, Z.Q.; Li, Q.; Si, Y.L.; Huang, K.; Ba, J.M.; Morinaga, H.; et al. Estrogenically regulated lrp16 interacts with estrogen receptor alpha and enhances the receptor’s transcriptional activity. Endocr. Relat. Cancer 2007, 14, 741–753. [Google Scholar] [CrossRef]
- Wu, Z.; Wang, C.; Bai, M.; Li, X.; Mei, Q.; Li, X.; Wang, Y.; Fu, X.; Luo, G.; Han, W. An lrp16-containing preassembly complex contributes to nf-kappab activation induced by DNA double-strand breaks. Nucleic Acids Res. 2015, 43, 3167–3179. [Google Scholar] [CrossRef]
- Wu, Z.; Li, Y.; Li, X.; Ti, D.; Zhao, Y.; Si, Y.; Mei, Q.; Zhao, P.; Fu, X.; Han, W. Lrp16 integrates into nf-kappab transcriptional complex and is required for its functional activation. PLoS ONE 2011, 6, e18157. [Google Scholar]
- Ahmed, S.; Bott, D.; Gomez, A.; Tamblyn, L.; Rasheed, A.; MacPherson, L.; Sugamori, K.S.; Cho, T.; Yang, Y.; Grant, D.M.; et al. Loss of the mono-adp-ribosyltransferase, tiparp, increases sensitivity to dioxin-induced steatohepatitis and lethality. J. Biol. Chem. 2015, 290, 16824–16840. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Hu, B.; Shi, X.; Weidert, E.R.; Lu, P.; Xu, M.; Huang, M.; Kelley, E.E.; Xie, W. Activation of the aryl hydrocarbon receptor sensitizes mice to nonalcoholic steatohepatitis by deactivating mitochondrial sirtuin deacetylase sirt3. Mol. Cell Biol. 2013, 33, 2047–2055. [Google Scholar] [CrossRef]
- Lautrup, S.; Sinclair, D.A.; Mattson, M.P.; Fang, E.F. Nad(+) in brain aging and neurodegenerative disorders. Cell Metab. 2019, 30, 630–655. [Google Scholar] [CrossRef]
- Beal, M.F. Mitochondria and neurodegeneration. Novartis Found. Symp. 2007, 287, 192–196. [Google Scholar]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Morishita, R.; Mizuno, M.; Kawamura, N.; Tabata, H.; Nagata, K.I. Biochemical and morphological characterization of a neurodevelopmental disorder-related mono-adp-ribosylhydrolase, macro domain containing 2. Dev. Neurosci. 2018, 40, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.M.; Cadby, G.; Blangero, J.; Abraham, L.J.; Whitehouse, A.J.O.; Moses, E.K. Macrod2 gene associated with autistic-like traits in a general population sample. Psychiatr. Genet. 2014, 24, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Woodroffe, A.; Rodriguez-Murillo, L.; Roos, J.L.; van Rensburg, E.J.; Abecasis, G.R.; Gogos, J.A.; Karayiorgou, M. Elucidating the genetic architecture of familial schizophrenia using rare copy number variant and linkage scans. Proc. Natl. Acad. Sci. USA 2009, 106, 16746–16751. [Google Scholar] [CrossRef]
- Lionel, A.C.; Crosbie, J.; Barbosa, N.; Goodale, T.; Thiruvahindrapuram, B.; Rickaby, J.; Gazzellone, M.; Carson, A.R.; Howe, J.L.; Wang, Z.; et al. Rare copy number variation discovery and cross-disorder comparisons identify risk genes for adhd. Sci. Transl. Med. 2011, 3, 95ra75. [Google Scholar] [CrossRef]
- Lahm, H.; Jia, M.; Dreßen, M.; Wirth, F.F.M.; Puluca, N.; Gilsbach, R.; Keavney, B.; Cleuziou, J.; Beck, N.; Bondareva, O.; et al. Congenital heart disease risk loci identified by genome-wide association study in european patients. J. Clin. Investig. 2020. [Google Scholar] [CrossRef]
- Jin, S.C.; Homsy, J.; Zaidi, S.; Lu, Q.; Morton, S.; DePalma, S.R.; Zeng, X.; Qi, H.; Chang, W.; Sierant, M.C.; et al. Contribution of rare inherited and de novo variants in 2871 congenital heart disease probands. Nat. Genet. 2017, 49, 1593–1601. [Google Scholar] [CrossRef]
- Bradley, W.E.; Raelson, J.V.; Dubois, D.Y.; Godin, E.; Fournier, H.; Privé, C.; Allard, R.; Pinchuk, V.; Lapalme, M.; Paulussen, R.J.; et al. Hotspots of large rare deletions in the human genome. PLoS ONE 2010, 5, e9401. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, J.; Lu, T.; Zhang, T.; Jia, M.; Ruan, Y.; Zhang, D.; Li, J.; Wang, L. Replication of previous gwas hits suggests the association between rs4307059 near msnp1as and autism in a chinese han population. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 92, 194–198. [Google Scholar] [CrossRef]
- Golia, B.; Moeller, G.K.; Jankevicius, G.; Schmidt, A.; Hegele, A.; Preißer, J.; Tran, M.L.; Imhof, A.; Timinszky, G. Atm induces macrod2 nuclear export upon DNA damage. Nucleic Acids Res. 2017, 45, 244–254. [Google Scholar] [CrossRef]
- Sakthianandeswaren, A.; Parsons, M.J.; Mouradov, D.; MacKinnon, R.N.; Catimel, B.; Liu, S.; Palmieri, M.; Love, C.; Jorissen, R.N.; Li, S.; et al. Macrod2 haploinsufficiency impairs catalytic activity of parp1 and promotes chromosome instability and growth of intestinal tumors. Cancer Discov. 2018, 8, 988–1005. [Google Scholar] [CrossRef] [PubMed]
- Mohseni, M.; Cidado, J.; Croessmann, S.; Cravero, K.; Cimino-Mathews, A.; Wong, H.Y.; Scharpf, R.; Zabransky, D.J.; Abukhdeir, A.M.; Garay, J.P.; et al. Macrod2 overexpression mediates estrogen independent growth and tamoxifen resistance in breast cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 17606–17611. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.J.; Luo, C.B.; Xin, H.Y.; Hu, Z.Q.; Zhu, G.Q.; Li, J.; Zhou, S.L. Macrod2 deficiency promotes hepatocellular carcinoma growth and metastasis by activating gsk-3β/β-catenin signaling. NPJ Genom. Med. 2020, 5, 15. [Google Scholar] [CrossRef] [PubMed]
- Feijs, K.A.-O.; Cooper, C.A.-O.; Žaja, R. The controversial roles of adp-ribosyl hydrolases macrod1, macrod2 and targ1 in carcinogenesis. Cancers (Basel) 2020, 12, 604. [Google Scholar] [CrossRef]
- Dickinson, M.E.; Flenniken, A.M.; Ji, X.; Teboul, L.; Wong, M.D.; White, J.K.; Meehan, T.F.; Weninger, W.J.; Westerberg, H.; Adissu, H.; et al. High-throughput discovery of novel developmental phenotypes. Nature 2016, 537, 508–514. [Google Scholar] [CrossRef]
- Joshi, S.S.; Sethi, M.; Striz, M.; Cole, N.; Denegre, J.M.; Ryan, J.; Lhamon, M.E.; Agarwal, A.; Murray, S.; Braun, R.E.; et al. Noninvasive sleep monitoring in large-scale screening of knock-out mice reveals novel sleep-related genes. bioRxiv 2019, 517680. [Google Scholar] [CrossRef]
- Justice, J.N.; Carter, C.S.; Beck, H.J.; Gioscia-Ryan, R.A.; Mcqueen, M.; Enoka, R.M.; Seals, D.R. Battery of behavioral tests in mice that models age-associated changes in human motor function. Age (Dordr.) 2014, 36, 583–592. [Google Scholar] [CrossRef]
- Chung, T.; Tian, Y.; Walston, J.; Hoke, A. Increased single-fiber jitter level is associated with reduction in motor function with aging. Am. J. Phys. Med. Rehabil. 2018, 97, 551–556. [Google Scholar] [CrossRef]
- Albin, R.L.; Miller, R.A. Mini-review: Retarding aging in murine genetic models of neurodegeneration. Neurobiol. Dis. 2016, 85, 73–80. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sharma, A.; Couture, J. A review of the pathophysiology, etiology, and treatment of attention-deficit hyperactivity disorder (adhd). Ann. Pharmacother. 2014, 48, 209–225. [Google Scholar] [CrossRef]
- Meehan, T.F.; Conte, N.; West, D.B.; Jacobsen, J.O.; Mason, J.; Warren, J.; Chen, C.-K.; Tudose, I.; Relac, M.; Matthews, P.; et al. Disease model discovery from 3328 gene knockouts by the international mouse phenotyping consortium. Nat. Genet. 2017, 49, 1231–1238. [Google Scholar] [CrossRef] [PubMed]
- Collins, F.S.; Rossant, J.; Wurst, W. A mouse for all reasons. Cell 2007, 128, 9–13. [Google Scholar]
- Vincent, S.D.; Robertson, E.J. Highly efficient transgene-independent recombination directed by a maternally derived sox2cre transgene. Genesis 2003, 37, 54–56. [Google Scholar] [CrossRef]
- Oliver, P.L.; Chodroff, R.A.; Gosal, A.; Edwards, B.; Cheung, A.F.; Gomez-Rodriguez, J.; Elliot, G.; Garrett, L.J.; Lickiss, T.; Szele, F.; et al. Disruption of visc-2, a brain-expressed conserved long noncoding rna, does not elicit an overt anatomical or behavioral phenotype. Cereb. Cortex. 2015, 25, 3572–3585. [Google Scholar] [CrossRef] [PubMed]
- Deacon, R.M.J. Measuring the strength of mice. J. Vis. Exp. 2013, 76, 2610. [Google Scholar] [CrossRef] [PubMed]
- Schellinck, H.M.; Cyr, D.P.; Brown, R.E. Chapter 7—How many ways can mouse behavioral experiments go wrong? Confounding variables in mouse models of neurodegenerative diseases and how to control them. In Advances in the Study of Behavior; Brockmann, H.J., Roper, T.J., Naguib, M., Wynne-Edwards, K.E., Mitani, J.C., Simmons, L.W., Eds.; Academic Press: Cambridge, MA, USA, 2010; Volume 41, pp. 255–366. [Google Scholar]
- McFadyen, M.P.; Kusek, G.; Bolivar, V.J.; Flaherty, L. Differences among eight inbred strains of mice in motor ability and motor learning on a rotorod. Genes Brain Behav. 2003, 2, 214–219. [Google Scholar] [CrossRef]
- Martinez-Huenchullan, S.F.; McLennan, S.V.; Ban, L.A.; Morsch, M.; Twigg, S.M.; Tam, C.S. Utility and reliability of non-invasive muscle function tests in high-fat-fed mice. Exp. Physiol. 2017, 102, 773–778. [Google Scholar] [CrossRef] [PubMed]
- Hartog, A.; Hulsman, J.; Garssen, J. Locomotion and muscle mass measures in a murine model of collagen-induced arthritis. BMC Musculoskelet. Disord. 2009, 10, 59. [Google Scholar] [CrossRef]
- Lo Re, O.; Mazza, T.; Vinciguerra, M. Mono-adp-ribosylhydrolase macrod2 is dispensable for murine responses to metabolic and genotoxic insults. Front. Genet. 2018, 9, 654. [Google Scholar] [CrossRef] [PubMed]
- The Jackson Laboratory. Body Weight Information for c57bl/6j (000664). Available online: https://www.jax.org/jax-mice-and-services/strain-data-sheet-pages/body-weight-chart-000664 (accessed on 4 January 2021).
- The Jackson Laboratory. Body weight information for aged c57bl/6j (000664). Available online: https://www.jax.org/jax-mice-and-services/strain-data-sheet-pages/body-weight-chart-aged-b6 (accessed on 4 January 2021).
- Ribeiro, R.P.; Santos, D.B.; Colle, D.; Naime, A.A.; Gonçalves, C.L.; Ghizoni, H.; Hort, M.A.; Godoi, M.; Dias, P.F.; Braga, A.L.; et al. Decreased forelimb ability in mice intracerebroventricularly injected with low dose 6-hydroxidopamine: A model on the dissociation of bradykinesia from hypokinesia. Behav. Brain Res. 2016, 15, 30–36. [Google Scholar] [CrossRef]
- Beastrom, N.; Lu, H.; Macke, A.; Canan, B.D.; Johnson, E.K.; Penton, C.M.; Kaspar, B.K.; Rodino-Klapac, L.R.; Zhou, L.; Janssen, P.M.L.; et al. Mdx5cv mice manifest more severe muscle dysfunction and diaphragm force deficits than do mdx mice. Am. J. Pathol. 2011, 179, 2464–2474. [Google Scholar] [CrossRef]
- Civiletto, G.; Varanita, T.; Cerutti, R.; Gorletta, T.; Barbaro, S.; Marchet, S.; Lamperti, C.; Viscomi, C.; Scorrano, L.; Zeviani, M. Opa1 overexpression ameliorates the phenotype of two mitochondrial disease mouse models. Cell Metab. 2015, 21, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Andrabi, S.S.; Tabassum, H.; Parveen, S.; Parvez, S. Ropinirole induces neuroprotection following reperfusion-promoted mitochondrial dysfunction after focal cerebral ischemia in wistar rats. Neurotoxicology 2020, 77, 94–104. [Google Scholar] [CrossRef] [PubMed]
- Freyssenet, D. Energy sensing and regulation of gene expression in skeletal muscle. J. Appl. Physiol. (1985) 2007, 102, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Lavorato, M.; Loro, E.; Debattisti, V.; Khurana, T.S.; Franzini-Armstrong, C. Elongated mitochondrial constrictions and fission in muscle fatigue. J. Cell Sci. 2018, 131, jcs221028. [Google Scholar] [CrossRef]
- Seo, D.Y.; Yoon, C.S.; Dizon, L.A.; Lee, S.R.; Youm, J.B.; Yang, W.S.; Kwak, H.B.; Ko, T.H.; Kim, H.K.; Han, J.; et al. Circadian modulation of the cardiac proteome underpins differential adaptation to morning and evening exercise training: An lc-ms/ms analysis. Pflug. Arch. 2020, 472, 259–269. [Google Scholar] [CrossRef]
- McEwen, B.S.; Milner, T.A. Understanding the broad influence of sex hormones and sex differences in the brain. J. Neurosci. Res. 2017, 95, 24–39. [Google Scholar] [CrossRef]
- Milner, T.A.; Ayoola, K.; Drake, C.T.; Herrick, S.P.; Tabori, N.E.; McEwen, B.S.; Warrier, S.; Alves, S.E. Ultrastructural localization of estrogen receptor beta immunoreactivity in the rat hippocampal formation. J. Comp. Neurol. 2005, 491, 81–95. [Google Scholar] [CrossRef]
- Watanabe, D.; Hatakeyama, K.; Ikegami, R.; Eshima, H.; Yagishita, K.; Poole, D.C.; Kano, Y. Sex differences in mitochondrial ca(2+) handling in mouse fast-twitch skeletal muscle in vivo. J. Appl. Physiol. (1985) 2020, 128, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Wolff, B.S.; Raheem, S.A.; Saligan, L.N. Comparing passive measures of fatigue-like behavior in mice. Sci. Rep. 2018, 8, 14238. [Google Scholar] [CrossRef]
- Delwig, A.; Logan, A.M.; Copenhagen, D.R.; Ahn, A.H. Light evokes melanopsin-dependent vocalization and neural activation associated with aversive experience in neonatal mice. PLoS ONE 2012, 7, e43787. [Google Scholar] [CrossRef]
- Altimus, C.M.; Güler, A.D.; Villa, K.I.; McNeill, D.S.; Legates, T.A.; Hattar, S. Rods-cones and melanopsin detect light and dark to modulate sleep independent of image formation. Proc. Natl. Acad. Sci. USA 2008, 105, 19998–20003. [Google Scholar] [CrossRef]
- Benaroya-Milshtein, N.; Hollander, N.; Apter, A.; Kukulansky, T.; Raz, N.; Wilf, A.; Yaniv, I.; Pick, C.G. Environmental enrichment in mice decreases anxiety, attenuates stress responses and enhances natural killer cell activity. Eur. J. Neurosci. 2004, 20, 1341–1347. [Google Scholar] [CrossRef] [PubMed]
- Bjerrum, M.B.; Pedersen, P.U.; Larsen, P. Living with symptoms of attention deficit hyperactivity disorder in adulthood: A systematic review of qualitative evidence. Jbi Database Syst. Rev. Implement. Rep. 2017, 15, 1080–1153. [Google Scholar] [CrossRef] [PubMed]
- von Kortzfleisch, V.T.; Kästner, N.; Prange, L.; Kaiser, S.; Sachser, N.; Richter, S.H. Have i been here before? Complex interactions of age and test experience modulate the results of behavioural tests. Behav. Brain Res. 2019, 23, 143–148. [Google Scholar] [CrossRef]
- Isaacs, A.M.; Oliver, P.L.; Jones, E.I.; Jeans, A.; Potter, A.; Hovik, B.H.; Nolan, P.M.; Vizor, L.; Glenister, P.; Simon, A.K.; et al. A mutation in af4 is predicted to cause cerebellar ataxia and cataracts in the robotic mouse. J. Neurosci. 2003, 23, 1631–1637. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jeans, A.F.; Oliver, P.L.; Johnson, R.; Capogna, M.; Vikman, J.; Molnár, Z.; Babbs, A.; Partridge, C.J.; Salehi, A.; Bengtsson, M.; et al. A dominant mutation in snap25 causes impaired vesicle trafficking, sensorimotor gating, and ataxia in the blind-drunk mouse. Proc. Nat.l Acad. Sci. USA 2007, 104, 2431–2436. [Google Scholar] [CrossRef] [PubMed]
- Bădescu, G.M.; Fîlfan, M.; Sandu, R.E.; Surugiu, R.; Ciobanu, O.; Popa-Wagner, A. Molecular mechanisms underlying neurodevelopmental disorders, adhd and autism. Rom. J. Morphol. Embryol. 2016, 57, 361–366. [Google Scholar] [PubMed]
- Albrecht, U.; Ripperger, J.A. Circadian clocks and sleep: Impact of rhythmic metabolism and waste clearance on the brain. Trends Neurosci. 2018, 47, 677–688. [Google Scholar] [CrossRef]
- Foster, R.G. Sleep, circadian rhythms and health. Interface Focus. 2020, 10, 20190098. [Google Scholar] [CrossRef]
- Jagannath, A.; Peirson, S.N.; Foster, R.G. Sleep and circadian rhythm disruption in neuropsychiatric illness. Curr. Opin. Neurobiol. 2013, 23, 888–894. [Google Scholar] [CrossRef] [PubMed]
- Bijlenga, D.A.-O.; Vollebregt, M.A.; Kooij, J.J.S.; Arns, M. The role of the circadian system in the etiology and pathophysiology of adhd: Time to redefine adhd? Atten. Defic. Hyperact. Disord. 2019, 11, 5–19. [Google Scholar] [CrossRef]
- Singh, K.; Zimmerman, A.W. Sleep in autism spectrum disorder and attention deficit hyperactivity disorder. Semin. Pediatr. Neurol. 2015, 22, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Koh, D.W.; Lawler, A.M.; Poitras, M.F.; Sasaki, M.; Wattler, S.; Nehls, M.C.; Stoger, T.; Poirier, G.G.; Dawson, V.L.; Dawson, T.M. Failure to degrade poly(adp-ribose) causes increased sensitivity to cytotoxicity and early embryonic lethality. Proc. Natl. Acad. Sci. USA 2004, 101, 17699–17704. [Google Scholar] [CrossRef]
- Rack, J.G.M.; Zorzini, V.; Zhu, Z.; Schuller, M.; Ahel, D.; Ahel, I. Viral macrodomains: A structural and evolutionary assessment of the pharmacological potential. Open Biol. 2020, 10, 200237. [Google Scholar] [CrossRef] [PubMed]
- Fehr, A.R.; Channappanavar, R.; Jankevicius, G.; Fett, C.; Zhao, J.; Athmer, J.; Meyerholz, D.K.; Ahel, I.; Perlman, S. The conserved coronavirus macrodomain promotes virulence and suppresses the innate immune response during severe acute respiratory syndrome coronavirus infection. MBio 2016, 7. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).