
Act Locally, Act Globally—Microbiota, Barriers, and Cytokines in Atherosclerosis


Abstract
1. Introduction
2. The Role of Selected Cytokines in Atherosclerosis
2.1. Cytokines of the Interleukin (IL)-1 Family in Atherosclerosis
2.2. Cytokines of the IL-17 Family in Atherosclerosis
2.3. Cytokines of the IL-10 Family in Atherosclerosis
2.4. Cytokines of the IL-6/IL-12 Superfamily in Atherosclerosis
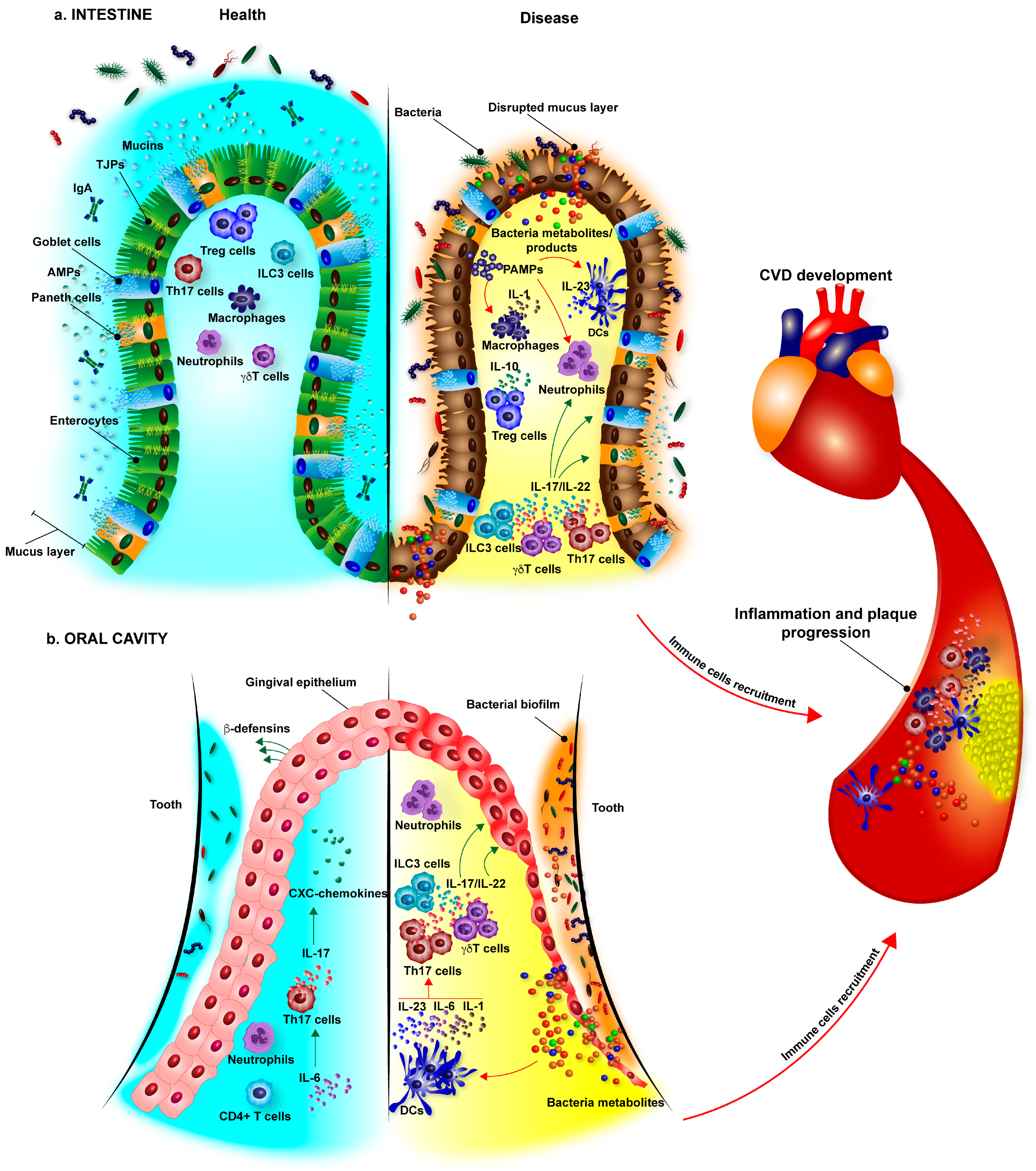
3. Mucosal Barriers as Gatekeepers for Microbiota: Potential Implication for Atherosclerosis
4. Microbiota in CVD Development
4.1. Intestinal Microbiota in CVD Development
4.2. Oral Microbiota in CVD Development
5. How Cytokines Regulate Barriers and Change Host–Microbiota Interactions
5.1. Cytokines of the IL-1 Family in Barrier Tissue Inflammation
5.2. Cytokines of the IL-17 Family in Barrier Tissue Inflammation
5.3. Cytokines of the IL-10 Family in Barrier Tissue Inflammation
5.4. Cytokines of the IL-6/IL-12 Superfamily in Barrier Tissue Inflammation
6. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgozoglu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Primers 2019, 5, 56. [Google Scholar] [CrossRef] [PubMed]
- Ait-Oufella, H.; Taleb, S.; Mallat, Z.; Tedgui, A. Recent advances on the role of cytokines in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 969–979. [Google Scholar] [CrossRef]
- Artis, D. Epithelial-cell recognition of commensal bacteria and maintenance of immune homeostasis in the gut. Nat. Rev. Immunol. 2008, 8, 411–420. [Google Scholar] [CrossRef]
- O’Neill, L.A.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef]
- Pan, W.; Wang, Q.; Chen, Q. The cytokine network involved in the host immune response to periodontitis. Int. J. Oral Sci. 2019, 11, 30. [Google Scholar] [CrossRef] [PubMed]
- Perez-Lopez, A.; Behnsen, J.; Nuccio, S.P.; Raffatellu, M. Mucosal immunity to pathogenic intestinal bacteria. Nat. Rev. Immunol. 2016, 16, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.S.; Davies, S.S. Microbial metabolism of dietary components to bioactive metabolites: Opportunities for new therapeutic interventions. Genome Med. 2016, 8, 46. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef]
- Dinarello, C.A. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 2018, 281, 8–27. [Google Scholar] [CrossRef]
- Frostegard, J.; Ulfgren, A.K.; Nyberg, P.; Hedin, U.; Swedenborg, J.; Andersson, U.; Hansson, G.K. Cytokine expression in advanced human atherosclerotic plaques: Dominance of pro-inflammatory (Th1) and macrophage-stimulating cytokines. Atherosclerosis 1999, 145, 33–43. [Google Scholar] [CrossRef]
- Galea, J.; Armstrong, J.; Gadsdon, P.; Holden, H.; Francis, S.E.; Holt, C.M. Interleukin-1 beta in coronary arteries of patients with ischemic heart disease. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 1000–1006. [Google Scholar] [CrossRef]
- Alexander, M.R.; Murgai, M.; Moehle, C.W.; Owens, G.K. Interleukin-1beta modulates smooth muscle cell phenotype to a distinct inflammatory state relative to PDGF-DD via NF-kappaB-dependent mechanisms. Physiol. Genom. 2012, 44, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Bhaskar, V.; Yin, J.; Mirza, A.M.; Phan, D.; Vanegas, S.; Issafras, H.; Michelson, K.; Hunter, J.J.; Kantak, S.S. Monoclonal antibodies targeting IL-1 beta reduce biomarkers of atherosclerosis in vitro and inhibit atherosclerotic plaque formation in Apolipoprotein E-deficient mice. Atherosclerosis 2011, 216, 313–320. [Google Scholar] [CrossRef]
- Chi, H.; Messas, E.; Levine, R.A.; Graves, D.T.; Amar, S. Interleukin-1 receptor signaling mediates atherosclerosis associated with bacterial exposure and/or a high-fat diet in a murine apolipoprotein E heterozygote model: Pharmacotherapeutic implications. Circulation 2004, 110, 1678–1685. [Google Scholar] [CrossRef] [PubMed]
- Devlin, C.M.; Kuriakose, G.; Hirsch, E.; Tabas, I. Genetic alterations of IL-1 receptor antagonist in mice affect plasma cholesterol level and foam cell lesion size. Proc. Natl. Acad. Sci. USA 2002, 99, 6280–6285. [Google Scholar] [CrossRef]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nunez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [PubMed]
- Freigang, S.; Ampenberger, F.; Weiss, A.; Kanneganti, T.D.; Iwakura, Y.; Hersberger, M.; Kopf, M. Fatty acid-induced mitochondrial uncoupling elicits inflammasome-independent IL-1alpha and sterile vascular inflammation in atherosclerosis. Nat. Immunol. 2013, 14, 1045–1053. [Google Scholar] [CrossRef]
- Gomez, D.; Baylis, R.A.; Durgin, B.G.; Newman, A.A.C.; Alencar, G.F.; Mahan, S.; St Hilaire, C.; Muller, W.; Waisman, A.; Francis, S.E.; et al. Interleukin-1beta has atheroprotective effects in advanced atherosclerotic lesions of mice. Nat. Med. 2018, 24, 1418–1429. [Google Scholar] [CrossRef]
- Kamari, Y.; Shaish, A.; Shemesh, S.; Vax, E.; Grosskopf, I.; Dotan, S.; White, M.; Voronov, E.; Dinarello, C.A.; Apte, R.N.; et al. Reduced atherosclerosis and inflammatory cytokines in apolipoprotein-E-deficient mice lacking bone marrow-derived interleukin-1alpha. Biochem. Biophys. Res. Commun. 2011, 405, 197–203. [Google Scholar] [CrossRef]
- Kirii, H.; Niwa, T.; Yamada, Y.; Wada, H.; Saito, K.; Iwakura, Y.; Asano, M.; Moriwaki, H.; Seishima, M. Lack of interleukin-1beta decreases the severity of atherosclerosis in ApoE-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 656–660. [Google Scholar] [CrossRef]
- Elhage, R.; Maret, A.; Pieraggi, M.T.; Thiers, J.C.; Arnal, J.F.; Bayard, F. Differential effects of interleukin-1 receptor antagonist and tumor necrosis factor binding protein on fatty-streak formation in apolipoprotein E-deficient mice. Circulation 1998, 97, 242–244. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, N.; Sukhova, G.K.; Libby, P.; Reynolds, R.S.; Young, J.L.; Schonbeck, U. Expression of interleukin (IL)-18 and functional IL-18 receptor on human vascular endothelial cells, smooth muscle cells, and macrophages: Implications for atherogenesis. J. Exp. Med. 2002, 195, 245–257. [Google Scholar] [CrossRef]
- Mallat, Z.; Corbaz, A.; Scoazec, A.; Besnard, S.; Leseche, G.; Chvatchko, Y.; Tedgui, A. Expression of interleukin-18 in human atherosclerotic plaques and relation to plaque instability. Circulation 2001, 104, 1598–1603. [Google Scholar] [CrossRef]
- Troseid, M.; Seljeflot, I.; Arnesen, H. The role of interleukin-18 in the metabolic syndrome. Cardiovasc. Diabetol. 2010, 9, 11. [Google Scholar] [CrossRef]
- Whitman, S.C.; Ravisankar, P.; Daugherty, A. Interleukin-18 enhances atherosclerosis in apolipoprotein E(−/−) mice through release of interferon-gamma. Circ. Res. 2002, 90, E34–E38. [Google Scholar] [CrossRef]
- Mallat, Z.; Corbaz, A.; Scoazec, A.; Graber, P.; Alouani, S.; Esposito, B.; Humbert, Y.; Chvatchko, Y.; Tedgui, A. Interleukin-18/interleukin-18 binding protein signaling modulates atherosclerotic lesion development and stability. Circ. Res. 2001, 89, E41–E45. [Google Scholar] [CrossRef] [PubMed]
- Elhage, R.; Jawien, J.; Rudling, M.; Ljunggren, H.G.; Takeda, K.; Akira, S.; Bayard, F.; Hansson, G.K. Reduced atherosclerosis in interleukin-18 deficient apolipoprotein E-knockout mice. Cardiovasc. Res. 2003, 59, 234–240. [Google Scholar] [CrossRef]
- Tenger, C.; Sundborger, A.; Jawien, J.; Zhou, X. IL-18 accelerates atherosclerosis accompanied by elevation of IFN-gamma and CXCL16 expression independently of T cells. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 791–796. [Google Scholar] [CrossRef]
- Wang, J.; Sun, C.; Gerdes, N.; Liu, C.; Liao, M.; Liu, J.; Shi, M.A.; He, A.; Zhou, Y.; Sukhova, G.K.; et al. Interleukin 18 function in atherosclerosis is mediated by the interleukin 18 receptor and the Na-Cl co-transporter. Nat. Med. 2015, 21, 820–826. [Google Scholar] [CrossRef]
- Levy, M.; Thaiss, C.A.; Zeevi, D.; Dohnalova, L.; Zilberman-Schapira, G.; Mahdi, J.A.; David, E.; Savidor, A.; Korem, T.; Herzig, Y.; et al. Microbiota-Modulated Metabolites Shape the Intestinal Microenvironment by Regulating NLRP6 Inflammasome Signaling. Cell 2015, 163, 1428–1443. [Google Scholar] [CrossRef]
- Henao-Mejia, J.; Elinav, E.; Jin, C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012, 482, 179–185. [Google Scholar] [CrossRef]
- Jarret, A.; Jackson, R.; Duizer, C.; Healy, M.E.; Zhao, J.; Rone, J.M.; Bielecki, P.; Sefik, E.; Roulis, M.; Rice, T.; et al. Enteric Nervous System-Derived IL-18 Orchestrates Mucosal Barrier Immunity. Cell 2020, 180, 50–63.e12. [Google Scholar] [CrossRef]
- McGeachy, M.J.; Cua, D.J.; Gaffen, S.L. The IL-17 Family of Cytokines in Health and Disease. Immunity 2019, 50, 892–906. [Google Scholar] [CrossRef] [PubMed]
- Gaffen, S.L. Structure and signalling in the IL-17 receptor family. Nat. Rev. Immunol. 2009, 9, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Bechara, R.; Zhao, J.; McGeachy, M.J.; Gaffen, S.L. IL-17 receptor-based signaling and implications for disease. Nat. Immunol. 2019, 20, 1594–1602. [Google Scholar] [CrossRef]
- Ishigame, H.; Kakuta, S.; Nagai, T.; Kadoki, M.; Nambu, A.; Komiyama, Y.; Fujikado, N.; Tanahashi, Y.; Akitsu, A.; Kotaki, H.; et al. Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity 2009, 30, 108–119. [Google Scholar] [CrossRef]
- Su, Y.; Huang, J.; Zhao, X.; Lu, H.; Wang, W.; Yang, X.O.; Shi, Y.; Wang, X.; Lai, Y.; Dong, C. Interleukin-17 receptor D constitutes an alternative receptor for interleukin-17A important in psoriasis-like skin inflammation. Sci. Immunol. 2019, 4. [Google Scholar] [CrossRef]
- Brauner, S.; Jiang, X.; Thorlacius, G.E.; Lundberg, A.M.; Ostberg, T.; Yan, Z.Q.; Kuchroo, V.K.; Hansson, G.K.; Wahren-Herlenius, M. Augmented Th17 differentiation in Trim21 deficiency promotes a stable phenotype of atherosclerotic plaques with high collagen content. Cardiovasc. Res. 2018, 114, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Butcher, M.J.; Gjurich, B.N.; Phillips, T.; Galkina, E.V. The IL-17A/IL-17RA axis plays a proatherogenic role via the regulation of aortic myeloid cell recruitment. Circ. Res. 2012, 110, 675–687. [Google Scholar] [CrossRef]
- Cheng, X.; Taleb, S.; Wang, J.; Tang, T.T.; Chen, J.; Gao, X.L.; Yao, R.; Xie, J.J.; Yu, X.; Xia, N.; et al. Inhibition of IL-17A in atherosclerosis. Atherosclerosis 2011, 215, 471–474. [Google Scholar] [CrossRef] [PubMed]
- Danzaki, K.; Matsui, Y.; Ikesue, M.; Ohta, D.; Ito, K.; Kanayama, M.; Kurotaki, D.; Morimoto, J.; Iwakura, Y.; Yagita, H.; et al. Interleukin-17A deficiency accelerates unstable atherosclerotic plaque formation in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Erbel, C.; Chen, L.; Bea, F.; Wangler, S.; Celik, S.; Lasitschka, F.; Wang, Y.; Bockler, D.; Katus, H.A.; Dengler, T.J. Inhibition of IL-17A attenuates atherosclerotic lesion development in apoE-deficient mice. J. Immunol. 2009, 183, 8167–8175. [Google Scholar] [CrossRef]
- Gistera, A.; Robertson, A.K.; Andersson, J.; Ketelhuth, D.F.; Ovchinnikova, O.; Nilsson, S.K.; Lundberg, A.M.; Li, M.O.; Flavell, R.A.; Hansson, G.K. Transforming growth factor-beta signaling in T cells promotes stabilization of atherosclerotic plaques through an interleukin-17-dependent pathway. Sci. Transl. Med. 2013, 5, 196ra100. [Google Scholar] [CrossRef]
- Ma, T.; Gao, Q.; Zhu, F.; Guo, C.; Wang, Q.; Gao, F.; Zhang, L. Th17 cells and IL-17 are involved in the disruption of vulnerable plaques triggered by short-term combination stimulation in apolipoprotein E-knockout mice. Cell. Mol. Immunol. 2013, 10, 338–348. [Google Scholar] [CrossRef]
- Taleb, S.; Romain, M.; Ramkhelawon, B.; Uyttenhove, C.; Pasterkamp, G.; Herbin, O.; Esposito, B.; Perez, N.; Yasukawa, H.; Van Snick, J.; et al. Loss of SOCS3 expression in T cells reveals a regulatory role for interleukin-17 in atherosclerosis. J. Exp. Med. 2009, 206, 2067–2077. [Google Scholar] [CrossRef]
- von Vietinghoff, S.; Koltsova, E.K.; Mestas, J.; Diehl, C.J.; Witztum, J.L.; Ley, K. Mycophenolate mofetil decreases atherosclerotic lesion size by depression of aortic T-lymphocyte and interleukin-17-mediated macrophage accumulation. J. Am. Coll. Cardiol. 2011, 57, 2194–2204. [Google Scholar] [CrossRef]
- Xie, J.J.; Wang, J.; Tang, T.T.; Chen, J.; Gao, X.L.; Yuan, J.; Zhou, Z.H.; Liao, M.Y.; Yao, R.; Yu, X.; et al. The Th17/Treg functional imbalance during atherogenesis in ApoE(−/−) mice. Cytokine 2010, 49, 185–193. [Google Scholar] [CrossRef]
- Smith, E.; Prasad, K.M.; Butcher, M.; Dobrian, A.; Kolls, J.K.; Ley, K.; Galkina, E. Blockade of interleukin-17A results in reduced atherosclerosis in apolipoprotein E-deficient mice. Circulation 2010, 121, 1746–1755. [Google Scholar] [CrossRef] [PubMed]
- Butcher, M.J.; Waseem, T.C.; Galkina, E.V. Smooth Muscle Cell-Derived Interleukin-17C Plays an Atherogenic Role via the Recruitment of Proinflammatory Interleukin-17A+ T Cells to the Aorta. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1496–1506. [Google Scholar] [CrossRef]
- Gelfand, J.M.; Dommasch, E.D.; Shin, D.B.; Azfar, R.S.; Kurd, S.K.; Wang, X.; Troxel, A.B. The risk of stroke in patients with psoriasis. J. Investig. Dermatol. 2009, 129, 2411–2418. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, B.; Kivelevitch, D.; Natarajan, B.; Joshi, A.A.; Ryan, C.; Benjegerdes, K.; Schussler, J.M.; Rader, D.J.; Reilly, M.P.; Menter, A.; et al. Comparison of Coronary Artery Calcium Scores Between Patients With Psoriasis and Type 2 Diabetes. JAMA Dermatol. 2016, 152, 1244–1253. [Google Scholar] [CrossRef]
- Elnabawi, Y.A.; Dey, A.K.; Goyal, A.; Groenendyk, J.W.; Chung, J.H.; Belur, A.D.; Rodante, J.; Harrington, C.L.; Teague, H.L.; Baumer, Y.; et al. Coronary artery plaque characteristics and treatment with biologic therapy in severe psoriasis: Results from a prospective observational study. Cardiovasc. Res. 2019, 115, 721–728. [Google Scholar] [CrossRef]
- Ouyang, W.; O’Garra, A. IL-10 Family Cytokines IL-10 and IL-22: From Basic Science to Clinical Translation. Immunity 2019, 50, 871–891. [Google Scholar] [CrossRef]
- Shouval, D.S.; Ouahed, J.; Biswas, A.; Goettel, J.A.; Horwitz, B.H.; Klein, C.; Muise, A.M.; Snapper, S.B. Interleukin 10 receptor signaling: Master regulator of intestinal mucosal homeostasis in mice and humans. Adv. Immunol. 2014, 122, 177–210. [Google Scholar] [CrossRef] [PubMed]
- Mallat, Z.; Heymes, C.; Ohan, J.; Faggin, E.; Leseche, G.; Tedgui, A. Expression of interleukin-10 in advanced human atherosclerotic plaques: Relation to inducible nitric oxide synthase expression and cell death. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 611–616. [Google Scholar] [CrossRef] [PubMed]
- Mallat, Z.; Besnard, S.; Duriez, M.; Deleuze, V.; Emmanuel, F.; Bureau, M.F.; Soubrier, F.; Esposito, B.; Duez, H.; Fievet, C.; et al. Protective role of interleukin-10 in atherosclerosis. Circ. Res. 1999, 85, e17–e24. [Google Scholar] [CrossRef] [PubMed]
- Caligiuri, G.; Rudling, M.; Ollivier, V.; Jacob, M.P.; Michel, J.B.; Hansson, G.K.; Nicoletti, A. Interleukin-10 deficiency increases atherosclerosis, thrombosis, and low-density lipoproteins in apolipoprotein E knockout mice. Mol. Med. 2003, 9, 10–17. [Google Scholar] [CrossRef]
- Potteaux, S.; Esposito, B.; van Oostrom, O.; Brun, V.; Ardouin, P.; Groux, H.; Tedgui, A.; Mallat, Z. Leukocyte-derived interleukin 10 is required for protection against atherosclerosis in low-density lipoprotein receptor knockout mice. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1474–1478. [Google Scholar] [CrossRef]
- Von Der Thusen, J.H.; Kuiper, J.; Fekkes, M.L.; De Vos, P.; Van Berkel, T.J.; Biessen, E.A. Attenuation of atherogenesis by systemic and local adenovirus-mediated gene transfer of interleukin-10 in LDLr−/− mice. FASEB J. 2001, 15, 2730–2732. [Google Scholar] [CrossRef]
- Pinderski, L.J.; Fischbein, M.P.; Subbanagounder, G.; Fishbein, M.C.; Kubo, N.; Cheroutre, H.; Curtiss, L.K.; Berliner, J.A.; Boisvert, W.A. Overexpression of interleukin-10 by activated T lymphocytes inhibits atherosclerosis in LDL receptor-deficient Mice by altering lymphocyte and macrophage phenotypes. Circ. Res. 2002, 90, 1064–1071. [Google Scholar] [CrossRef]
- Mallat, Z.; Gojova, A.; Brun, V.; Esposito, B.; Fournier, N.; Cottrez, F.; Tedgui, A.; Groux, H. Induction of a regulatory T cell type 1 response reduces the development of atherosclerosis in apolipoprotein E-knockout mice. Circulation 2003, 108, 1232–1237. [Google Scholar] [CrossRef]
- Sage, A.P.; Nus, M.; Baker, L.L.; Finigan, A.J.; Masters, L.M.; Mallat, Z. Regulatory B cell-specific interleukin-10 is dispensable for atherosclerosis development in mice. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1770–1773. [Google Scholar] [CrossRef]
- Eberl, G.; Di Santo, J.P.; Vivier, E. The brave new world of innate lymphoid cells. Nat. Immunol. 2015, 16, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Rutz, S.; Wang, X.; Ouyang, W. The IL-20 subfamily of cytokines--from host defence to tissue homeostasis. Nat. Rev. Immunol. 2014, 14, 783–795. [Google Scholar] [CrossRef] [PubMed]
- Behnsen, J.; Jellbauer, S.; Wong, C.P.; Edwards, R.A.; George, M.D.; Ouyang, W.; Raffatellu, M. The cytokine IL-22 promotes pathogen colonization by suppressing related commensal bacteria. Immunity 2014, 40, 262–273. [Google Scholar] [CrossRef]
- Qiu, J.; Guo, X.; Chen, Z.M.; He, L.; Sonnenberg, G.F.; Artis, D.; Fu, Y.X.; Zhou, L. Group 3 innate lymphoid cells inhibit T-cell-mediated intestinal inflammation through aryl hydrocarbon receptor signaling and regulation of microflora. Immunity 2013, 39, 386–399. [Google Scholar] [CrossRef] [PubMed]
- Shui, J.W.; Larange, A.; Kim, G.; Vela, J.L.; Zahner, S.; Cheroutre, H.; Kronenberg, M. HVEM signalling at mucosal barriers provides host defence against pathogenic bacteria. Nature 2012, 488, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ota, N.; Manzanillo, P.; Kates, L.; Zavala-Solorio, J.; Eidenschenk, C.; Zhang, J.; Lesch, J.; Lee, W.P.; Ross, J.; et al. Interleukin-22 alleviates metabolic disorders and restores mucosal immunity in diabetes. Nature 2014, 514, 237–241. [Google Scholar] [CrossRef]
- Rattik, S.; Hultman, K.; Rauch, U.; Soderberg, I.; Sundius, L.; Ljungcrantz, I.; Hultgardh-Nilsson, A.; Wigren, M.; Bjorkbacka, H.; Fredrikson, G.N.; et al. IL-22 affects smooth muscle cell phenotype and plaque formation in apolipoprotein E knockout mice. Atherosclerosis 2015, 242, 506–514. [Google Scholar] [CrossRef]
- Fatkhullina, A.R.; Peshkova, I.O.; Dzutsev, A.; Aghayev, T.; McCulloch, J.A.; Thovarai, V.; Badger, J.H.; Vats, R.; Sundd, P.; Tang, H.Y.; et al. An Interleukin-23-Interleukin-22 Axis Regulates Intestinal Microbial Homeostasis to Protect from Diet-Induced Atherosclerosis. Immunity 2018, 49, 943–957.e9. [Google Scholar] [CrossRef]
- Tait Wojno, E.D.; Hunter, C.A.; Stumhofer, J.S. The Immunobiology of the Interleukin-12 Family: Room for Discovery. Immunity 2019, 50, 851–870. [Google Scholar] [CrossRef]
- Hunter, C.A.; Jones, S.A. IL-6 as a keystone cytokine in health and disease. Nat. Immunol. 2015, 16, 448–457. [Google Scholar] [CrossRef]
- Luc, G.; Bard, J.M.; Juhan-Vague, I.; Ferrieres, J.; Evans, A.; Amouyel, P.; Arveiler, D.; Fruchart, J.C.; Ducimetiere, P.; Group, P.S. C-reactive protein, interleukin-6, and fibrinogen as predictors of coronary heart disease: The PRIME Study. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1255–1261. [Google Scholar] [CrossRef]
- Huber, S.A.; Sakkinen, P.; Conze, D.; Hardin, N.; Tracy, R. Interleukin-6 exacerbates early atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2364–2367. [Google Scholar] [CrossRef] [PubMed]
- Schieffer, B.; Selle, T.; Hilfiker, A.; Hilfiker-Kleiner, D.; Grote, K.; Tietge, U.J.; Trautwein, C.; Luchtefeld, M.; Schmittkamp, C.; Heeneman, S.; et al. Impact of interleukin-6 on plaque development and morphology in experimental atherosclerosis. Circulation 2004, 110, 3493–3500. [Google Scholar] [CrossRef] [PubMed]
- Elhage, R.; Clamens, S.; Besnard, S.; Mallat, Z.; Tedgui, A.; Arnal, J.; Maret, A.; Bayard, F. Involvement of interleukin-6 in atherosclerosis but not in the prevention of fatty streak formation by 17beta-estradiol in apolipoprotein E-deficient mice. Atherosclerosis 2001, 156, 315–320. [Google Scholar] [CrossRef]
- Rose-John, S. IL-6 trans-signaling via the soluble IL-6 receptor: Importance for the pro-inflammatory activities of IL-6. Int. J. Biol. Sci. 2012, 8, 1237–1247. [Google Scholar] [CrossRef] [PubMed]
- Schuett, H.; Oestreich, R.; Waetzig, G.H.; Annema, W.; Luchtefeld, M.; Hillmer, A.; Bavendiek, U.; von Felden, J.; Divchev, D.; Kempf, T.; et al. Transsignaling of interleukin-6 crucially contributes to atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Trinchieri, G.; Pflanz, S.; Kastelein, R.A. The IL-12 family of heterodimeric cytokines: New players in the regulation of T cell responses. Immunity 2003, 19, 641–644. [Google Scholar] [CrossRef]
- Lee, T.S.; Yen, H.C.; Pan, C.C.; Chau, L.Y. The role of interleukin 12 in the development of atherosclerosis in ApoE-deficient mice. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 734–742. [Google Scholar] [CrossRef]
- Davenport, P.; Tipping, P.G. The role of interleukin-4 and interleukin-12 in the progression of atherosclerosis in apolipoprotein E-deficient mice. Am. J. Pathol. 2003, 163, 1117–1125. [Google Scholar] [CrossRef]
- Gaffen, S.L.; Jain, R.; Garg, A.V.; Cua, D.J. The IL-23-IL-17 immune axis: From mechanisms to therapeutic testing. Nat. Rev. Immunol. 2014, 14, 585–600. [Google Scholar] [CrossRef] [PubMed]
- Abbas, A.; Gregersen, I.; Holm, S.; Daissormont, I.; Bjerkeli, V.; Krohg-Sorensen, K.; Skagen, K.R.; Dahl, T.B.; Russell, D.; Almas, T.; et al. Interleukin 23 levels are increased in carotid atherosclerosis: Possible role for the interleukin 23/interleukin 17 axis. Stroke 2015, 46, 793–799. [Google Scholar] [CrossRef]
- Gordon, K.B.; Langley, R.G.; Gottlieb, A.B.; Papp, K.A.; Krueger, G.G.; Strober, B.E.; Williams, D.A.; Gu, Y.; Valdes, J.M. A phase III, randomized, controlled trial of the fully human IL-12/23 mAb briakinumab in moderate-to-severe psoriasis. J. Investig. Dermatol. 2012, 132, 304–314. [Google Scholar] [CrossRef]
- Poizeau, F.; Nowak, E.; Kerbrat, S.; Le Nautout, B.; Droitcourt, C.; Drici, M.D.; Sbidian, E.; Guillot, B.; Bachelez, H.; Ait-Oufella, H.; et al. Association Between Early Severe Cardiovascular Events and the Initiation of Treatment With the Anti-Interleukin 12/23p40 Antibody Ustekinumab. JAMA Dermatol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Birchenough, G.M.; Nystrom, E.E.; Johansson, M.E.; Hansson, G.C. A sentinel goblet cell guards the colonic crypt by triggering Nlrp6-dependent Muc2 secretion. Science 2016, 352, 1535–1542. [Google Scholar] [CrossRef]
- Johansson, M.E.; Hansson, G.C. Immunological aspects of intestinal mucus and mucins. Nat. Rev. Immunol. 2016, 16, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Loonen, L.M.; Stolte, E.H.; Jaklofsky, M.T.; Meijerink, M.; Dekker, J.; van Baarlen, P.; Wells, J.M. REG3gamma-deficient mice have altered mucus distribution and increased mucosal inflammatory responses to the microbiota and enteric pathogens in the ileum. Mucosal. Immunol. 2014, 7, 939–947. [Google Scholar] [CrossRef] [PubMed]
- Vaishnava, S.; Yamamoto, M.; Severson, K.M.; Ruhn, K.A.; Yu, X.; Koren, O.; Ley, R.; Wakeland, E.K.; Hooper, L.V. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science 2011, 334, 255–258. [Google Scholar] [CrossRef]
- Slack, E.; Balmer, M.L.; Macpherson, A.J. B cells as a critical node in the microbiota-host immune system network. Immunol. Rev. 2014, 260, 50–66. [Google Scholar] [CrossRef] [PubMed]
- Reikvam, D.H.; Derrien, M.; Islam, R.; Erofeev, A.; Grcic, V.; Sandvik, A.; Gaustad, P.; Meza-Zepeda, L.A.; Jahnsen, F.L.; Smidt, H.; et al. Epithelial-microbial crosstalk in polymeric Ig receptor deficient mice. Eur. J. Immunol. 2012, 42, 2959–2970. [Google Scholar] [CrossRef]
- Van der Sluis, M.; De Koning, B.A.; De Bruijn, A.C.; Velcich, A.; Meijerink, J.P.; Van Goudoever, J.B.; Buller, H.A.; Dekker, J.; Van Seuningen, I.; Renes, I.B.; et al. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology 2006, 131, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Slifer, Z.M.; Blikslager, A.T. The Integral Role of Tight Junction Proteins in the Repair of Injured Intestinal Epithelium. Int. J. Mol. Sci. 2020, 21, 972. [Google Scholar] [CrossRef] [PubMed]
- Gaffen, S.L.; Moutsopoulos, N.M. Regulation of host-microbe interactions at oral mucosal barriers by type 17 immunity. Sci. Immunol. 2020, 5. [Google Scholar] [CrossRef]
- Moutsopoulos, N.M.; Konkel, J.E. Tissue-Specific Immunity at the Oral Mucosal Barrier. Trends Immunol. 2018, 39, 276–287. [Google Scholar] [CrossRef]
- Moutsopoulos, N.M.; Moutsopoulos, H.M. The oral mucosa: A barrier site participating in tissue-specific and systemic immunity. Oral Dis. 2018, 24, 22–25. [Google Scholar] [CrossRef]
- Forner, L.; Larsen, T.; Kilian, M.; Holmstrup, P. Incidence of bacteremia after chewing, tooth brushing and scaling in individuals with periodontal inflammation. J. Clin. Periodontol. 2006, 33, 401–407. [Google Scholar] [CrossRef]
- Marsh, P.D.; Do, T.; Beighton, D.; Devine, D.A. Influence of saliva on the oral microbiota. Periodontol. 2000 2016, 70, 80–92. [Google Scholar] [CrossRef]
- Fak, F.; Tremaroli, V.; Bergstrom, G.; Backhed, F. Oral microbiota in patients with atherosclerosis. Atherosclerosis 2015, 243, 573–578. [Google Scholar] [CrossRef]
- Hayashi, C.; Papadopoulos, G.; Gudino, C.V.; Weinberg, E.O.; Barth, K.R.; Madrigal, A.G.; Chen, Y.; Ning, H.; LaValley, M.; Gibson, F.C., 3rd; et al. Protective role for TLR4 signaling in atherosclerosis progression as revealed by infection with a common oral pathogen. J. Immunol. 2012, 189, 3681–3688. [Google Scholar] [CrossRef]
- Hayashi, C.; Viereck, J.; Hua, N.; Phinikaridou, A.; Madrigal, A.G.; Gibson, F.C., 3rd; Hamilton, J.A.; Genco, C.A. Porphyromonas gingivalis accelerates inflammatory atherosclerosis in the innominate artery of ApoE deficient mice. Atherosclerosis 2011, 215, 52–59. [Google Scholar] [CrossRef]
- Kitamoto, S.; Nagao-Kitamoto, H.; Jiao, Y.; Gillilland, M.G., 3rd; Hayashi, A.; Imai, J.; Sugihara, K.; Miyoshi, M.; Brazil, J.C.; Kuffa, P.; et al. The Intermucosal Connection between the Mouth and Gut in Commensal Pathobiont-Driven Colitis. Cell 2020, 182, 447–462.e14. [Google Scholar] [CrossRef] [PubMed]
- Konig, M.F.; Abusleme, L.; Reinholdt, J.; Palmer, R.J.; Teles, R.P.; Sampson, K.; Rosen, A.; Nigrovic, P.A.; Sokolove, J.; Giles, J.T.; et al. Aggregatibacter actinomycetemcomitans-induced hypercitrullination links periodontal infection to autoimmunity in rheumatoid arthritis. Sci. Transl. Med. 2016, 8, 369ra176. [Google Scholar] [CrossRef] [PubMed]
- Yakob, M.; Soder, B.; Meurman, J.H.; Jogestrand, T.; Nowak, J.; Soder, P.O. Prevotella nigrescens and Porphyromonas gingivalis are associated with signs of carotid atherosclerosis in subjects with and without periodontitis. J. Periodontal. Res. 2011, 46, 749–755. [Google Scholar] [CrossRef] [PubMed]
- Dutzan, N.; Konkel, J.E.; Greenwell-Wild, T.; Moutsopoulos, N.M. Characterization of the human immune cell network at the gingival barrier. Mucosal. Immunol. 2016, 9, 1163–1172. [Google Scholar] [CrossRef]
- Singh, R.K.; Chang, H.W.; Yan, D.; Lee, K.M.; Ucmak, D.; Wong, K.; Abrouk, M.; Farahnik, B.; Nakamura, M.; Zhu, T.H.; et al. Influence of diet on the gut microbiome and implications for human health. J. Transl. Med. 2017, 15, 73. [Google Scholar] [CrossRef]
- Ohira, H.; Tsutsui, W.; Fujioka, Y. Are Short Chain Fatty Acids in Gut Microbiota Defensive Players for Inflammation and Atherosclerosis? J. Atheroscler. Thromb. 2017, 24, 660–672. [Google Scholar] [CrossRef]
- Araujo, J.R.; Tazi, A.; Burlen-Defranoux, O.; Vichier-Guerre, S.; Nigro, G.; Licandro, H.; Demignot, S.; Sansonetti, P.J. Fermentation Products of Commensal Bacteria Alter Enterocyte Lipid Metabolism. Cell Host Microbe 2020, 27, 358–375.e357. [Google Scholar] [CrossRef]
- Zhu, W.; Gregory, J.C.; Org, E.; Buffa, J.A.; Gupta, N.; Wang, Z.; Li, L.; Fu, X.; Wu, Y.; Mehrabian, M.; et al. Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell 2016, 165, 111–124. [Google Scholar] [CrossRef]
- Marques, F.Z.; Nelson, E.; Chu, P.Y.; Horlock, D.; Fiedler, A.; Ziemann, M.; Tan, J.K.; Kuruppu, S.; Rajapakse, N.W.; El-Osta, A.; et al. High-Fiber Diet and Acetate Supplementation Change the Gut Microbiota and Prevent the Development of Hypertension and Heart Failure in Hypertensive Mice. Circulation 2017, 135, 964–977. [Google Scholar] [CrossRef]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef]
- Jonsson, A.L.; Backhed, F. Role of gut microbiota in atherosclerosis. Nat. Rev. Cardiol. 2017, 14, 79–87. [Google Scholar] [CrossRef]
- Tang, W.H.; Kitai, T.; Hazen, S.L. Gut Microbiota in Cardiovascular Health and Disease. Circ. Res. 2017, 120, 1183–1196. [Google Scholar] [CrossRef]
- Johnson, E.L.; Heaver, S.L.; Walters, W.A.; Ley, R.E. Microbiome and metabolic disease: Revisiting the bacterial phylum Bacteroidetes. J. Mol. Med. 2017, 95, 1–8. [Google Scholar] [CrossRef]
- Hildebrandt, M.A.; Hoffmann, C.; Sherrill-Mix, S.A.; Keilbaugh, S.A.; Hamady, M.; Chen, Y.Y.; Knight, R.; Ahima, R.S.; Bushman, F.; Wu, G.D. High-fat diet determines the composition of the murine gut microbiome independently of obesity. Gastroenterology 2009, 137, 1716–1724.e2. [Google Scholar] [CrossRef]
- Lee, J.Y.; Cevallos, S.A.; Byndloss, M.X.; Tiffany, C.R.; Olsan, E.E.; Butler, B.P.; Young, B.M.; Rogers, A.W.L.; Nguyen, H.; Kim, K.; et al. High-Fat Diet and Antibiotics Cooperatively Impair Mitochondrial Bioenergetics to Trigger Dysbiosis that Exacerbates Pre-inflammatory Bowel Disease. Cell Host Microbe 2020, 28, 273–284.e276. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Wolf, P.G.; Carbonero, F.; Zhong, W.; Reid, T.; Gaskins, H.R.; McIntosh, M.K. Intestinal and systemic inflammatory responses are positively associated with sulfidogenic bacteria abundance in high-fat-fed male C57BL/6J mice. J. Nutr. 2014, 144, 1181–1187. [Google Scholar] [CrossRef]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef]
- Griffiths, E.A.; Duffy, L.C.; Schanbacher, F.L.; Qiao, H.; Dryja, D.; Leavens, A.; Rossman, J.; Rich, G.; Dirienzo, D.; Ogra, P.L. In vivo effects of bifidobacteria and lactoferrin on gut endotoxin concentration and mucosal immunity in Balb/c mice. Dig. Dis. Sci. 2004, 49, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Byndloss, M.X.; Olsan, E.E.; Rivera-Chavez, F.; Tiffany, C.R.; Cevallos, S.A.; Lokken, K.L.; Torres, T.P.; Byndloss, A.J.; Faber, F.; Gao, Y.; et al. Microbiota-activated PPAR-gamma signaling inhibits dysbiotic Enterobacteriaceae expansion. Science 2017, 357, 570–575. [Google Scholar] [CrossRef]
- Litvak, Y.; Byndloss, M.X.; Baumler, A.J. Colonocyte metabolism shapes the gut microbiota. Science 2018, 362. [Google Scholar] [CrossRef]
- Slocum, C.; Kramer, C.; Genco, C.A. Immune dysregulation mediated by the oral microbiome: Potential link to chronic inflammation and atherosclerosis. J. Intern. Med. 2016, 280, 114–128. [Google Scholar] [CrossRef]
- Stoll, L.L.; Denning, G.M.; Weintraub, N.L. Potential role of endotoxin as a proinflammatory mediator of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2227–2236. [Google Scholar] [CrossRef]
- Wiesner, P.; Choi, S.H.; Almazan, F.; Benner, C.; Huang, W.; Diehl, C.J.; Gonen, A.; Butler, S.; Witztum, J.L.; Glass, C.K.; et al. Low doses of lipopolysaccharide and minimally oxidized low-density lipoprotein cooperatively activate macrophages via nuclear factor kappa B and activator protein-1: Possible mechanism for acceleration of atherosclerosis by subclinical endotoxemia. Circ. Res. 2010, 107, 56–65. [Google Scholar] [CrossRef]
- Li, H.; Sun, B. Toll-like receptor 4 in atherosclerosis. J. Cell Mol. Med. 2007, 11, 88–95. [Google Scholar] [CrossRef]
- Majdalawieh, A.; Ro, H.S. LPS-induced suppression of macrophage cholesterol efflux is mediated by adipocyte enhancer-binding protein 1. Int. J. Biochem. Cell Biol. 2009, 41, 1518–1525. [Google Scholar] [CrossRef]
- Li, X.Y.; Wang, C.; Xiang, X.R.; Chen, F.C.; Yang, C.M.; Wu, J. Porphyromonas gingivalis lipopolysaccharide increases lipid accumulation by affecting CD36 and ATP-binding cassette transporter A1 in macrophages. Oncol. Rep. 2013, 30, 1329–1336. [Google Scholar] [CrossRef]
- Wang, J.; Si, Y.; Wu, C.; Sun, L.; Ma, Y.; Ge, A.; Li, B. Lipopolysaccharide promotes lipid accumulation in human adventitial fibroblasts via TLR4-NF-kappaB pathway. Lipids Health Dis. 2012, 11, 139. [Google Scholar] [CrossRef]
- Liu, H.; Chen, X.; Hu, X.; Niu, H.; Tian, R.; Wang, H.; Pang, H.; Jiang, L.; Qiu, B.; Chen, X.; et al. Alterations in the gut microbiome and metabolism with coronary artery disease severity. Microbiome 2019, 7, 68. [Google Scholar] [CrossRef]
- Karlsson, F.H.; Fak, F.; Nookaew, I.; Tremaroli, V.; Fagerberg, B.; Petranovic, D.; Backhed, F.; Nielsen, J. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat. Commun. 2012, 3, 1245. [Google Scholar] [CrossRef] [PubMed]
- Al-Obaide, M.A.I.; Singh, R.; Datta, P.; Rewers-Felkins, K.A.; Salguero, M.V.; Al-Obaidi, I.; Kottapalli, K.R.; Vasylyeva, T.L. Gut Microbiota-Dependent Trimethylamine-N-oxide and Serum Biomarkers in Patients with T2DM and Advanced CKD. J. Clin. Med. 2017, 6, 86. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Li, Q.; Jiang, H. Gut microbiota in atherosclerosis: Focus on trimethylamine N-oxide. APMIS 2020, 128, 353–366. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Henderson, A.; Petriello, M.C.; Romano, K.A.; Gearing, M.; Miao, J.; Schell, M.; Sandoval-Espinola, W.J.; Tao, J.; Sha, B.; et al. Trimethylamine N-Oxide Binds and Activates PERK to Promote Metabolic Dysfunction. Cell Metab. 2019, 30, 1141–1151. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Roberts, A.B.; Buffa, J.A.; Levison, B.S.; Zhu, W.; Org, E.; Gu, X.; Huang, Y.; Zamanian-Daryoush, M.; Culley, M.K.; et al. Non-lethal Inhibition of Gut Microbial Trimethylamine Production for the Treatment of Atherosclerosis. Cell 2015, 163, 1585–1595. [Google Scholar] [CrossRef]
- Falony, G.; Vieira-Silva, S.; Raes, J. Microbiology Meets Big Data: The Case of Gut Microbiota-Derived Trimethylamine. Annu. Rev. Microbiol. 2015, 69, 305–321. [Google Scholar] [CrossRef]
- Romano, K.A.; Vivas, E.I.; Amador-Noguez, D.; Rey, F.E. Intestinal microbiota composition modulates choline bioavailability from diet and accumulation of the proatherogenic metabolite trimethylamine-N-oxide. mBio 2015, 6, e02481. [Google Scholar] [CrossRef] [PubMed]
- Brandsma, E.; Kloosterhuis, N.J.; Koster, M.; Dekker, D.C.; Gijbels, M.J.J.; van der Velden, S.; Rios-Morales, M.; van Faassen, M.J.R.; Loreti, M.G.; de Bruin, A.; et al. A Proinflammatory Gut Microbiota Increases Systemic Inflammation and Accelerates Atherosclerosis. Circ. Res. 2019, 124, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; van der Veeken, J.; deRoos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef]
- Chang, P.V.; Hao, L.; Offermanns, S.; Medzhitov, R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc. Natl. Acad. Sci. USA 2014, 111, 2247–2252. [Google Scholar] [CrossRef] [PubMed]
- Nooromid, M.; Chen, E.B.; Xiong, L.; Shapiro, K.; Jiang, Q.; Demsas, F.; Eskandari, M.; Priyadarshini, M.; Chang, E.B.; Layden, B.T.; et al. Microbe-Derived Butyrate and Its Receptor, Free Fatty Acid Receptor 3, But Not Free Fatty Acid Receptor 2, Mitigate Neointimal Hyperplasia Susceptibility After Arterial Injury. J. Am. Heart Assoc. 2020, 9, e016235. [Google Scholar] [CrossRef]
- Peng, L.; Li, Z.R.; Green, R.S.; Holzman, I.R.; Lin, J. Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in Caco-2 cell monolayers. J. Nutr. 2009, 139, 1619–1625. [Google Scholar] [CrossRef] [PubMed]
- Jie, Z.; Xia, H.; Zhong, S.L.; Feng, Q.; Li, S.; Liang, S.; Zhong, H.; Liu, Z.; Gao, Y.; Zhao, H.; et al. The gut microbiome in atherosclerotic cardiovascular disease. Nat. Commun. 2017, 8, 845. [Google Scholar] [CrossRef]
- Kasahara, K.; Krautkramer, K.A.; Org, E.; Romano, K.A.; Kerby, R.L.; Vivas, E.I.; Mehrabian, M.; Denu, J.M.; Backhed, F.; Lusis, A.J.; et al. Interactions between Roseburia intestinalis and diet modulate atherogenesis in a murine model. Nat. Microbiol. 2018, 3, 1461–1471. [Google Scholar] [CrossRef]
- Vacca, M.; Celano, G.; Calabrese, F.M.; Portincasa, P.; Gobbetti, M.; De Angelis, M. The Controversial Role of Human Gut Lachnospiraceae. Microorganisms 2020, 8, 573. [Google Scholar] [CrossRef] [PubMed]
- Belzer, C.; Chia, L.W.; Aalvink, S.; Chamlagain, B.; Piironen, V.; Knol, J.; de Vos, W.M. Microbial Metabolic Networks at the Mucus Layer Lead to Diet-Independent Butyrate and Vitamin B12 Production by Intestinal Symbionts. mBio 2017, 8. [Google Scholar] [CrossRef]
- Desai, M.S.; Seekatz, A.M.; Koropatkin, N.M.; Kamada, N.; Hickey, C.A.; Wolter, M.; Pudlo, N.A.; Kitamoto, S.; Terrapon, N.; Muller, A.; et al. A Dietary Fiber-Deprived Gut Microbiota Degrades the Colonic Mucus Barrier and Enhances Pathogen Susceptibility. Cell 2016, 167, 1339–1353.e21. [Google Scholar] [CrossRef]
- Parada Venegas, D.; De la Fuente, M.K.; Landskron, G.; Gonzalez, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.M.; Faber, K.N.; Hermoso, M.A. Short Chain Fatty Acids (SCFAs)-Mediated Gut Epithelial and Immune Regulation and Its Relevance for Inflammatory Bowel Diseases. Front. Immunol. 2019, 10, 277. [Google Scholar] [CrossRef]
- Larsen, J.M. The immune response to Prevotella bacteria in chronic inflammatory disease. Immunology 2017, 151, 363–374. [Google Scholar] [CrossRef]
- Liu, Z.; Li, J.; Liu, H.; Tang, Y.; Zhan, Q.; Lai, W.; Ao, L.; Meng, X.; Ren, H.; Xu, D.; et al. The intestinal microbiota associated with cardiac valve calcification differs from that of coronary artery disease. Atherosclerosis 2019, 284, 121–128. [Google Scholar] [CrossRef]
- Wang, Y.; Ames, N.P.; Tun, H.M.; Tosh, S.M.; Jones, P.J.; Khafipour, E. High Molecular Weight Barley beta-Glucan Alters Gut Microbiota Toward Reduced Cardiovascular Disease Risk. Front. Microbiol. 2016, 7, 129. [Google Scholar] [CrossRef]
- Deo, P.N.; Deshmukh, R. Oral microbiome: Unveiling the fundamentals. J. Oral Maxillofac. Pathol. 2019, 23, 122–128. [Google Scholar] [CrossRef]
- Koren, O.; Spor, A.; Felin, J.; Fak, F.; Stombaugh, J.; Tremaroli, V.; Behre, C.J.; Knight, R.; Fagerberg, B.; Ley, R.E.; et al. Human oral, gut, and plaque microbiota in patients with atherosclerosis. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. S1), 4592–4598. [Google Scholar] [CrossRef]
- Sanz, M.; Marco Del Castillo, A.; Jepsen, S.; Gonzalez-Juanatey, J.R.; D’Aiuto, F.; Bouchard, P.; Chapple, I.; Dietrich, T.; Gotsman, I.; Graziani, F.; et al. Periodontitis and cardiovascular diseases: Consensus report. J. Clin. Periodontol. 2020, 47, 268–288. [Google Scholar] [CrossRef] [PubMed]
- Montenegro, M.M.; Ribeiro, I.W.J.; Kampits, C.; Saffi, M.A.L.; Furtado, M.V.; Polanczyk, C.A.; Haas, A.N.; Rosing, C.K. Randomized controlled trial of the effect of periodontal treatment on cardiovascular risk biomarkers in patients with stable coronary artery disease: Preliminary findings of 3 months. J. Clin. Periodontol. 2019, 46, 321–331. [Google Scholar] [CrossRef]
- Santisteban, M.M.; Qi, Y.; Zubcevic, J.; Kim, S.; Yang, T.; Shenoy, V.; Cole-Jeffrey, C.T.; Lobaton, G.O.; Stewart, D.C.; Rubiano, A.; et al. Hypertension-Linked Pathophysiological Alterations in the Gut. Circ. Res. 2017, 120, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.S.; Bie, J.; Wang, J.; Ghosh, S. Oral supplementation with non-absorbable antibiotics or curcumin attenuates western diet-induced atherosclerosis and glucose intolerance in LDLR−/− mice—Role of intestinal permeability and macrophage activation. PLoS ONE 2014, 9, e108577. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, F.; Wang, J.; Wang, Y.; Fang, Y. Intestinal fatty acid-binding protein mediates atherosclerotic progress through increasing intestinal inflammation and permeability. J. Cell Mol. Med. 2020, 24, 5205–5212. [Google Scholar] [CrossRef] [PubMed]
- Biondi, R.B.; Salmazo, P.S.; Bazan, S.G.Z.; Hueb, J.C.; de Paiva, S.A.R.; Sassaki, L.Y. Cardiovascular Risk in Individuals with Inflammatory Bowel Disease. Clin. Exp. Gastroenterol. 2020, 13, 107–113. [Google Scholar] [CrossRef]
- Cappello, M.; Licata, A.; Calvaruso, V.; Bravata, I.; Aiello, A.; Torres, D.; Della Corte, V.; Tuttolomondo, A.; Perticone, M.; Licata, G.; et al. Increased expression of markers of early atherosclerosis in patients with inflammatory bowel disease. Eur. J. Intern. Med. 2017, 37, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Sinh, P.; Cross, R. Cardiovascular Risk Assessment and Impact of Medications on Cardiovascular Disease in Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2020. [Google Scholar] [CrossRef]
- Wald, D.; Qin, J.; Zhao, Z.; Qian, Y.; Naramura, M.; Tian, L.; Towne, J.; Sims, J.E.; Stark, G.R.; Li, X. SIGIRR, a negative regulator of Toll-like receptor-interleukin 1 receptor signaling. Nat. Immunol. 2003, 4, 920–927. [Google Scholar] [CrossRef]
- Garlanda, C.; Riva, F.; Polentarutti, N.; Buracchi, C.; Sironi, M.; De Bortoli, M.; Muzio, M.; Bergottini, R.; Scanziani, E.; Vecchi, A.; et al. Intestinal inflammation in mice deficient in Tir8, an inhibitory member of the IL-1 receptor family. Proc. Natl. Acad. Sci. USA 2004, 101, 3522–3526. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Gulen, M.F.; Qin, J.; Yao, J.; Bulek, K.; Kish, D.; Altuntas, C.Z.; Wald, D.; Ma, C.; Zhou, H.; et al. The Toll-interleukin-1 receptor member SIGIRR regulates colonic epithelial homeostasis, inflammation, and tumorigenesis. Immunity 2007, 26, 461–475. [Google Scholar] [CrossRef]
- Bersudsky, M.; Luski, L.; Fishman, D.; White, R.M.; Ziv-Sokolovskaya, N.; Dotan, S.; Rider, P.; Kaplanov, I.; Aychek, T.; Dinarello, C.A.; et al. Non-redundant properties of IL-1alpha and IL-1beta during acute colon inflammation in mice. Gut 2014, 63, 598–609. [Google Scholar] [CrossRef]
- Nunberg, M.; Werbner, N.; Neuman, H.; Bersudsky, M.; Braiman, A.; Ben-Shoshan, M.; Ben Izhak, M.; Louzoun, Y.; Apte, R.N.; Voronov, E.; et al. Interleukin 1alpha-Deficient Mice Have an Altered Gut Microbiota Leading to Protection from Dextran Sodium Sulfate-Induced Colitis. mSystems 2018, 3. [Google Scholar] [CrossRef]
- Siegmund, B.; Lehr, H.A.; Fantuzzi, G.; Dinarello, C.A. IL-1 beta -converting enzyme (caspase-1) in intestinal inflammation. Proc. Natl. Acad. Sci. USA 2001, 98, 13249–13254. [Google Scholar] [CrossRef] [PubMed]
- Coccia, M.; Harrison, O.J.; Schiering, C.; Asquith, M.J.; Becher, B.; Powrie, F.; Maloy, K.J. IL-1beta mediates chronic intestinal inflammation by promoting the accumulation of IL-17A secreting innate lymphoid cells and CD4(+) Th17 cells. J. Exp. Med. 2012, 209, 1595–1609. [Google Scholar] [CrossRef] [PubMed]
- Nowarski, R.; Jackson, R.; Gagliani, N.; de Zoete, M.R.; Palm, N.W.; Bailis, W.; Low, J.S.; Harman, C.C.; Graham, M.; Elinav, E.; et al. Epithelial IL-18 Equilibrium Controls Barrier Function in Colitis. Cell 2015, 163, 1444–1456. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.I.; Atarashi, K.; Manel, N.; Brodie, E.L.; Shima, T.; Karaoz, U.; Wei, D.; Goldfarb, K.C.; Santee, C.A.; Lynch, S.V.; et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 2009, 139, 485–498. [Google Scholar] [CrossRef]
- Khosravi, A.; Mazmanian, S.K. Disruption of the gut microbiome as a risk factor for microbial infections. Curr Opin Microbiol 2013, 16, 221–227. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Lebeis, S.L.; Powell, K.R.; Merlin, D.; Sherman, M.A.; Kalman, D. Interleukin-1 receptor signaling protects mice from lethal intestinal damage caused by the attaching and effacing pathogen Citrobacter rodentium. Infect. Immun. 2009, 77, 604–614. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zaki, M.H.; Vogel, P.; Gurung, P.; Finlay, B.B.; Deng, W.; Lamkanfi, M.; Kanneganti, T.D. Role of inflammasomes in host defense against Citrobacter rodentium infection. J. Biol. Chem. 2012, 287, 16955–16964. [Google Scholar] [CrossRef]
- Moyes, D.L.; Wilson, D.; Richardson, J.P.; Mogavero, S.; Tang, S.X.; Wernecke, J.; Hofs, S.; Gratacap, R.L.; Robbins, J.; Runglall, M.; et al. Candidalysin is a fungal peptide toxin critical for mucosal infection. Nature 2016, 532, 64–68. [Google Scholar] [CrossRef]
- Verma, A.H.; Richardson, J.P.; Zhou, C.; Coleman, B.M.; Moyes, D.L.; Ho, J.; Huppler, A.R.; Ramani, K.; McGeachy, M.J.; Mufazalov, I.A.; et al. Oral epithelial cells orchestrate innate type 17 responses to Candida albicans through the virulence factor candidalysin. Sci. Immunol. 2017, 2. [Google Scholar] [CrossRef]
- Netea, M.G.; Vonk, A.G.; van den Hoven, M.; Verschueren, I.; Joosten, L.A.; van Krieken, J.H.; van den Berg, W.B.; Van der Meer, J.W.; Kullberg, B.J. Differential role of IL-18 and IL-12 in the host defense against disseminated Candida albicans infection. Eur. J. Immunol. 2003, 33, 3409–3417. [Google Scholar] [CrossRef]
- Stuyt, R.J.; Netea, M.G.; van Krieken, J.H.; van der Meer, J.W.; Kullberg, B.J. Recombinant interleukin-18 protects against disseminated Candida albicans infection in mice. J. Infect. Dis. 2004, 189, 1524–1527. [Google Scholar] [CrossRef]
- Verma, A.H.; Zafar, H.; Ponde, N.O.; Hepworth, O.W.; Sihra, D.; Aggor, F.E.Y.; Ainscough, J.S.; Ho, J.; Richardson, J.P.; Coleman, B.M.; et al. IL-36 and IL-1/IL-17 Drive Immunity to Oral Candidiasis via Parallel Mechanisms. J. Immunol. 2018, 201, 627–634. [Google Scholar] [CrossRef]
- Figueredo, C.M.; Rescala, B.; Teles, R.P.; Teles, F.P.; Fischer, R.G.; Haffajee, A.D.; Socransky, S.S.; Gustafsson, A. Increased interleukin-18 in gingival crevicular fluid from periodontitis patients. Oral Microbiol. Immunol. 2008, 23, 173–176. [Google Scholar] [CrossRef]
- Gilowski, L.; Wiench, R.; Plocica, I.; Krzeminski, T.F. Amount of interleukin-1beta and interleukin-1 receptor antagonist in periodontitis and healthy patients. Arch. Oral Biol. 2014, 59, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Ozcaka, O.; Nalbantsoy, A.; Buduneli, N. Interleukin-17 and interleukin-18 levels in saliva and plasma of patients with chronic periodontitis. J. Periodontal. Res. 2011, 46, 592–598. [Google Scholar] [CrossRef]
- Sanchez-Hernandez, P.E.; Zamora-Perez, A.L.; Fuentes-Lerma, M.; Robles-Gomez, C.; Mariaud-Schmidt, R.P.; Guerrero-Velazquez, C. IL-12 and IL-18 levels in serum and gingival tissue in aggressive and chronic periodontitis. Oral Dis. 2011, 17, 522–529. [Google Scholar] [CrossRef]
- Hueber, W.; Sands, B.E.; Lewitzky, S.; Vandemeulebroecke, M.; Reinisch, W.; Higgins, P.D.; Wehkamp, J.; Feagan, B.G.; Yao, M.D.; Karczewski, M.; et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: Unexpected results of a randomised, double-blind placebo-controlled trial. Gut 2012, 61, 1693–1700. [Google Scholar] [CrossRef]
- Lee, J.S.; Tato, C.M.; Joyce-Shaikh, B.; Gulen, M.F.; Cayatte, C.; Chen, Y.; Blumenschein, W.M.; Judo, M.; Ayanoglu, G.; McClanahan, T.K.; et al. Interleukin-23-Independent IL-17 Production Regulates Intestinal Epithelial Permeability. Immunity 2015, 43, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, J.R.; Zhang, Y.; Brown, W.A.; Smith, C.L.; Byrne, F.R.; Fiorino, M.; Stevens, E.; Bigler, J.; Davis, J.A.; Rottman, J.B.; et al. Differential Roles for Interleukin-23 and Interleukin-17 in Intestinal Immunoregulation. Immunity 2015, 43, 739–750. [Google Scholar] [CrossRef]
- Cao, A.T.; Yao, S.; Gong, B.; Elson, C.O.; Cong, Y. Th17 cells upregulate polymeric Ig receptor and intestinal IgA and contribute to intestinal homeostasis. J. Immunol. 2012, 189, 4666–4673. [Google Scholar] [CrossRef] [PubMed]
- Raffatellu, M.; Santos, R.L.; Verhoeven, D.E.; George, M.D.; Wilson, R.P.; Winter, S.E.; Godinez, I.; Sankaran, S.; Paixao, T.A.; Gordon, M.A.; et al. Simian immunodeficiency virus-induced mucosal interleukin-17 deficiency promotes Salmonella dissemination from the gut. Nat. Med. 2008, 14, 421–428. [Google Scholar] [CrossRef]
- Kumar, P.; Monin, L.; Castillo, P.; Elsegeiny, W.; Horne, W.; Eddens, T.; Vikram, A.; Good, M.; Schoenborn, A.A.; Bibby, K.; et al. Intestinal Interleukin-17 Receptor Signaling Mediates Reciprocal Control of the Gut Microbiota and Autoimmune Inflammation. Immunity 2016, 44, 659–671. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Dela Cruz, P.; Wanek, A.G.; Kumar, P.; An, X.; Elsegeiny, W.; Horne, W.; Fitch, A.; Burr, A.H.P.; Gopalakrishna, K.P.; Chen, K.; et al. Intestinal IL-17R Signaling Constrains IL-18-Driven Liver Inflammation by the Regulation of Microbiome-Derived Products. Cell Rep. 2019, 29, 2270–2283.e2277. [Google Scholar] [CrossRef] [PubMed]
- Conti, H.R.; Peterson, A.C.; Brane, L.; Huppler, A.R.; Hernandez-Santos, N.; Whibley, N.; Garg, A.V.; Simpson-Abelson, M.R.; Gibson, G.A.; Mamo, A.J.; et al. Oral-resident natural Th17 cells and gammadelta T cells control opportunistic Candida albicans infections. J. Exp. Med. 2014, 211, 2075–2084. [Google Scholar] [CrossRef]
- Gladiator, A.; Wangler, N.; Trautwein-Weidner, K.; LeibundGut-Landmann, S. Cutting edge: IL-17-secreting innate lymphoid cells are essential for host defense against fungal infection. J. Immunol. 2013, 190, 521–525. [Google Scholar] [CrossRef]
- Boisson, B.; Wang, C.; Pedergnana, V.; Wu, L.; Cypowyj, S.; Rybojad, M.; Belkadi, A.; Picard, C.; Abel, L.; Fieschi, C.; et al. An ACT1 mutation selectively abolishes interleukin-17 responses in humans with chronic mucocutaneous candidiasis. Immunity 2013, 39, 676–686. [Google Scholar] [CrossRef] [PubMed]
- Ling, Y.; Cypowyj, S.; Aytekin, C.; Galicchio, M.; Camcioglu, Y.; Nepesov, S.; Ikinciogullari, A.; Dogu, F.; Belkadi, A.; Levy, R.; et al. Inherited IL-17RC deficiency in patients with chronic mucocutaneous candidiasis. J. Exp. Med. 2015, 212, 619–631. [Google Scholar] [CrossRef]
- Puel, A.; Cypowyj, S.; Bustamante, J.; Wright, J.F.; Liu, L.; Lim, H.K.; Migaud, M.; Israel, L.; Chrabieh, M.; Audry, M.; et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science 2011, 332, 65–68. [Google Scholar] [CrossRef]
- Trautwein-Weidner, K.; Gladiator, A.; Nur, S.; Diethelm, P.; LeibundGut-Landmann, S. IL-17-mediated antifungal defense in the oral mucosa is independent of neutrophils. Mucosal. Immunol. 2015, 8, 221–231. [Google Scholar] [CrossRef]
- Conti, H.R.; Bruno, V.M.; Childs, E.E.; Daugherty, S.; Hunter, J.P.; Mengesha, B.G.; Saevig, D.L.; Hendricks, M.R.; Coleman, B.M.; Brane, L.; et al. IL-17 Receptor Signaling in Oral Epithelial Cells Is Critical for Protection against Oropharyngeal Candidiasis. Cell Host Microbe 2016, 20, 606–617. [Google Scholar] [CrossRef]
- Conti, H.R.; Shen, F.; Nayyar, N.; Stocum, E.; Sun, J.N.; Lindemann, M.J.; Ho, A.W.; Hai, J.H.; Yu, J.J.; Jung, J.W.; et al. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J. Exp. Med. 2009, 206, 299–311. [Google Scholar] [CrossRef]
- Dutzan, N.; Kajikawa, T.; Abusleme, L.; Greenwell-Wild, T.; Zuazo, C.E.; Ikeuchi, T.; Brenchley, L.; Abe, T.; Hurabielle, C.; Martin, D.; et al. A dysbiotic microbiome triggers TH17 cells to mediate oral mucosal immunopathology in mice and humans. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Tsukasaki, M.; Komatsu, N.; Nagashima, K.; Nitta, T.; Pluemsakunthai, W.; Shukunami, C.; Iwakura, Y.; Nakashima, T.; Okamoto, K.; Takayanagi, H. Host defense against oral microbiota by bone-damaging T cells. Nat. Commun. 2018, 9, 701. [Google Scholar] [CrossRef]
- Kuhn, R.; Lohler, J.; Rennick, D.; Rajewsky, K.; Muller, W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell 1993, 75, 263–274. [Google Scholar] [CrossRef]
- Davidson, N.J.; Leach, M.W.; Fort, M.M.; Thompson-Snipes, L.; Kuhn, R.; Muller, W.; Berg, D.J.; Rennick, D.M. T helper cell 1-type CD4+ T cells, but not B cells, mediate colitis in interleukin 10-deficient mice. J. Exp. Med. 1996, 184, 241–251. [Google Scholar] [CrossRef]
- Zigmond, E.; Bernshtein, B.; Friedlander, G.; Walker, C.R.; Yona, S.; Kim, K.W.; Brenner, O.; Krauthgamer, R.; Varol, C.; Muller, W.; et al. Macrophage-restricted interleukin-10 receptor deficiency, but not IL-10 deficiency, causes severe spontaneous colitis. Immunity 2014, 40, 720–733. [Google Scholar] [CrossRef] [PubMed]
- Bernshtein, B.; Curato, C.; Ioannou, M.; Thaiss, C.A.; Gross-Vered, M.; Kolesnikov, M.; Wang, Q.; David, E.; Chappell-Maor, L.; Harmelin, A.; et al. IL-23-producing IL-10Ralpha-deficient gut macrophages elicit an IL-22-driven proinflammatory epithelial cell response. Sci. Immunol. 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Kirchner, F.R.; Littringer, K.; Altmeier, S.; Tran, V.D.T.; Schonherr, F.; Lemberg, C.; Pagni, M.; Sanglard, D.; Joller, N.; LeibundGut-Landmann, S. Persistence of Candida albicans in the Oral Mucosa Induces a Curbed Inflammatory Host Response That Is Independent of Immunosuppression. Front. Immunol. 2019, 10, 330. [Google Scholar] [CrossRef]
- Dutzan, N.; Gamonal, J.; Silva, A.; Sanz, M.; Vernal, R. Over-expression of forkhead box P3 and its association with receptor activator of nuclear factor-kappa B ligand, interleukin (IL) -17, IL-10 and transforming growth factor-beta during the progression of chronic periodontitis. J. Clin. Periodontol. 2009, 36, 396–403. [Google Scholar] [CrossRef]
- Kobayashi, R.; Kono, T.; Bolerjack, B.A.; Fukuyama, Y.; Gilbert, R.S.; Fujihashi, K.; Ruby, J.; Kataoka, K.; Wada, M.; Yamamoto, M.; et al. Induction of IL-10-producing CD4+ T-cells in chronic periodontitis. J. Dent. Res. 2011, 90, 653–658. [Google Scholar] [CrossRef]
- Sasaki, H.; Okamatsu, Y.; Kawai, T.; Kent, R.; Taubman, M.; Stashenko, P. The interleukin-10 knockout mouse is highly susceptible to Porphyromonas gingivalis-induced alveolar bone loss. J. Periodontal. Res. 2004, 39, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Girnary, M.; Wang, L.; Jiao, Y.; Zeng, E.; Mercer, K.; Zhang, J.; Marchesan, J.T.; Yu, N.; Moss, K.; et al. IL-10 Dampens an IL-17-Mediated Periodontitis-Associated Inflammatory Network. J. Immunol. 2020, 204, 2177–2191. [Google Scholar] [CrossRef] [PubMed]
- Sonnenberg, G.F.; Monticelli, L.A.; Alenghat, T.; Fung, T.C.; Hutnick, N.A.; Kunisawa, J.; Shibata, N.; Grunberg, S.; Sinha, R.; Zahm, A.M.; et al. Innate lymphoid cells promote anatomical containment of lymphoid-resident commensal bacteria. Science 2012, 336, 1321–1325. [Google Scholar] [CrossRef]
- Zheng, Y.; Valdez, P.A.; Danilenko, D.M.; Hu, Y.; Sa, S.M.; Gong, Q.; Abbas, A.R.; Modrusan, Z.; Ghilardi, N.; de Sauvage, F.J.; et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat. Med. 2008, 14, 282–289. [Google Scholar] [CrossRef]
- Zwarycz, B.; Gracz, A.D.; Rivera, K.R.; Williamson, I.A.; Samsa, L.A.; Starmer, J.; Daniele, M.A.; Salter-Cid, L.; Zhao, Q.; Magness, S.T. IL22 Inhibits Epithelial Stem Cell Expansion in an Ileal Organoid Model. Cell Mol. Gastroenterol. Hepatol. 2019, 7, 1–17. [Google Scholar] [CrossRef]
- Goto, Y.; Obata, T.; Kunisawa, J.; Sato, S.; Ivanov, I.I.; Lamichhane, A.; Takeyama, N.; Kamioka, M.; Sakamoto, M.; Matsuki, T.; et al. Innate lymphoid cells regulate intestinal epithelial cell glycosylation. Science 2014, 345, 1254009. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, K.; Ogawa, A.; Mizoguchi, E.; Shimomura, Y.; Andoh, A.; Bhan, A.K.; Blumberg, R.S.; Xavier, R.J.; Mizoguchi, A. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J. Clin. Investig. 2008, 118, 534–544. [Google Scholar] [CrossRef]
- De Luca, A.; Zelante, T.; D’Angelo, C.; Zagarella, S.; Fallarino, F.; Spreca, A.; Iannitti, R.G.; Bonifazi, P.; Renauld, J.C.; Bistoni, F.; et al. IL-22 defines a novel immune pathway of antifungal resistance. Mucosal. Immunol. 2010, 3, 361–373. [Google Scholar] [CrossRef]
- Hendrikx, T.; Duan, Y.; Wang, Y.; Oh, J.H.; Alexander, L.M.; Huang, W.; Starkel, P.; Ho, S.B.; Gao, B.; Fiehn, O.; et al. Bacteria engineered to produce IL-22 in intestine induce expression of REG3G to reduce ethanol-induced liver disease in mice. Gut 2019, 68, 1504–1515. [Google Scholar] [CrossRef]
- Aggor, F.E.Y.; Break, T.J.; Trevejo-Nunez, G.; Whibley, N.; Coleman, B.M.; Bailey, R.D.; Kaplan, D.H.; Naglik, J.R.; Shan, W.; Shetty, A.C.; et al. Oral epithelial IL-22/STAT3 signaling licenses IL-17-mediated immunity to oral mucosal candidiasis. Sci. Immunol. 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S.; Winthrop, K.; Calabrese, L. The role of IL-6 in host defence against infections: Immunobiology and clinical implications. Nat. Rev. Rheumatol. 2017, 13, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Dann, S.M.; Spehlmann, M.E.; Hammond, D.C.; Iimura, M.; Hase, K.; Choi, L.J.; Hanson, E.; Eckmann, L. IL-6-dependent mucosal protection prevents establishment of a microbial niche for attaching/effacing lesion-forming enteric bacterial pathogens. J. Immunol. 2008, 180, 6816–6826. [Google Scholar] [CrossRef]
- Tebbutt, N.C.; Giraud, A.S.; Inglese, M.; Jenkins, B.; Waring, P.; Clay, F.J.; Malki, S.; Alderman, B.M.; Grail, D.; Hollande, F.; et al. Reciprocal regulation of gastrointestinal homeostasis by SHP2 and STAT-mediated trefoil gene activation in gp130 mutant mice. Nat. Med. 2002, 8, 1089–1097. [Google Scholar] [CrossRef]
- Kuhn, C.; Rezende, R.M.; M’Hamdi, H.; da Cunha, A.P.; Weiner, H.L. IL-6 Inhibits Upregulation of Membrane-Bound TGF-beta 1 on CD4+ T Cells and Blocking IL-6 Enhances Oral Tolerance. J. Immunol. 2017, 198, 1202–1209. [Google Scholar] [CrossRef]
- Kuhn, K.A.; Manieri, N.A.; Liu, T.C.; Stappenbeck, T.S. IL-6 stimulates intestinal epithelial proliferation and repair after injury. PLoS ONE 2014, 9, e114195. [Google Scholar] [CrossRef] [PubMed]
- Neurath, M.F.; Fuss, I.; Kelsall, B.; Meyer zum Buschenfelde, K.H.; Strober, W. Effect of IL-12 and antibodies to IL-12 on established granulomatous colitis in mice. Ann. N. Y. Acad. Sci. 1996, 795, 368–370. [Google Scholar] [CrossRef] [PubMed]
- Simpson, S.J.; Shah, S.; Comiskey, M.; de Jong, Y.P.; Wang, B.; Mizoguchi, E.; Bhan, A.K.; Terhorst, C. T cell-mediated pathology in two models of experimental colitis depends predominantly on the interleukin 12/Signal transducer and activator of transcription (Stat)-4 pathway, but is not conditional on interferon gamma expression by T cells. J. Exp. Med. 1998, 187, 1225–1234. [Google Scholar] [CrossRef] [PubMed]
- Mencacci, A.; Cenci, E.; Del Sero, G.; Fe d’Ostiani, C.; Mosci, P.; Trinchieri, G.; Adorini, L.; Romani, L. IL-10 is required for development of protective Th1 responses in IL-12-deficient mice upon Candida albicans infection. J. Immunol. 1998, 161, 6228–6237. [Google Scholar]
- Simmons, C.P.; Goncalves, N.S.; Ghaem-Maghami, M.; Bajaj-Elliott, M.; Clare, S.; Neves, B.; Frankel, G.; Dougan, G.; MacDonald, T.T. Impaired resistance and enhanced pathology during infection with a noninvasive, attaching-effacing enteric bacterial pathogen, Citrobacter rodentium, in mice lacking IL-12 or IFN-gamma. J. Immunol. 2002, 168, 1804–1812. [Google Scholar] [CrossRef]
- Zelante, T.; De Luca, A.; Bonifazi, P.; Montagnoli, C.; Bozza, S.; Moretti, S.; Belladonna, M.L.; Vacca, C.; Conte, C.; Mosci, P.; et al. IL-23 and the Th17 pathway promote inflammation and impair antifungal immune resistance. Eur. J. Immunol. 2007, 37, 2695–2706. [Google Scholar] [CrossRef]
- Hue, S.; Ahern, P.; Buonocore, S.; Kullberg, M.C.; Cua, D.J.; McKenzie, B.S.; Powrie, F.; Maloy, K.J. Interleukin-23 drives innate and T cell-mediated intestinal inflammation. J. Exp. Med. 2006, 203, 2473–2483. [Google Scholar] [CrossRef]
- Kullberg, M.C.; Jankovic, D.; Feng, C.G.; Hue, S.; Gorelick, P.L.; McKenzie, B.S.; Cua, D.J.; Powrie, F.; Cheever, A.W.; Maloy, K.J.; et al. IL-23 plays a key role in Helicobacter hepaticus-induced T cell-dependent colitis. J. Exp. Med. 2006, 203, 2485–2494. [Google Scholar] [CrossRef]
- Uhlig, H.H.; McKenzie, B.S.; Hue, S.; Thompson, C.; Joyce-Shaikh, B.; Stepankova, R.; Robinson, N.; Buonocore, S.; Tlaskalova-Hogenova, H.; Cua, D.J.; et al. Differential activity of IL-12 and IL-23 in mucosal and systemic innate immune pathology. Immunity 2006, 25, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Eftychi, C.; Schwarzer, R.; Vlantis, K.; Wachsmuth, L.; Basic, M.; Wagle, P.; Neurath, M.F.; Becker, C.; Bleich, A.; Pasparakis, M. Temporally Distinct Functions of the Cytokines IL-12 and IL-23 Drive Chronic Colon Inflammation in Response to Intestinal Barrier Impairment. Immunity 2019, 51, 367–380.e4. [Google Scholar] [CrossRef] [PubMed]
- Aychek, T.; Mildner, A.; Yona, S.; Kim, K.W.; Lampl, N.; Reich-Zeliger, S.; Boon, L.; Yogev, N.; Waisman, A.; Cua, D.J.; et al. IL-23-mediated mononuclear phagocyte crosstalk protects mice from Citrobacter rodentium-induced colon immunopathology. Nat. Commun. 2015, 6, 6525. [Google Scholar] [CrossRef] [PubMed]
- Kinnebrew, M.A.; Buffie, C.G.; Diehl, G.E.; Zenewicz, L.A.; Leiner, I.; Hohl, T.M.; Flavell, R.A.; Littman, D.R.; Pamer, E.G. Interleukin 23 production by intestinal CD103(+)CD11b(+) dendritic cells in response to bacterial flagellin enhances mucosal innate immune defense. Immunity 2012, 36, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Ngo, V.L.; Abo, H.; Kuczma, M.; Szurek, E.; Moore, N.; Medina-Contreras, O.; Nusrat, A.; Merlin, D.; Gewirtz, A.T.; Ignatowicz, L.; et al. IL-36R signaling integrates innate and adaptive immune-mediated protection against enteropathogenic bacteria. Proc. Natl. Acad. Sci. USA 2020, 117, 27540–27548. [Google Scholar] [CrossRef] [PubMed]
- Aden, K.; Rehman, A.; Falk-Paulsen, M.; Secher, T.; Kuiper, J.; Tran, F.; Pfeuffer, S.; Sheibani-Tezerji, R.; Breuer, A.; Luzius, A.; et al. Epithelial IL-23R Signaling Licenses Protective IL-22 Responses in Intestinal Inflammation. Cell Rep. 2016, 16, 2208–2218. [Google Scholar] [CrossRef] [PubMed]
- Stadler, A.F.; Angst, P.D.; Arce, R.M.; Gomes, S.C.; Oppermann, R.V.; Susin, C. Gingival crevicular fluid levels of cytokines/chemokines in chronic periodontitis: A meta-analysis. J. Clin. Periodontol. 2016, 43, 727–745. [Google Scholar] [CrossRef]


{kind=link}
| Cytokine | Effect | Model | Reference |
|---|---|---|---|
| IL-1⍺, IL-1β | Promote atherosclerosis |
| [17,19] [20] [13] [14] [21] [18] |
| Suppress atherosclerosis or have no effect |
| [18] | |
| IL-6 | Promote atherosclerosis |
| [75] [79] |
| Suppress atherosclerosis or have no effect |
| [76,77] | |
| IL-10 | Promote atherosclerosis | ||
| Suppress atherosclerosis |
| [58] [59] [60] [61] | |
| IL-12 | Promote atherosclerosis |
| [81] [82] |
| Suppress atherosclerosis | |||
| IL-17 | Promote atherosclerosis |
| [40] [43] [49] [50] |
| Suppress atherosclerosis |
| [42] [46] [46] [39] [44] | |
| IL-18 | Promote atherosclerosis |
| [26] [27] [28] [29] [30] |
| Suppress atherosclerosis | |||
| IL-22 | Promote atherosclerosis |
| [70] |
| Suppress atherosclerosis |
| [71] | |
| IL-23 | Promote atherosclerosis |
| [84] |
| Suppress atherosclerosis |
| [71] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kurilenko, N.; Fatkhullina, A.R.; Mazitova, A.; Koltsova, E.K. Act Locally, Act Globally—Microbiota, Barriers, and Cytokines in Atherosclerosis. Cells 2021, 10, 348. https://doi.org/10.3390/cells10020348
Kurilenko N, Fatkhullina AR, Mazitova A, Koltsova EK. Act Locally, Act Globally—Microbiota, Barriers, and Cytokines in Atherosclerosis. Cells. 2021; 10(2):348. https://doi.org/10.3390/cells10020348
Chicago/Turabian StyleKurilenko, Natalia, Aliia R. Fatkhullina, Aleksandra Mazitova, and Ekaterina K. Koltsova. 2021. "Act Locally, Act Globally—Microbiota, Barriers, and Cytokines in Atherosclerosis" Cells 10, no. 2: 348. https://doi.org/10.3390/cells10020348
APA StyleKurilenko, N., Fatkhullina, A. R., Mazitova, A., & Koltsova, E. K. (2021). Act Locally, Act Globally—Microbiota, Barriers, and Cytokines in Atherosclerosis. Cells, 10(2), 348. https://doi.org/10.3390/cells10020348