
Genetic Modifiers of Hereditary Neuromuscular Disorders and Cardiomyopathy

 , ,
, , 
Abstract
1. Introduction
2. Methods
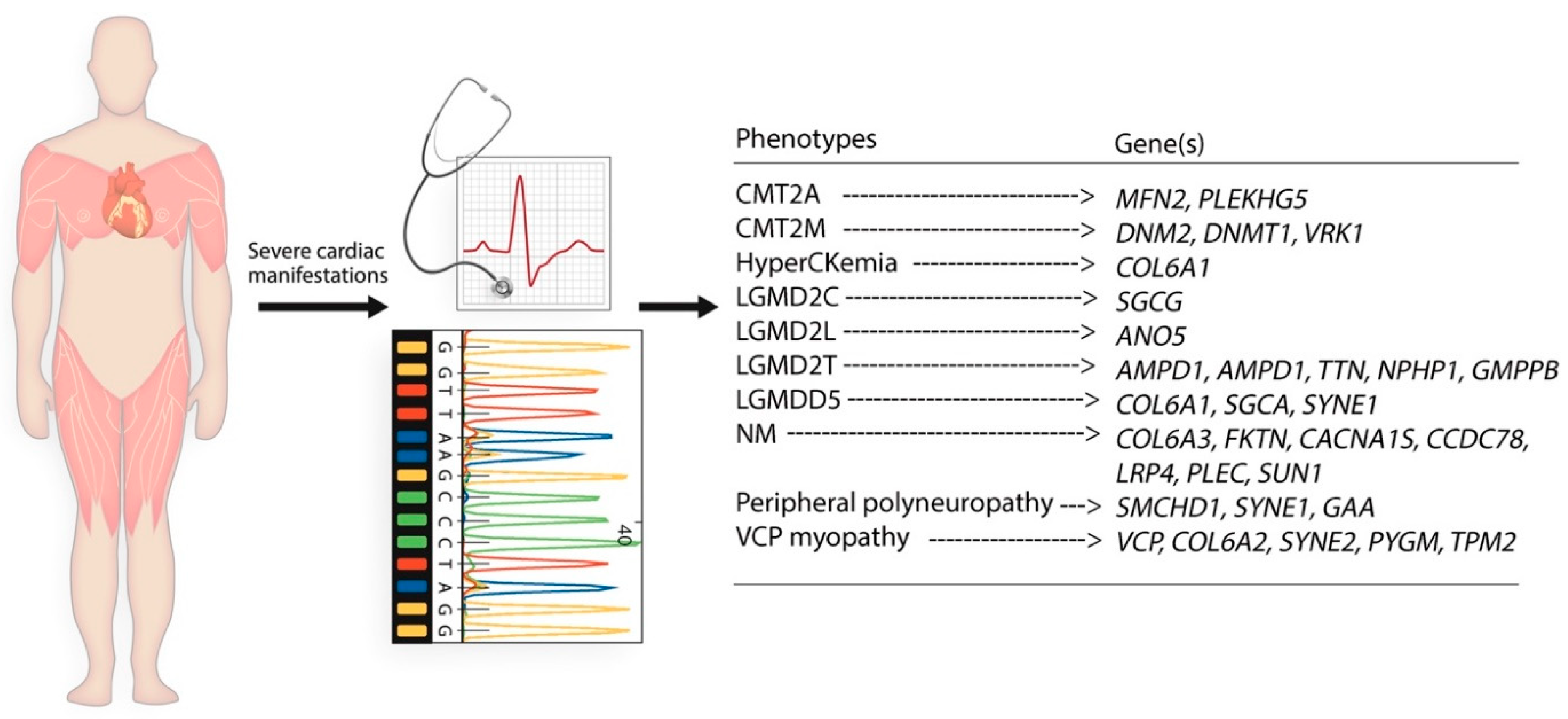
3. Results
Cases with NMD in Present Study
4. Discussion
Study Limitation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Glossary
| AMPD1 | Adenosine monophosphate deaminase 1 |
| ANO5 | Anoctamin 5 |
| AV | Atrioventricular |
| BMD | Becker muscular dystrophy |
| CACNA1S | Calcium voltage-gated channel subunit alpha1 S |
| CCDC78 | Coiled-coil domain containing 78 |
| CK | Creatine kinase |
| CMT | Charcot-Marie-Tooth |
| CMT2A | Charcot-Marie-Tooth type 2A |
| CMT2M | Charcot-Marie-Tooth type 2M |
| COL6A1 | Collagen Type VI Alpha 1 chain |
| COL6A2 | Collagen Type VI Alpha 2 chain |
| COL6A3 | Collagen Type VI Alpha 3 chain |
| DES | Desmin |
| DCM | Dilated cardiomyopathy |
| DMD | Dystrophin |
| DNM2 | Dynamin 2 |
| DNMT1 | DNA methyltransferase 1 |
| DSCAM | DS cell adhesion molecule |
| DYSF | Dysferlin |
| ECG | Electrocardiogram |
| EF | Ejection fraction |
| EMR | Electronic medical records |
| FKTN | Fukutin |
| GMPPB | GDP-mannose pyrophosphorylase B |
| HCM | Hypertrophic cardiomyopathy |
| HF | Heart failure |
| LGMD | Limb-girdle muscular dystrophy |
| LGMD2C | Limb-girdle muscular dystrophy type 2C |
| LGMD2L | Limb-girdle muscular dystrophy type 2L |
| LGMD2T | Limb-girdle muscular dystrophy type 2T |
| LGMDD5 | Limb-girdle muscular dystrophy D5/Bethlem Myopathy |
| LRP4 | LDL receptor related protein 4 |
| LV | Left ventricle |
| LVH | Left ventricular hypertrophy |
| MD | Muscular dystrophy |
| MDA | Muscular Dystrophy Association |
| MFN2 | Mitofusin 2 |
| MMD | Miyoshi muscular dystrophy |
| NGS | Next-generation sequencing |
| NMD | Hereditary neuromuscular disorders |
| NM | Necrotizing Myopathy |
| NPHP1 | Nephrocystin 1 |
| PLEC | Plectin |
| PLEKHG5 | Pleckstrin homology and RhoGEF domain containing G5 |
| PYGM | Glycogen phosphorylase, muscle associated |
| RBBB | Right bundle branch block |
| SCD | Sudden cardiac death |
| SGCA | Sarcoglycan alpha |
| SGCB | Sarcoglycan beta |
| SGCG | Sarcoglycan gamma |
| SMCHD1 | Structural maintenance of chromosomes flexible hinge domain containing 1 |
| SUN1 | Sad1 and UNC84 domain containing 1 |
| SYNE1 | Spectrin repeat containing nuclear envelope protein 1 |
| SYNE2 | Spectrin repeat containing nuclear envelope protein 2 |
| TPM2 | Tropomyosin 2 |
| TTE | Transthoracic echocardiography |
| TTN | Titin |
| UCMC | University of Cincinnati Medical Center |
| VCP | Valosin containing protein |
| VCP myopathy | Mutant valosin containing protein related myopathy |
| VUS | Variant of unknown significance |
| VRK1 | VRK serine/threonine kinase 1 |
References
- Pagola-Lorz, I.; Vicente, E.; Ibanez, B.; Torne, L.; Elizalde-Beiras, I.; Garcia-Solaesa, V.; Garcia, F.; Delfrade, J.; Jerico, I. Epidemiological study and genetic characterization of inherited muscle diseases in a northern Spanish region. Orphanet J. Rare Dis. 2019, 14, 276. [Google Scholar] [CrossRef] [PubMed]
- Theadom, A.; Rodrigues, M.; Poke, G.; O’Grady, G.; Love, D.; Hammond-Tooke, G.; Parmar, P.; Baker, R.; Feigin, V.; Jones, K.; et al. A Nationwide, Population-Based Prevalence Study of Genetic Muscle Disorders. Neuroepidemiology 2019, 52, 128–135. [Google Scholar] [CrossRef]
- Emery, A.E.H. The muscular dystrophies. Lancet 2002, 359, 687–695. [Google Scholar] [CrossRef]
- Kaplan, J.C.; Hamroun, D. The 2016 version of the gene table of monogenic neuromuscular disorders (nuclear genome). Neuromuscul. Disord. 2015, 25, 991–1020. [Google Scholar] [CrossRef]
- Mah, J.K.; Korngut, L.; Dykeman, J.; Day, L.; Pringsheim, T.; Jette, N. A systematic review and meta-analysis on the epidemiology of Duchenne and Becker muscular dystrophy. Neuromuscul. Disord. 2014, 24, 482–491. [Google Scholar] [CrossRef]
- Mah, J.K.; Korngut, L.; Fiest, K.M.; Dykeman, J.; Day, L.J.; Pringsheim, T.; Jette, N. A Systematic Review and Meta-analysis on the Epidemiology of the Muscular Dystrophies. Can. J. Neurol. Sci. 2016, 43, 163–177. [Google Scholar] [CrossRef] [PubMed]
- Blagova, O.; Nedostup, A.; Shumakov, D.; Poptsov, V.; Shestak, A.; Zaklyasminskaya, E. Dilated cardiomyopathy with severe arrhythmias in Emery-Dreifuss muscular dystrophy: From ablation to heart transplantation. J. Atr. Fibrillation 2016, 9, 1468. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, P.; Nandakumar, R.; Greig, H.; Broadhurst, P.; Dean, J.; Puranik, R.; Celermajer, D.S.; Hillis, G.S. Structural and electrical cardiac abnormalities are prevalent in asymptomatic adults with myotonic dystrophy. Heart 2016, 102, 1472–1478. [Google Scholar] [CrossRef]
- Kulach, A.; Majewski, M.; Gasior, Z.; Gardas, R.; Goscinska-Bis, K.; Golba, K.S. Dilated cardiomyopathy with severe arrhythmias in Emery-Dreifuss muscular dystrophy. Cardiol. J. 2020, 27, 93–94. [Google Scholar] [CrossRef] [PubMed]
- Ho, R.; Nguyen, M.L.; Mather, P. Cardiomyopathy in becker muscular dystrophy: Overview. World J. Cardiol. 2016, 8, 356–361. [Google Scholar] [CrossRef]
- McNally, E.M.; Kaltman, J.R.; Benson, D.W.; Canter, C.E.; Cripe, L.H.; Duan, D.; Finder, J.D.; Groh, W.J.; Hoffman, E.P.; Judge, D.P.; et al. Contemporary cardiac issues in Duchenne muscular dystrophy. Working Group of the National Heart, Lung, and Blood Institute in collaboration with Parent Project Muscular Dystrophy. Circulation 2015, 131, 1590–1598. [Google Scholar] [CrossRef]
- Sveen, M.L.; Thune, J.J.; Køber, L.; Vissing, J. Cardiac involvement in patients with limb-girdle muscular dystrophy type 2 and becker muscular dystrophy. Arch. Neurol. 2008, 65, 1196–1201. [Google Scholar] [CrossRef]
- Tandon, A.; Taylor, M.D.; Cripe, L.H. Co-occurring Duchenne muscular dystrophy and hypertrophic cardiomyopathy in an adult with atypical cardiac phenotype. Cardiol. Young 2015, 25, 355–357. [Google Scholar] [CrossRef]
- Feingold, B.; Mahle, W.T.; Auerbach, S.; Clemens, P.; Domenighetti, A.A.; Jefferies, J.L.; Judge, D.P.; Lal, A.K.; Markham, L.W.; Parks, W.J.; et al. Management of Cardiac Involvement Associated With Neuromuscular Diseases: A Scientific Statement from the American Heart Association. Circulation 2017, 136, e200–e231. [Google Scholar] [CrossRef]
- Chardon, J.W.; Smith, A.C.; Woulfe, J.; Pena, E.; Rakhra, K.; Dennie, C.; Beaulieu, C.; Huang, L.; Schwartzentruber, J.; Hawkins, C.; et al. LIMS2 mutations are associated with a novel muscular dystrophy, severe cardiomyopathy and triangular tongues. Clin. Genet. 2015, 88, 558–564. [Google Scholar] [CrossRef]
- Ribeiro, J.; Rebelo, O.; Fernández-Marmiesse, A.; Negrão, L. Novel mosaic mutation in the dystrophin gene causing distal asymmetric muscle weakness of the upper limbs and dilated cardiomyopathy. Acta Myol. 2018, 37, 117–120. [Google Scholar]
- Chen, L.; Ren, J.; Chen, X.; Chen, K.; Rao, M.; Zhang, N.; Yu, W.; Song, J. A novel mutation of dystrophin in a Becker muscular dystrophy family with severe cardiac involvement: From genetics to clinicopathology. Cardiovasc. Pathol. 2018, 36, 64–70. [Google Scholar] [CrossRef]
- Tsuda, T.; Fitzgerald, K.; Scavena, M.; Gidding, S.; Cox, M.O.; Marks, H.; Flanigan, K.M.; Moore, S.A. Early-progressive dilated cardiomyopathy in a family with Becker muscular dystrophy related to a novel frameshift mutation in the dystrophin gene exon 27. J. Hum. Genet. 2015, 60, 151–155. [Google Scholar] [CrossRef]
- Kong, D.; Zhan, Y.; Liu, C.; Hu, Y.; Zhou, Y.; Luo, J.; Gu, L.; Zhou, X.; Zhang, Z. A Novel Mutation Of The EMD Gene In A Family With Cardiac Conduction Abnormalities And A High Incidence Of Sudden Cardiac Death. Pharmgenom. Pers. Med. 2019, 12, 319–327. [Google Scholar] [CrossRef]
- SNP–NCBI. Available online: https://www.ncbi.nlm.nih.gov/snp (accessed on 6 February 2021).
- Wahbi, K.; Béhin, A.; Bécane, H.M.; Leturcq, F.; Cossée, M.; Laforêt, P.; Stojkovic, T.; Carlier, P.; Toussaint, M.; Gaxotte, V.; et al. Dilated cardiomyopathy in patients with mutations in anoctamin 5. Int. J. Cardiol. 2013, 168, 76–79. [Google Scholar] [CrossRef]
- Barresi, R.; Di Blasi, C.; Negri, T.; Brugnoni, R.; Vitali, A.; Felisari, G.; Salandi, A.; Daniel, S.; Cornelio, F.; Morandi, L.; et al. Disruption of heart sarcoglycan complex and severe cardiomyopathy caused by beta sarcoglycan mutations. J. Med. Genet. 2000, 37, 102–107. [Google Scholar] [CrossRef]
- Ben Hamida, M.; Ben Hamida, C.; Zouari, M.; Belal, S.; Hentati, F. Limb-girdle muscular dystrophy 2C: Clinical aspects. Neuromuscul. Disord. 1996, 6, 493–494. [Google Scholar] [CrossRef]
- Nigro, V.; Okazaki, Y.; Belsito, A.; Piluso, G.; Matsuda, Y.; Politano, L.; Nigro, G.; Ventura, C.; Abbondanza, C.; Molinari, A.M.; et al. Identification of the Syrian hamster cardiomyopathy gene. Hum. Mol. Genet. 1997, 6, 601–607. [Google Scholar] [CrossRef]
- Piccolo, F.; Roberds, S.L.; Jeanpierre, M.; Leturcq, F.; Azibi, K.; Beldjord, C.; Carrie, A.; Recan, D.; Chaouch, M.; Reghis, A.; et al. Primary adhalinopathy: A common cause of autosomal recessive muscular dystrophy of variable severity. Nat. Genet. 1995, 10, 243–245. [Google Scholar] [CrossRef]
- Tsubata, S.; Bowles, K.R.; Vatta, M.; Zintz, C.; Titus, J.; Muhonen, L.; Bowles, N.E.; Towbin, J.A. Mutations in the human δ-sarcoglycan gene in familial and sporadic dilated cardiomyopathy. J. Clin. Investig. 2000, 106, 655–662. [Google Scholar] [CrossRef]
- McNally, E.M.; Mestroni, L. Dilated cardiomyopathy: Genetic determinants and mechanisms. Circ. Res. 2017, 121, 731–748. [Google Scholar] [CrossRef]
- Sandra, M.; Maria Pia, L.; Stefano, C.; Pietro, P.; Crociani, P.; Aldo, R.; Giuseppe, D.S.; Massimo, C. Emery-Dreifuss muscular dystrophy type 4: A new SYNE1 mutation associated with hypertrophic cardiomyopathy masked by a perinatal distress-related spastic diplegia. Clin. Case Rep. 2019, 7, 1078–1082. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Y.; Gong, Y.J.; Zhong, B.F.; Zhou, Y.; Gong, L. Bioinformatics method identifies potential biomarkers of dilated cardiomyopathy in a human induced pluripotent stem cell-derived cardiomyocyte model. Exp. Ther. Med. 2017, 14, 2771–2778. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Hayashi, Y.K.; Noguchi, S.; Ogawa, M.; Nonaka, I.; Tanabe, Y.; Ogino, M.; Takada, F.; Eriguchi, M.; Kotooka, N.; et al. Fukutin gene mutations cause dilated cardiomyopathy with minimal muscle weakness. Ann. Neurol. 2006, 60, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Anandan, C.; Cipriani, M.A.; Laughlin, R.S.; Niu, Z.; Milone, M. Rhabdomyolysis and fluctuating asymptomatic hyperCKemia associated with CACNA1S variant. Eur. J. Neurol. 2018, 25, 417–419. [Google Scholar] [CrossRef]
- Zuchner, S.; Mersiyanova, I.V.; Muglia, M.; Bissar-Tadmouri, N.; Rochelle, J.; Dadali, E.L.; Zappia, M.; Nelis, E.; Patitucci, A.; Senderek, J.; et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 2004, 36, 449–451. [Google Scholar] [CrossRef]
- Dorn, G.W., 2nd; Clark, C.F.; Eschenbacher, W.H.; Kang, M.Y.; Engelhard, J.T.; Warner, S.J.; Matkovich, S.J.; Jowdy, C.C. MARF and Opa1 control mitochondrial and cardiac function in Drosophila. Circ. Res. 2011, 108, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Dorn, G.W. PINK1-phosphorylated mitofusin 2 is a parkin receptor for culling damaged mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef]
- Papanicolaou, K.N.; Khairallah, R.J.; Ngoh, G.A.; Chikando, A.; Luptak, I.; O’Shea, K.M.; Riley, D.D.; Lugus, J.J.; Colucci, W.S.; Lederer, W.J.; et al. Mitofusin-2 Maintains Mitochondrial Structure and Contributes to Stress-Induced Permeability Transition in Cardiac Myocytes. Mol. Cell Biol. 2011, 31, 1309–1328. [Google Scholar] [CrossRef]
- Stojkovic, T.; Richard, P.; Charron, P.; Rondeau, S.; JeanPierre, M. Identification of both LMNA and SMCHD1 mutations in a case with overlapping phenotypes. Neuromuscul. Disord. 2014, 24, 844. [Google Scholar] [CrossRef]
- Gal, A.; Inczedy-Farkas, G.; Pal, E.; Remenyi, V.; Bereznai, B.; Geller, L.; Szelid, Z.; Merkely, B.; Molnar, M.J. The coexistence of dynamin 2 mutation and multiple mitochondrial DNA (mtDNA) deletions in the background of severe cardiomyopathy and centronuclear myopathy. Clin. Neuropathol. 2015, 34, 89–95. [Google Scholar] [CrossRef]
- Watts, G.D.; Wymer, J.; Kovach, M.J.; Mehta, S.G.; Mumm, S.; Darvish, D.; Pestronk, A.; Whyte, M.P.; Kimonis, V.E. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet. 2004, 36, 377–381. [Google Scholar] [CrossRef]
- Mizuta, K.; Tsutsumi, S.; Inoue, H.; Sakamoto, Y.; Miyatake, K.; Miyawaki, K.; Noji, S.; Kamata, N.; Itakura, M. Molecular characterization of GDD1/TMEM16E, the gene product responsible for autosomal dominant gnathodiaphyseal dysplasia. Biochem. Biophys. Res. Commun. 2007, 357, 126–132. [Google Scholar] [CrossRef]
- Bolduc, V.; Marlow, G.; Boycott, K.M.; Saleki, K.; Inoue, H.; Kroon, J.; Itakura, M.; Robitaille, Y.; Parent, L.; Baas, F.; et al. Recessive mutations in the putative calcium-activated chloride channel Anoctamin 5 cause proximal LGMD2L and distal MMD3 muscular dystrophies. Am. J. Hum. Genet. 2010, 86, 213–221. [Google Scholar] [CrossRef]
- Hicks, D.; Sarkozy, A.; Muelas, N.; Koehler, K.; Huebner, A.; Hudson, G.; Chinnery, P.F.; Barresi, R.; Eagle, M.; Polvikoski, T.; et al. A founder mutation in Anoctamin 5 is a major cause of limb-girdle muscular dystrophy. Brain 2011, 134, 171–182. [Google Scholar] [CrossRef]
- Campbell, K.P.; Kahl, S.D. Association of dystrophin and an integral membrane glycoprotein. Nature 1989, 338, 259–262. [Google Scholar] [CrossRef]
- Yoshida, M.; Ozawa, E. Glycoprotein complex anchoring dystrophin to sarcolemma. J. Biochem. 1990, 108, 748–752. [Google Scholar] [CrossRef]
- Bonnemann, C.G.; Modi, R.; Noguchi, S.; Mizuno, Y.; Yoshida, M.; Gussoni, E.; McNally, E.M.; Duggan, D.J.; Angelini, C.; Hoffman, E.P. Beta-sarcoglycan (A3b) mutations cause autosomal recessive muscular dystrophy with loss of the sarcoglycan complex. Nat. Genet. 1995, 11, 266–273. [Google Scholar] [CrossRef]
- Lim, L.E.; Duclos, F.; Broux, O.; Bourg, N.; Sunada, Y.; Allamand, V.; Meyer, J.; Richard, I.; Moomaw, C.; Slaughter, C.; et al. Beta-sarcoglycan: Characterization and role in limb-girdle muscular dystrophy linked to 4q12. Nat. Genet. 1995, 11, 257–265. [Google Scholar] [CrossRef]
- Noguchi, S.; McNally, E.M.; Ben Othmane, K.; Hagiwara, Y.; Mizuno, Y.; Yoshida, M.; Yamamoto, H.; Bonnemann, C.G.; Gussoni, E.; Denton, P.H.; et al. Mutations in the dystrophin-associated protein gamma-sarcoglycan in chromosome 13 muscular dystrophy. Science 1995, 270, 819–822. [Google Scholar] [CrossRef]
- Roberds, S.L.; Leturcq, F.; Allamand, V.; Piccolo, F.; Jeanpierre, M.; Anderson, R.D.; Lim, L.E.; Lee, J.C.; Tome, F.M.; Romero, N.B.; et al. Missense mutations in the adhalin gene linked to autosomal recessive muscular dystrophy. Cell 1994, 78, 625–633. [Google Scholar] [CrossRef]
- Barresi, R.; Confalonieri, V.; Lanfossi, M.; Di Blasi, C.; Torchiana, E.; Mantegazza, R.; Jarre, L.; Nardocci, N.; Boffi, P.; Tezzon, F.; et al. Concomitant deficiency of beta- and gamma-sarcoglycans in 20 alpha-sarcoglycan (adhalin)-deficient patients: Immunohistochemical analysis and clinical aspects. Acta Neuropathol. 1997, 94, 28–35. [Google Scholar] [CrossRef]
- Ben Othmane, K.; Ben Hamida, M.; Pericak-Vance, M.A.; Ben Hamida, C.; Blel, S.; Carter, S.C.; Bowcock, A.M.; Petruhkin, K.; Gilliam, T.C.; Roses, A.D.; et al. Linkage of Tunisian autosomal recessive Duchenne-like muscular dystrophy to the pericentromeric region of chromosome 13q. Nat. Genet. 1992, 2, 315–317. [Google Scholar] [CrossRef]
- Van der Kooi, A.J.; de Visser, M.; van Meegen, M.; Ginjaar, H.B.; van Essen, A.J.; Jennekens, F.G.; Jongen, P.J.; Leschot, N.J.; Bolhuis, P.A. A novel gamma-sarcoglycan mutation causing childhood onset, slowly progressive limb girdle muscular dystrophy. Neuromuscul. Disord. 1998, 8, 305–308. [Google Scholar] [CrossRef]
- Townsend, D.; Yasuda, S.; McNally, E.; Metzger, J.M. Distinct pathophysiological mechanisms of cardiomyopathy in hearts lacking dystrophin or the sarcoglycan complex. FASEB J. 2011, 25, 3106–3114. [Google Scholar] [CrossRef]
- Bonnemann, C.G.; Wong, J.; Jones, K.J.; Lidov, H.G.; Feener, C.A.; Shapiro, F.; Darras, B.T.; Kunkel, L.M.; North, K.N. Primary gamma-sarcoglycanopathy (LGMD 2C): Broadening of the mutational spectrum guided by the immunohistochemical profile. Neuromuscul. Disord. 2002, 12, 273–280. [Google Scholar] [CrossRef]
- Lucioli, S.; Giusti, B.; Mercuri, E.; Vanegas, O.C.; Lucarini, L.; Pietroni, V.; Urtizberea, A.; Ben Yaou, R.; de Visser, M.; van der Kooi, A.J.; et al. Detection of common and private mutations in the COL6A1 gene of patients with Bethlem myopathy. Neurology 2005, 64, 1931–1937. [Google Scholar] [CrossRef]
- Okada, M.; Kawahara, G.; Noguchi, S.; Sugie, K.; Murayama, K.; Nonaka, I.; Hayashi, Y.K.; Nishino, I. Primary collagen VI deficiency is the second most common congenital muscular dystrophy in Japan. Neurology 2007, 69, 1035–1042. [Google Scholar] [CrossRef]
- Van der Kooi, A.J.; de Voogt, W.G.; Bertini, E.; Merlini, L.; Talim, F.B.; Ben Yaou, R.; Urtziberea, A.; de Visser, M. Cardiac and pulmonary investigations in Bethlem myopathy. Arch. Neurol. 2006, 63, 1617–1621. [Google Scholar] [CrossRef]
- Rutschow, D.; Bauer, R.; Gohringer, C.; Bekeredjian, R.; Schinkel, S.; Straub, V.; Koenen, M.; Weichenhan, D.; Katus, H.A.; Muller, O.J. S151A delta-sarcoglycan mutation causes a mild phenotype of cardiomyopathy in mice. Eur. J. Hum. Genet. 2014, 22, 119–125. [Google Scholar] [CrossRef][Green Version]
- Zhou, C.; Li, C.; Zhou, B.; Sun, H.; Koullourou, V.; Holt, I.; Puckelwartz, M.J.; Warren, D.T.; Hayward, R.; Lin, Z.; et al. Novel nesprin-1 mutations associated with dilated cardiomyopathy cause nuclear envelope disruption and defects in myogenesis. Hum. Mol. Genet. 2017, 26, 2258–2276. [Google Scholar] [CrossRef]
- Lee, J.H.; Shin, H.Y.; Park, H.J.; Kim, S.H.; Kim, S.M.; Choi, Y.C. Clinical, Pathologic, and Genetic Features of Collagen VI-Related Myopathy in Korea. J. Clin. Neurol. 2017, 13, 331–339. [Google Scholar] [CrossRef]
- Tucker, W.S., Jr.; Hubbard, W.H.; Stryker, T.D.; Morgan, S.W.; Evans, O.B.; Freemon, F.R.; Theil, G.B. A new familial disorder of combined lower motor neuron degeneration and skeletal disorganization. Trans. Assoc. Am. Physicians 1982, 95, 126–134. [Google Scholar]
- Kimonis, V.E.; Kovach, M.J.; Waggoner, B.; Leal, S.; Salam, A.; Rimer, L.; Davis, K.; Khardori, R.; Gelber, D. Clinical and molecular studies in a unique family with autosomal dominant limb-girdle muscular dystrophy and Paget disease of bone. Genet. Med. 2000, 2, 232–241. [Google Scholar] [CrossRef]
- Kovach, M.J.; Waggoner, B.; Leal, S.M.; Gelber, D.; Khardori, R.; Levenstien, M.A.; Shanks, C.A.; Gregg, G.; Al-Lozi, M.T.; Miller, T.; et al. Clinical delineation and localization to chromosome 9p13.3–p12 of a unique dominant disorder in four families: Hereditary inclusion body myopathy, Paget disease of bone, and frontotemporal dementia. Mol. Genet. Metab. 2001, 74, 458–475. [Google Scholar] [CrossRef]
- Brody, M.J.; Vanhoutte, D.; Bakshi, C.V.; Liu, R.; Correll, R.N.; Sargent, M.A.; Molkentin, J.D. Disruption of valosin-containing protein activity causes cardiomyopathy and reveals pleiotropic functions in cardiac homeostasis. J. Biol. Chem. 2019, 294, 8918–8929. [Google Scholar] [CrossRef]
- Grossman, T.R.; Gamliel, A.; Wessells, R.J.; Taghli-Lamallem, O.; Jepsen, K.; Ocorr, K.; Korenberg, J.R.; Peterson, K.L.; Rosenfeld, M.G.; Bodmer, R.; et al. Over-expression of DSCAM and COL6A2 cooperatively generates congenital heart defects. PLoS Genet. 2011, 7, e1002344. [Google Scholar] [CrossRef]
- Connell, P.S.; Jeewa, A.; Kearney, D.L.; Tunuguntla, H.; Denfield, S.W.; Allen, H.D.; Landstrom, A.P. A 14-year-old in heart failure with multiple cardiomyopathy variants illustrates a role for signal-to-noise analysis in gene test re-interpretation. Clin. Case Rep. 2019, 7, 211–217. [Google Scholar] [CrossRef]
- Jones, D.M.; Lopes, L.; Quinlivan, R.; Elliott, P.M.; Khanji, M.Y. Cardiac manifestations of McArdle disease. Eur. Heart J. 2019, 40, 397–398. [Google Scholar] [CrossRef]
- Brodehl, A.; Gaertner-Rommel, A.; Milting, H. Molecular insights into cardiomyopathies associated with desmin (DES) mutations. Biophys. Rev. 2018, 10, 983–1006. [Google Scholar] [CrossRef]


{kind=link}
| Variables | n = 14 |
|---|---|
| Age (year) | 44.00 ± 13.77 * |
| Female, n (%) | 7 (50) |
| African-American ethnicity, n (%) | 3 (21.42) |
| BMI kg/m2 | 31.02 ± 7.78 |
| BSA m2 | 2.1 ± 0.39 |
| Current smoker, n (%) | 3 (21.42) |
| Hypertension, n (%) | 6 (42.85) |
| Hyperlipidemia, n (%) | 3 (21.42) |
| Diabetes mellitus, n (%) | 3 (21.42) |
| Coronary artery disease, n (%) | 2 (14.28) |
| 1 | 2 | 3 | 4 | 5 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diagnosis | LGMD2L | LGMD2C | LGMDD5 | NM | CMT2A | CMT2M | HyperCKemia | LGMD2T | VCP myopathy | Unknown | Unknown | Unknown | Unknown |
| Baseline echocardiographic characteristics | |||||||||||||
| LAD/BSA | 1.6 | 1.6 | 1.53 | 2.28 a | 1.6 | 1.5 | 1.7 | 1.8 | 1.5 | NA | 1.6 | 1.64 | 1.3 |
| LVES ID mm | 26 | 24 | 22.3 a | 27.15 | 29 | 32 | 23 | 24.4 | 28.96 | 34.73 | 34 | 29.49 | 29 |
| LVED ID mm | 39 | 35 | 36.7 a | 39.94 | 45 | 48 | 41 | 37.9 | 49.04 | 53.17 | 50 | 42.68 | 44 |
| EF% | 70 | 60 | 77.5 | 60.72 | 74 | 62.5 | 74 | 62.5 | 71.59 | 63.38 | 59 | 58.88 | 72 |
| FS% | 33 | 31 | 39 | 32 | 36 | 34 | 43 | 36 | 41 | 35 | 31 | 31 | 34 |
| Longitudinal strain % | −20.96 | −12.9 | NA | NA | −18.6 | NA | −17.6 | NA | NA | NA | −21.05 | NA | NA |
| IVS ED mm | 13 a | 9.0 | 10.2 | 15.99 a | 9.0 | 7.0 | 0.7 | 8.1 | 11.32 a | 12.66 a | 14 a | 11.42 a | 12 a |
| LVPW ED mm | 12 a | 9.0 | 10.9 | 15.48 a | 7.0 | 9.0 | 8.0 | 9.2 | 10.68 | 11.44 a | 10 | 8.97 | 14 a |
| IVS/LVPW | 1.08 | 1.06 | 0.94 | 1.03 | 1.26 | 0.77 | 0.96 | 0.88 | 1.06 | 1.11 | 1.4 | 1.27 | 0.9 |
| DD grade | 1.0 | - | 1.0 | - | - | - | - | - | - | 1.0 | 1.0 | - | - |
| LVH/HCM/DCM | mild LVH | - | - | mod LVH | - | - | - | - | - | mild LVH | DCM | - | mild LVH |
| Doppler echocardiographic characteristics | |||||||||||||
| E/A | 1.0 | 1.26 | 0.7 | 1.11 | 1.44 | 1.2 | 2.23 | 1.89 | 1.27 | 0.65 | 0.72 | 2.43 | 1.0 |
| E/e’ | 14 | 8.5 | 6.45 | 8.75 | 6.0 | 6.7 | 4.72 | 6.4 | 6.1 | 4.6 | 8.17 | 6.5 | 6.0 |
| DT | 313 | 93 | 243 | 214 | 206 | 217 | 150 | 260 | 240 | 220 | 150 | 264 a | 109 |
| AV peak velocity cm/s | 130 | 90 | NA | 117.86 | 110 | 118 | 100 | 125.16 | 98.72 | 118.22 | 120 | 130.94 | 80 |
| AV mean velocity cm/s | 84 a | NA | NA | 64.7 a | NA | NA | NA | NA | 73.04 a | 87.64 | NA | NA | NA |
| PV peak velocity cm/s | 100 | 100 | NA | NA | 100 | 52 | 100 | NA | 88.79 a | 98.31 a | 100 | 52.01 a | 200 |
| AR | mild | - | - | - | - | - | - | trivial | - | - | - | - | - |
| MR | mild | - | - | trivial | - | - | - | trivial | - | - | - | trivial | - |
| TR | mild | - | - | trivial | - | - | - | trivial | trivial | - | trivial | - | - |
| PR | mild | - | - | - | - | - | - | trivial | - | - | - | - | trivial |
| Diagnosis | Lab | Variant 1 | Variant 2 | Variant 3 | Variant 4 | Variant 5 | Variant 6 | Variant 7 | |
|---|---|---|---|---|---|---|---|---|---|
| 1 | LGMD2L | EGL Genetics | ANO5 | ANO5 | |||||
| Exon 5 | Exon 8 | ||||||||
| c.191dupA | c.692G>T | ||||||||
| MAF: dupA = 0.001096 | T = 0.00102 | ||||||||
| p.N64KfsX15 | p.Gly231Val | ||||||||
| Duplication, Het | Missense, Het | ||||||||
| Pathogenic | Pathogenic | ||||||||
| MIM # 608662 | |||||||||
| 2 | LGMD2C | EGL Genetics | SGCG | SGCG | |||||
| IVS 2 | Exon 5 | ||||||||
| c.195+4_195+7delAGTA | c.452_458delTTACTGT | ||||||||
| MAF: delAGTA = 0.000008 | NA | ||||||||
| - | p.Leu150_Phe151insTer | ||||||||
| Deletion, Het | Deletion, Het | ||||||||
| Pathogenic | Pathogenic | ||||||||
| MIM # 608896 | |||||||||
| 3 | LGMDD5 | EGL Genetics | COL6A1 | SGCA | SYNE1 | ||||
| Exon 12 | Exon 2 | Exon 92 | |||||||
| c.956A>G | c.155T>G | c.17342G>A | |||||||
| MAF: NA | G = 0.000036 | NA | |||||||
| p.Lys319Arg | p.Val52Gly | p.Arg5781His | |||||||
| Missense, Het | Missense, Het | Missense, Het | |||||||
| VUS | VUS | VUS | |||||||
| MIM # 120220 | MIM # 600119 | MIM # 608441 | |||||||
| 4 | NM | EGL Genetics & Invitae | COL6A3 | FKTN | CACNA1S | CCDC78 | LRP4 | PLEC | SUN1 |
| Exon 4 | 5′UTR | Exon 26 | Exon 4 | Exon 38 | Exon 32 | Exon 1 | |||
| c.1214T>C | c.-7C>G | c.3398T>C | c.416C>T | c.5417_5419delAGA | c.4666C>T | c.13C>T | |||
| MAF: G = 0.00080 | G = 0.000004 | NA | NA | delTCT = 0.000004 | NA | T = 0.000032 | |||
| p.Phe405Ser | - | p.Ile1133Thr | p.Ser139Phe | p.Lys1806del | p.Arg1556Cys | p.Arg5Trp | |||
| Missense, Het | Missense, Het | Missense, Het | Missense, Het | Deletion | Missense, Het | Missense, Het | |||
| VUS | VUS | VUS | VUS | VUS | VUS | VUS | |||
| MIM # 120250 | MIM # 607440 | MIM # 114208 | MIM # 614666 | MIM # 604270 | MIM # 601282 | MIM # 607723 | |||
| 5 | CMT2A | Invitae | MFN2 | PLEKHG5 | |||||
| Exon 9 | Exon 5 | ||||||||
| c.839G>A | c.274G>A | ||||||||
| A = 0.000004 | T = 0.000049 | ||||||||
| p.Arg280His | p.Val92Ile | ||||||||
| Missense, Het | Missense, Het | ||||||||
| Pathogenic | VUS | ||||||||
| MIM # 608507 | MIM # 611101 | ||||||||
| 6 | Peripheral polyneuropathy | EGL Genetics | SMCHD1 | SYNE1 | GAA | ||||
| Exon 28 | Exon 91 | Exon 10 | |||||||
| c.3596T>C | c.17140G>A | c.1482A>G | |||||||
| MAF: NA | NA | G = 0.000080 | |||||||
| p.Val1199Ala | p.Ala5714Thr | p.Thr494= | |||||||
| Missense, Het | Missense, Het | Silent | |||||||
| VUS | VUS | VUS | |||||||
| MIM # 614982 | MIM # 608441 | MIM # 606800 | |||||||
| 7 | CMT2M | Invitae | DNM2 | DNMT1 | VRK1 | ||||
| Exon 21 | Exon 14 | Exon 2 | |||||||
| c.2576_2578delCCA | c.1043C>T | c.8G>A | |||||||
| MAF: NA | A = 0.000032 | NA | |||||||
| p.Thr859del | p.Pro348Leu | p.Arg3His | |||||||
| Deletion, Het | Missense, Het | Missense, Het | |||||||
| VUS | VUS | VUS | |||||||
| MIM # 602378 | MIM # 126375 | MIM # 602168 | |||||||
| 8 | HyperCKemia | EGL Genetics | COL6A1 | ||||||
| IVS 18 | |||||||||
| c.1273-4T>C | |||||||||
| MAF: C = 0.000082 | |||||||||
| - | |||||||||
| Missense, Het | |||||||||
| VUS | |||||||||
| MIM # 120220 | |||||||||
| 9 | LGMD2T | EGL Genetics | AMPD1 | AMPD1 | TTN | NPHP1 | GMPPB | ||
| Exon 7 | Exon 11 | Exon 206 | Exon 1-20 | Report not available | |||||
| c.959A>T | c.1531.A>G | c.40508C>T | Deletion | ||||||
| MAF: A = 0.028281 | C = 0.000016 | NA | NA | ||||||
| p.Lys320Ile | p.Met511Val | p.Ser13503Leu | - | ||||||
| Missense, Het | Missense, Het | Missense, Het | Deletion, Het | ||||||
| Pathogenic | VUS | VUS | Pathogenic | ||||||
| MIM # 102770 | MIM # 188840 | MIM # 607100 | |||||||
| 10 | VCP myopathy | EGL Genetics | VCP | COL6A2 | SYNE2 | PYGM | TPM2 | ||
| Exon 5 | IVS 25 | Exon 96 | Exon 5 | Exon 5 | |||||
| c.572G>A | c.1970-3C>A | c.17539G>A | c.580C>T | c.536C>T | |||||
| MAF: T = 0.000016 | A = 0.00060 | A = 0.000096 | NA | A = 0.000052 | |||||
| p.Arg191Gln | - | p.Glu5847Lys | p.Arg194Trp | p.Ser179Leu | |||||
| Missense, Het | Missense, Het | Missense, Het | Missense, Het | Missense, Het | |||||
| Pathogenic | VUS | VUS | VUS | VUS | |||||
| MIM # 601023 | MIM # 120240 | MIM # 608442 | MIM # 608455 | MIM # 190990 | |||||
| 11 | Unknown | EGL Genetics | DYSF | ||||||
| Exon 30 | |||||||||
| c.3213C>T | |||||||||
| MAF: T = 0.000347 | |||||||||
| p.Tyr1071= | |||||||||
| Missense, Het | |||||||||
| VUS | |||||||||
| MIM # 603,009 | |||||||||
| 12 | Unknown | EGL Genetics | SMCHD1 | ||||||
| Exon 38 | |||||||||
| c.4787G>A | |||||||||
| MAF: A = 0.000017 | |||||||||
| p.Arg1596Gln | |||||||||
| Missense, Het | |||||||||
| VUS | |||||||||
| MIM # 614982 | |||||||||
| 13 | Unknown | EGL Genetics | PLEC | PLEC | SGCB | TTN | |||
| Exon 32 | Exon 32 | Exon 1 | Exon 224 | ||||||
| c.4642G>A | c.5412G>C | c.21_23dupGGC | c.44832C>G | ||||||
| MAF: T = 0.000086 | NA | dupCGC = 0.000462 | C = 0.000332 | ||||||
| p.Val1548Met | p.Glu1804Asp | p.Ala9_Glu10insAla | p.Asn14944Lys | ||||||
| Missense, Het | Missense, Het | Duplication, Het | Missense, Het | ||||||
| VUS | VUS | VUS | VUS | ||||||
| MIM # 601282 | MIM # 600900 | MIM # 188840 | |||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bazrafshan, S.; Kushlaf, H.; Kakroo, M.; Quinlan, J.; Becker, R.C.; Sadayappan, S. Genetic Modifiers of Hereditary Neuromuscular Disorders and Cardiomyopathy. Cells 2021, 10, 349. https://doi.org/10.3390/cells10020349
Bazrafshan S, Kushlaf H, Kakroo M, Quinlan J, Becker RC, Sadayappan S. Genetic Modifiers of Hereditary Neuromuscular Disorders and Cardiomyopathy. Cells. 2021; 10(2):349. https://doi.org/10.3390/cells10020349
Chicago/Turabian StyleBazrafshan, Sholeh, Hani Kushlaf, Mashhood Kakroo, John Quinlan, Richard C. Becker, and Sakthivel Sadayappan. 2021. "Genetic Modifiers of Hereditary Neuromuscular Disorders and Cardiomyopathy" Cells 10, no. 2: 349. https://doi.org/10.3390/cells10020349
APA StyleBazrafshan, S., Kushlaf, H., Kakroo, M., Quinlan, J., Becker, R. C., & Sadayappan, S. (2021). Genetic Modifiers of Hereditary Neuromuscular Disorders and Cardiomyopathy. Cells, 10(2), 349. https://doi.org/10.3390/cells10020349