Interaction between Parkin and α-Synuclein in PARK2-Mediated Parkinson’s Disease




Abstract
1. Introduction
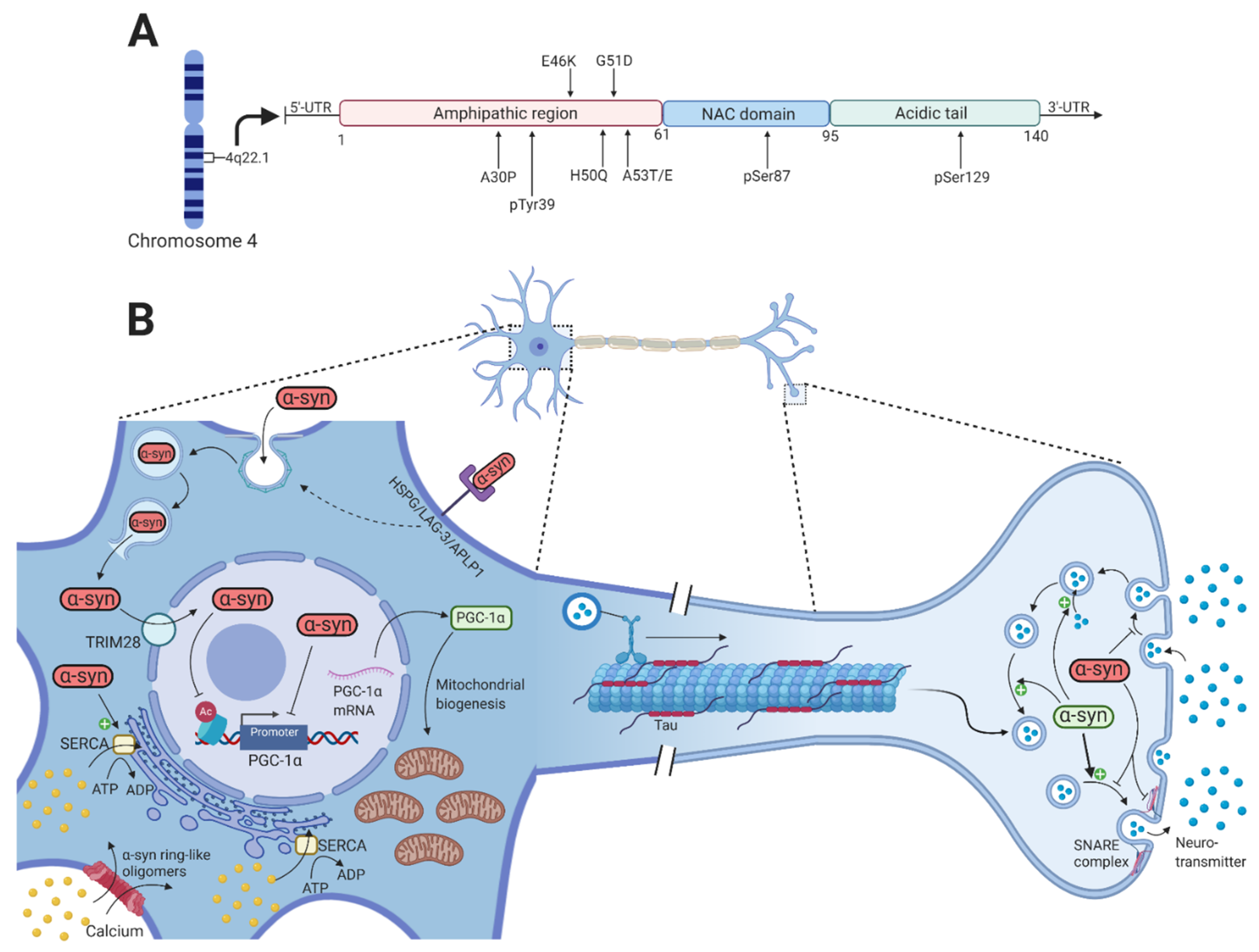
2. α-Synuclein
2.1. α-Synuclein Propagation and Seeding
2.2. α-Synuclein-Mediated Toxicity
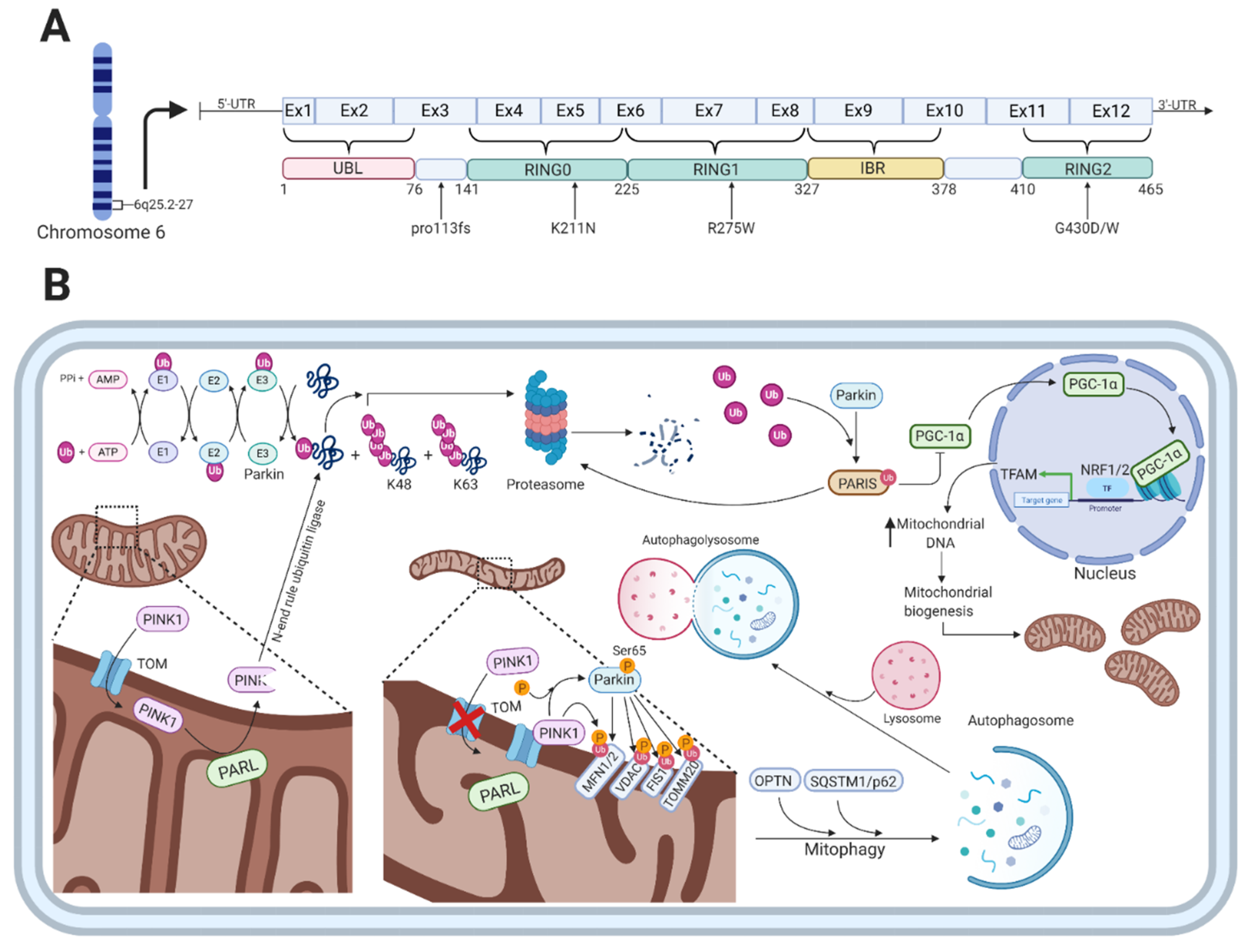
3. Parkin
Parkin Function
4. Lewy Body Pathology in PARK2-Related PD
4.1. What Could Be the Reason for the Ambiguous Post-Mortem Results?
4.2. Are Lewy Bodies Neuroprotective or Not?
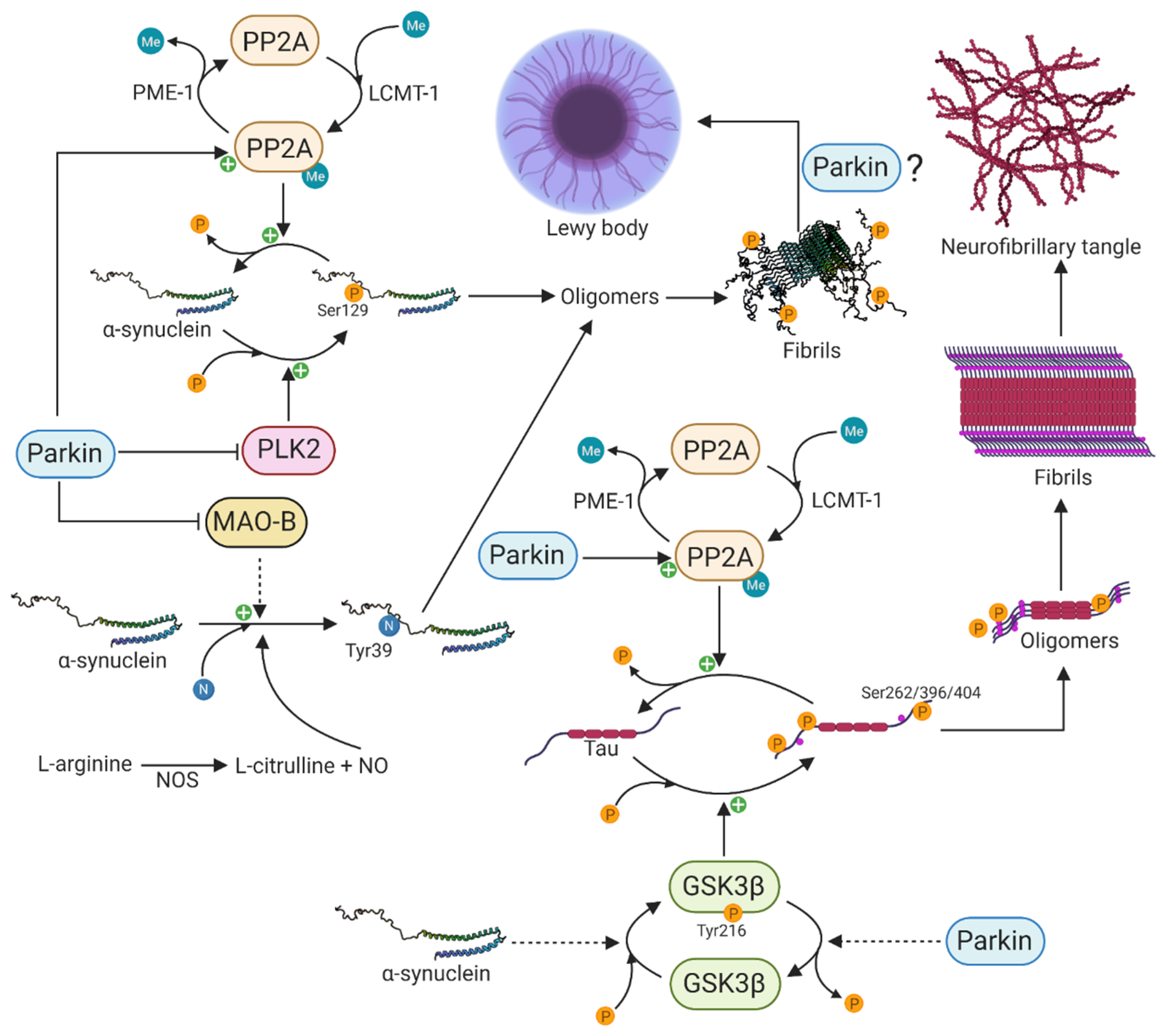
5. The Functional Interaction between Parkin and α-Synuclein
5.1. Parkin Influences Posttranslational Modifications of α-Synuclein
5.2. Parkin Function in α-Synuclein-Mediated Tau Pathology
5.3. The Regulation of Apoptosis by Parkin and α-Synuclein
5.4. Micro-Aggregates Instead of Lewy Bodies?
6. Concluding Remarks and Future Directions
Funding
Acknowledgments
Conflicts of Interest
References
- Balestrino, R.; Schapira, A.H.V. Parkinson disease. Eur. J. Neurol. 2020, 27, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Henderson, M.X.; Trojanowski, J.Q.; Lee, V.M.Y. α-Synuclein pathology in Parkinson’s disease and related α-synucleinopathies. Neurosci. Lett. 2019, 709, 134316. [Google Scholar] [CrossRef] [PubMed]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef]
- Lee, A.; Gilbert, R.M. Epidemiology of Parkinson Disease. Neurol. Clin. 2016, 34, 955–965. [Google Scholar] [CrossRef]
- Draoui, A.; El Hiba, O.; Aimrane, A.; El Khiat, A.; Gamrani, H. Parkinson’s disease: From bench to bedside. Rev. Neurol. (Paris) 2020, 176, 543–559. [Google Scholar] [CrossRef]
- Lunati, A.; Lesage, S.; Brice, A. The genetic landscape of Parkinson’s disease. Rev. Neurol. (Paris) 2018, 174, 628–643. [Google Scholar] [CrossRef]
- Del Rey, N.L.-G.; Quiroga-Varela, A.; Garbayo, E.; Carballo-Carbajal, I.; Fernández-Santiago, R.; Monje, M.H.G.; Trigo-Damas, I.; Blanco-Prieto, M.J.; Blesa, J. Advances in Parkinson’s disease: 200 years later. Front. Neuroanat. 2018, 12, 113. [Google Scholar] [CrossRef]
- Barodia, S.K.; Creed, R.B.; Goldberg, M.S. Parkin and PINK1 functions in oxidative stress and neurodegeneration. Brain Res. Bull. 2017, 133, 51–59. [Google Scholar] [CrossRef]
- Rodrigues e Silva, A.M.; Geldsetzer, F.; Holdorff, B.; Kielhorn, F.W.; Balzer-Geldsetzer, M.; Oertel, W.H.; Hurtig, H.; Dodel, R. Who was the man who discovered the “Lewy bodies”? Mov. Disord. 2010, 25, 1765–1773. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Shahmoradian, S.H.; Lewis, A.J.; Genoud, C.; Hench, J.; Moors, T.E.; Navarro, P.P.; Castaño-Díez, D.; Schweighauser, G.; Graff-Meyer, A.; Goldie, K.N.; et al. Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat. Neurosci. 2019, 22, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Savitt, J.M.; Dawson, V.L.; Dawson, T.M. Diagnosis and treatment of Parkinson disease: Molecules to medicine. J. Clin. Investig. 2006, 116, 1744–1754. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Pu, J. Alpha-synuclein in Parkinson’s disease: From pathogenetic dysfunction to potential clinical application. Parkinson’s Dis. 2016, 2016, 1720621. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Watanabe, Y.; Tsujimura, A.; Tanaka, M. Expression of α-synuclein is regulated in a neuronal cell type-dependent manner. Anat. Sci. Int. 2019, 94, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Beach, T.G.; Adler, C.H.; Sue, L.I.; Vedders, L.; Lue, L.; White Iii, C.L.; Akiyama, H.; Caviness, J.N.; Shill, H.A.; Sabbagh, M.N.; et al. Multi-organ distribution of phosphorylated alpha-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol. 2010, 119, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Fumimura, Y.; Ikemura, M.; Saito, Y.; Sengoku, R.; Kanemaru, K.; Sawabe, M.; Arai, T.; Ito, G.; Iwatsubo, T.; Fukayama, M.; et al. Analysis of the adrenal gland is useful for evaluating pathology of the peripheral autonomic nervous system in lewy body disease. J. Neuropathol. Exp. Neurol. 2007, 66, 354–362. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Mori, F.; Tanji, K.; Orimo, S.; Takahashi, H. Involvement of the peripheral nervous system in synucleinopathies, tauopathies and other neurodegenerative proteinopathies of the brain. Acta Neuropathol. 2010, 120, 1–12. [Google Scholar] [CrossRef]
- Del Tredici, K.; Hawkes, C.H.; Ghebremedhin, E.; Braak, H. Lewy pathology in the submandibular gland of individuals with incidental Lewy body disease and sporadic Parkinson’s disease. Acta Neuropathol. 2010, 119, 703–713. [Google Scholar] [CrossRef]
- Cersosimo, M.G. Gastrointestinal biopsies for the diagnosis of alpha-synuclein pathology in Parkinson’s disease. Gastroenterol. Res. Pract. 2015, 2015, 476041. [Google Scholar] [CrossRef]
- Emamzadeh, F.N. Alpha-synuclein structure, functions, and interactions. J. Res. Med. Sci. 2016, 21, 29. [Google Scholar] [CrossRef] [PubMed]
- Dehay, B.; Bourdenx, M.; Gorry, P.; Przedborski, S.; Vila, M.; Hunot, S.; Singleton, A.; Olanow, C.W.; Merchant, K.M.; Bezard, E.; et al. Targeting α-synuclein for treatment of Parkinson’s disease: Mechanistic and therapeutic considerations. Lancet Neurol. 2015, 14, 855–866. [Google Scholar] [CrossRef]
- Waxman, E.A.; Mazzulli, J.R.; Giasson, B.I. Characterization of hydrophobic residue requirements for alpha-synuclein fibrillization. Biochemistry 2009, 48, 9427–9436. [Google Scholar] [CrossRef] [PubMed]
- Mehra, S.; Sahay, S.; Maji, S.K. α-Synuclein misfolding and aggregation: Implications in Parkinson’s disease pathogenesis. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 890–908. [Google Scholar] [CrossRef] [PubMed]
- Allen Reish, H.E.; Standaert, D.G. Role of α-synuclein in inducing innate and adaptive immunity in Parkinson disease. J. Parkinsons’ Dis. 2015, 5, 1–19. [Google Scholar] [CrossRef]
- Kahle, P.J.; Neumann, M.; Ozmen, L.; Muller, V.; Jacobsen, H.; Schindzielorz, A.; Okochi, M.; Leimer, U.; van Der Putten, H.; Probst, A.; et al. Subcellular localization of wild-type and Parkinson’s disease-associated mutant alpha -synuclein in human and transgenic mouse brain. J. Neurosci. 2000, 20, 6365–6373. [Google Scholar] [CrossRef]
- Davidson, W.S.; Jonas, A.; Clayton, D.F.; George, J.M. Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J. Biol. Chem. 1998, 273, 9443–9449. [Google Scholar] [CrossRef]
- Varkey, J.; Isas, J.M.; Mizuno, N.; Jensen, M.B.; Bhatia, V.K.; Jao, C.C.; Petrlova, J.; Voss, J.C.; Stamou, D.G.; Steven, A.C.; et al. Membrane curvature induction and tubulation are common features of synucleins and apolipoproteins. J. Biol. Chem. 2010, 285, 32486–32493. [Google Scholar] [CrossRef]
- Burré, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Südhof, T.C. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef]
- Gould, N.; Mor, D.E.; Lightfoot, R.; Malkus, K.; Giasson, B.; Ischiropoulos, H. Evidence of native α-synuclein conformers in the human brain. J. Biol. Chem. 2014, 289, 7929–7934. [Google Scholar] [CrossRef]
- Bartels, T.; Choi, J.G.; Selkoe, D.J. α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 2011, 477, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Dettmer, U.; Newman, A.J.; Soldner, F.; Luth, E.S.; Kim, N.C.; von Saucken, V.E.; Sanderson, J.B.; Jaenisch, R.; Bartels, T.; Selkoe, D. Parkinson-causing α-synuclein missense mutations shift native tetramers to monomers as a mechanism for disease initiation. Nat. Commun. 2015, 6, 7314. [Google Scholar] [CrossRef] [PubMed]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed]
- Krüger, R.; Kuhn, W.; Müller, T.; Woitalla, D.; Graeber, M.; Kösel, S.; Przuntek, H.; Epplen, J.T.; Schöls, L.; Riess, O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef] [PubMed]
- Zarranz, J.J.; Alegre, J.; Gómez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atarés, B.; et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 2004, 55, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Appel-Cresswell, S.; Vilarino-Guell, C.; Encarnacion, M.; Sherman, H.; Yu, I.; Shah, B.; Weir, D.; Thompson, C.; Szu-Tu, C.; Trinh, J.; et al. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov. Disord. 2013, 28, 811–813. [Google Scholar] [CrossRef]
- Proukakis, C.; Dudzik, C.G.; Brier, T.; MacKay, D.S.; Cooper, J.M.; Millhauser, G.L.; Houlden, H.; Schapira, A.H. A novel α-synuclein missense mutation in Parkinson disease. Neurology 2013, 80, 1062–1064. [Google Scholar] [CrossRef]
- Lesage, S.; Anheim, M.; Letournel, F.; Bousset, L.; Honoré, A.; Rozas, N.; Pieri, L.; Madiona, K.; Dürr, A.; Melki, R.; et al. G51D α-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann. Neurol. 2013, 73, 459–471. [Google Scholar] [CrossRef]
- Pasanen, P.; Myllykangas, L.; Siitonen, M.; Raunio, A.; Kaakkola, S.; Lyytinen, J.; Tienari, P.J.; Pöyhönen, M.; Paetau, A. Novel α-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol. Aging 2014, 35, 2180.e1–2180.e5. [Google Scholar] [CrossRef]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. Alpha-synuclein locus triplication causes Parkinson’s disease. Science 2003, 302, 841. [Google Scholar] [CrossRef]
- Chartier-Harlin, M.-C.; Kachergus, J.; Roumier, C.; Mouroux, V.; Douay, X.; Lincoln, S.; Levecque, C.; Larvor, L.; Andrieux, J.; Hulihan, M.; et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 2004, 364, 1167–1169. [Google Scholar] [CrossRef]
- Iwatsubo, T. Pathological biochemistry of alpha-synucleinopathy. Neuropathology 2007, 27, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Gallegos, S.; Pacheco, C.; Peters, C.; Opazo, C.M.; Aguayo, L.G. Features of alpha-synuclein that could explain the progression and irreversibility of Parkinson’s disease. Front. Neurosci. 2015, 9, 59. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Li, J.Y.; Englund, E.; Holton, J.L.; Soulet, D.; Hagell, P.; Lees, A.J.; Lashley, T.; Quinn, N.P.; Rehncrona, S.; Bjorklund, A.; et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat. Med. 2008, 14, 501–503. [Google Scholar] [CrossRef]
- Kordower, J.H.; Chu, Y.; Hauser, R.A.; Freeman, T.B.; Olanow, C.W. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat. Med. 2008, 14, 504–506. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Patel, S.; Lee, S.J. Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J. Neurosci. 2005, 25, 6016–6024. [Google Scholar] [CrossRef]
- Danzer, K.M.; Kranich, L.R.; Ruf, W.P.; Cagsal-Getkin, O.; Winslow, A.R.; Zhu, L.; Vanderburg, C.R.; McLean, P.J. Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol. Neurodegener. 2012, 7, 42. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Seow, Y.; Schapira, A.H.; Gardiner, C.; Sargent, I.L.; Wood, M.J.; Cooper, J.M. Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol. Dis. 2011, 42, 360–367. [Google Scholar] [CrossRef]
- Emmanouilidou, E.; Melachroinou, K.; Roumeliotis, T.; Garbis, S.D.; Ntzouni, M.; Margaritis, L.H.; Stefanis, L.; Vekrellis, K. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J. Neurosci. 2010, 30, 6838–6851. [Google Scholar] [CrossRef]
- Tsigelny, I.F.; Sharikov, Y.; Wrasidlo, W.; Gonzalez, T.; Desplats, P.A.; Crews, L.; Spencer, B.; Masliah, E. Role of α-synuclein penetration into the membrane in the mechanisms of oligomer pore formation. FEBS J. 2012, 279, 1000–1013. [Google Scholar] [CrossRef] [PubMed]
- Jang, A.; Lee, H.J.; Suk, J.E.; Jung, J.W.; Kim, K.P.; Lee, S.J. Non-classical exocytosis of alpha-synuclein is sensitive to folding states and promoted under stress conditions. J. Neurochem. 2010, 113, 1263–1274. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, L.; Marano, M.M.; Tandon, A. Import and export of misfolded α-synuclein. Front. Neurosci. 2018, 12, 344. [Google Scholar] [CrossRef] [PubMed]
- Abounit, S.; Bousset, L.; Loria, F.; Zhu, S.; de Chaumont, F.; Pieri, L.; Olivo-Marin, J.C.; Melki, R.; Zurzolo, C. Tunneling nanotubes spread fibrillar α-synuclein by intercellular trafficking of lysosomes. EMBO J. 2016, 35, 2120–2138. [Google Scholar] [CrossRef]
- Flavin, W.P.; Bousset, L.; Green, Z.C.; Chu, Y.; Skarpathiotis, S.; Chaney, M.J.; Kordower, J.H.; Melki, R.; Campbell, E.M. Endocytic vesicle rupture is a conserved mechanism of cellular invasion by amyloid proteins. Acta Neuropathol. 2017, 134, 629–653. [Google Scholar] [CrossRef]
- Holmes, B.B.; DeVos, S.L.; Kfoury, N.; Li, M.; Jacks, R.; Yanamandra, K.; Ouidja, M.O.; Brodsky, F.M.; Marasa, J.; Bagchi, D.P.; et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. USA 2013, 110, E3138–E3147. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Ou, M.T.; Karuppagounder, S.S.; Kam, T.I.; Yin, X.; Xiong, Y.; Ge, P.; Umanah, G.E.; Brahmachari, S.; Shin, J.H.; et al. Pathological α-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 2016, 353. [Google Scholar] [CrossRef]
- Luk, K.C.; Song, C.; O’Brien, P.; Stieber, A.; Branch, J.R.; Brunden, K.R.; Trojanowski, J.Q.; Lee, V.M. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc. Natl. Acad. Sci. USA 2009, 106, 20051–20056. [Google Scholar] [CrossRef]
- Volpicelli-Daley, L.A.; Luk, K.C.; Patel, T.P.; Tanik, S.A.; Riddle, D.M.; Stieber, A.; Meaney, D.F.; Trojanowski, J.Q.; Lee, V.M. Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 2011, 72, 57–71. [Google Scholar] [CrossRef]
- Wong, Y.C.; Krainc, D. α-synuclein toxicity in neurodegeneration: Mechanism and therapeutic strategies. Nat. Med. 2017, 23, 1–13. [Google Scholar] [CrossRef]
- Choi, B.K.; Choi, M.G.; Kim, J.Y.; Yang, Y.; Lai, Y.; Kweon, D.H.; Lee, N.K.; Shin, Y.K. Large α-synuclein oligomers inhibit neuronal SNARE-mediated vesicle docking. Proc. Natl. Acad. Sci. USA 2013, 110, 4087–4092. [Google Scholar] [CrossRef]
- Nemani, V.M.; Lu, W.; Berge, V.; Nakamura, K.; Onoa, B.; Lee, M.K.; Chaudhry, F.A.; Nicoll, R.A.; Edwards, R.H. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron 2010, 65, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Maroteaux, L.; Campanelli, J.T.; Scheller, R.H. Synuclein: A neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 1988, 8, 2804–2815. [Google Scholar] [CrossRef]
- Xu, S.; Zhou, M.; Yu, S.; Cai, Y.; Zhang, A.; Uéda, K.; Chan, P. Oxidative stress induces nuclear translocation of C-terminus of alpha-synuclein in dopaminergic cells. Biochem. Biophys. Res. Commun. 2006, 342, 330–335. [Google Scholar] [CrossRef] [PubMed]
- Schell, H.; Hasegawa, T.; Neumann, M.; Kahle, P.J. Nuclear and neuritic distribution of serine-129 phosphorylated alpha-synuclein in transgenic mice. Neuroscience 2009, 160, 796–804. [Google Scholar] [CrossRef] [PubMed]
- Kontopoulos, E.; Parvin, J.D.; Feany, M.B. Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum. Mol. Genet. 2006, 15, 3012–3023. [Google Scholar] [CrossRef]
- Rousseaux, M.W.; de Haro, M.; Lasagna-Reeves, C.A.; De Maio, A.; Park, J.; Jafar-Nejad, P.; Al-Ramahi, I.; Sharma, A.; See, L.; Lu, N.; et al. TRIM28 regulates the nuclear accumulation and toxicity of both alpha-synuclein and tau. eLife 2016, 5. [Google Scholar] [CrossRef]
- Siddiqui, A.; Chinta, S.J.; Mallajosyula, J.K.; Rajagopolan, S.; Hanson, I.; Rane, A.; Melov, S.; Andersen, J.K. Selective binding of nuclear alpha-synuclein to the PGC1alpha promoter under conditions of oxidative stress may contribute to losses in mitochondrial function: Implications for Parkinson’s disease. Free Radic. Biol. Med. 2012, 53, 993–1003. [Google Scholar] [CrossRef]
- Eschbach, J.; von Einem, B.; Müller, K.; Bayer, H.; Scheffold, A.; Morrison, B.E.; Rudolph, K.L.; Thal, D.R.; Witting, A.; Weydt, P.; et al. Mutual exacerbation of peroxisome proliferator-activated receptor γ coactivator 1α deregulation and α-synuclein oligomerization. Ann. Neurol. 2015, 77, 15–32. [Google Scholar] [CrossRef]
- Luth, E.S.; Stavrovskaya, I.G.; Bartels, T.; Kristal, B.S.; Selkoe, D.J. Soluble, prefibrillar α-synuclein oligomers promote complex I-dependent, Ca2+-induced mitochondrial dysfunction. J. Biol. Chem. 2014, 289, 21490–21507. [Google Scholar] [CrossRef]
- Zaichick, S.V.; McGrath, K.M.; Caraveo, G. The role of Ca2+ signaling in Parkinson’s disease. Dis. Models Mech. 2017, 10, 519–535. [Google Scholar] [CrossRef] [PubMed]
- Betzer, C.; Lassen, L.B.; Olsen, A.; Kofoed, R.H.; Reimer, L.; Gregersen, E.; Zheng, J.; Cali, T.; Gai, W.P.; Chen, T.; et al. Alpha-synuclein aggregates activate calcium pump SERCA leading to calcium dysregulation. EMBO Rep. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Caraveo, G.; Auluck, P.K.; Whitesell, L.; Chung, C.Y.; Baru, V.; Mosharov, E.V.; Yan, X.; Ben-Johny, M.; Soste, M.; Picotti, P.; et al. Calcineurin determines toxic versus beneficial responses to α-synuclein. Proc. Natl. Acad. Sci. USA 2014, 111, E3544–E3552. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Sun, L.; Lin, X.; Liu, G.; Yu, J.; Parisiadou, L.; Xie, C.; Ding, J.; Cai, H. A calcineurin- and NFAT-dependent pathway is involved in α-synuclein-induced degeneration of midbrain dopaminergic neurons. Hum. Mol. Genet. 2014, 23, 6567–6574. [Google Scholar] [CrossRef] [PubMed]
- Volles, M.J.; Lansbury, P.T., Jr. Vesicle permeabilization by protofibrillar alpha-synuclein is sensitive to Parkinson’s disease-linked mutations and occurs by a pore-like mechanism. Biochemistry 2002, 41, 4595–4602. [Google Scholar] [CrossRef] [PubMed]
- Van Rooijen, B.D.; Claessens, M.M.; Subramaniam, V. Membrane permeabilization by oligomeric α-synuclein: In search of the mechanism. PLoS ONE 2010, 5, e14292. [Google Scholar] [CrossRef] [PubMed]
- Mazzulli, J.R.; Xu, Y.H.; Sun, Y.; Knight, A.L.; McLean, P.J.; Caldwell, G.A.; Sidransky, E.; Grabowski, G.A.; Krainc, D. Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011, 146, 37–52. [Google Scholar] [CrossRef]
- Mazzulli, J.R.; Zunke, F.; Isacson, O.; Studer, L.; Krainc, D. α-Synuclein-induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proc. Natl. Acad. Sci. USA 2016, 113, 1931–1936. [Google Scholar] [CrossRef]
- Wong, Y.C.; Krainc, D. Lysosomal trafficking defects link Parkinson’s disease with Gaucher’s disease. Mov. Disord. 2016, 31, 1610–1618. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2018, 46, 2699. [CrossRef]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Hedrich, K.; Eskelson, C.; Wilmot, B.; Marder, K.; Harris, J.; Garrels, J.; Meija-Santana, H.; Vieregge, P.; Jacobs, H.; Bressman, S.B.; et al. Distribution, type, and origin of Parkin mutations: Review and case studies. Mov. Disord. 2004, 19, 1146–1157. [Google Scholar] [CrossRef] [PubMed]
- Giguère, N.; Pacelli, C.; Saumure, C.; Bourque, M.-J.; Matheoud, D.; Levesque, D.; Slack, R.S.; Park, D.S.; Trudeau, L.-É. Comparative analysis of Parkinson’s disease-associated genes in mice reveals altered survival and bioenergetics of Parkin-deficient dopamine neurons. J. Biol. Chem. 2018, 293, 9580–9593. [Google Scholar] [CrossRef] [PubMed]
- Geisler, S.; Holmstrom, K.M.; Treis, A.; Skujat, D.; Weber, S.S.; Fiesel, F.C.; Kahle, P.J.; Springer, W. The PINK1/Parkin-mediated mitophagy is compromised by PD-associated mutations. Autophagy 2010, 6, 871–878. [Google Scholar] [CrossRef]
- Spratt, D.E.; Walden, H.; Shaw, G.S. RBR E3 ubiquitin ligases: New structures, new insights, new questions. Biochem. J. 2014, 458, 421–437. [Google Scholar] [CrossRef]
- Sarraf, S.A.; Raman, M.; Guarani-Pereira, V.; Sowa, M.E.; Huttlin, E.L.; Gygi, S.P.; Harper, J.W. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 2013, 496, 372–376. [Google Scholar] [CrossRef]
- Hristova, V.A.; Beasley, S.A.; Rylett, R.J.; Shaw, G.S. Identification of a novel Zn2+-binding domain in the autosomal recessive juvenile Parkinson-related E3 ligase parkin. J. Biol. Chem. 2009, 284, 14978–14986. [Google Scholar] [CrossRef]
- Truban, D.; Hou, X.; Caulfield, T.R.; Fiesel, F.C.; Springer, W. PINK1, Parkin, and mitochondrial quality control: What can we learn about Parkinson’s disease pathobiology? J. Parkinson’s Dis. 2017, 7, 13–29. [Google Scholar] [CrossRef]
- Lesage, S.; Magali, P.; Lohmann, E.; Lacomblez, L.; Teive, H.; Janin, S.; Cousin, P.-Y.; Dürr, A.; Brice, A.; French Parkinson Disease Genetics Study, G. Deletion of the parkin and PACRG gene promoter in early-onset parkinsonism. Hum. Mutat. 2007, 28, 27–32. [Google Scholar] [CrossRef]
- Chung, K.K.K.; Thomas, B.; Li, X.; Pletnikova, O.; Troncoso, J.C.; Marsh, L.; Dawson, V.L.; Dawson, T.M. S-nitrosylation of parkin regulates ubiquitination and compromises parkin’s protective function. Science 2004, 304, 1328–1331. [Google Scholar] [CrossRef]
- Imam, S.Z.; Zhou, Q.; Yamamoto, A.; Valente, A.J.; Ali, S.F.; Bains, M.; Roberts, J.L.; Kahle, P.J.; Clark, R.A.; Li, S. Novel regulation of parkin function through c-Abl-mediated tyrosine phosphorylation: Implications for Parkinson’s disease. J. Neurosci. 2011, 31, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.S.; Lee, Y.; Shin, J.-H.; Karuppagounder, S.S.; Gadad, B.S.; Koleske, A.J.; Pletnikova, O.; Troncoso, J.C.; Dawson, V.L.; Dawson, T.M. Phosphorylation by the c-Abl protein tyrosine kinase inhibits parkin’s ubiquitination and protective function. Proc. Natl. Acad. Sci. USA 2010, 107, 16691–16696. [Google Scholar] [CrossRef] [PubMed]
- LaVoie, M.J.; Ostaszewski, B.L.; Weihofen, A.; Schlossmacher, M.G.; Selkoe, D.J. Dopamine covalently modifies and functionally inactivates parkin. Nat. Med. 2005, 11, 1214–1221. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Yao, D.; Shi, Y.; Kabakoff, J.; Wu, W.; Reicher, J.; Ma, Y.; Moosmann, B.; Masliah, E.; Lipton, S.A.; et al. Oxidation of the cysteine-rich regions of parkin perturbs its E3 ligase activity and contributes to protein aggregation. Mol. Neurodegener. 2011, 6, 34. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Ko, H.S.; Thomas, B.; Tsang, F.; Chew, K.C.M.; Tay, S.-P.; Ho, M.W.L.; Lim, T.-M.; Soong, T.-W.; Pletnikova, O.; et al. Stress-induced alterations in parkin solubility promote parkin aggregation and compromise parkin’s protective function. Hum. Mol. Genet. 2005, 14, 3885–3897. [Google Scholar] [CrossRef]
- Yao, D.; Gu, Z.; Nakamura, T.; Shi, Z.-Q.; Ma, Y.; Gaston, B.; Palmer, L.A.; Rockenstein, E.M.; Zhang, Z.; Masliah, E.; et al. Nitrosative stress linked to sporadic Parkinson’s disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proc. Natl. Acad. Sci. USA 2004, 101, 10810–10814. [Google Scholar] [CrossRef]
- Corti, O.; Lesage, S.; Brice, A. What genetics tells us about the causes and mechanisms of Parkinson’s disease. Physiol. Rev. 2011, 91, 1161–1218. [Google Scholar] [CrossRef]
- Lücking, C.B.; Dürr, A.; Bonifati, V.; Vaughan, J.; De Michele, G.; Gasser, T.; Harhangi, B.S.; Meco, G.; Denèfle, P.; Wood, N.W.; et al. Association between early-onset Parkinson’s disease and mutations in the parkin gene. N. Engl. J. Med. 2000, 342, 1560–1567. [Google Scholar] [CrossRef]
- Schulte, C.; Gasser, T. Genetic basis of Parkinson’s disease: Inheritance, penetrance, and expression. Appl. Clin. Genet. 2011, 4, 67–80. [Google Scholar] [CrossRef]
- Lohmann, E.; Thobois, S.; Lesage, S.; Broussolle, E.; du Montcel, S.T.; Ribeiro, M.J.; Remy, P.; Pelissolo, A.; Dubois, B.; Mallet, L.; et al. A multidisciplinary study of patients with early-onset PD with and without parkin mutations. Neurology 2009, 72, 110–116. [Google Scholar] [CrossRef]
- Mori, H.; Kondo, T.; Yokochi, M.; Matsumine, H.; Nakagawa-Hattori, Y.; Miyake, T.; Suda, K.; Mizuno, Y. Pathologic and biochemical studies of juvenile parkinsonism linked to chromosome 6q. Neurology 1998, 51, 890–892. [Google Scholar] [CrossRef] [PubMed]
- Wasner, K.; Grünewald, A.; Klein, C. Parkin-linked Parkinson’s disease: From clinical insights to pathogenic mechanisms and novel therapeutic approaches. Neurosci. Res. 2020, 159, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.L.; Graham, E.; Critchley, P.; Schrag, A.E.; Wood, N.W.; Lees, A.J.; Bhatia, K.P.; Quinn, N. Parkin disease: A phenotypic study of a large case series. Brain J. Neurol. 2003, 126, 1279–1292. [Google Scholar] [CrossRef] [PubMed]
- Hampe, C.; Ardila-Osorio, H.; Fournier, M.; Brice, A.; Corti, O. Biochemical analysis of Parkinson’s disease-causing variants of Parkin, an E3 ubiquitin-protein ligase with monoubiquitylation capacity. Hum. Mol. Genet. 2006, 15, 2059–2075. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, N.; Kitami, T.; Suzuki, T.; Mizuno, Y.; Hattori, N.; Tanaka, K. Diverse effects of pathogenic mutations of Parkin that catalyze multiple monoubiquitylation in vitro. J. Biol. Chem. 2006, 281, 3204–3209. [Google Scholar] [CrossRef] [PubMed]
- Doss-Pepe, E.W.; Chen, L.; Madura, K. Alpha-synuclein and parkin contribute to the assembly of ubiquitin lysine 63-linked multiubiquitin chains. J. Biol. Chem. 2005, 280, 16619–16624. [Google Scholar] [CrossRef] [PubMed]
- Chan, N.C.; Salazar, A.M.; Pham, A.H.; Sweredoski, M.J.; Kolawa, N.J.; Graham, R.L.J.; Hess, S.; Chan, D.C. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum. Mol. Genet. 2011, 20, 1726–1737. [Google Scholar] [CrossRef]
- Zheng, X.; Hunter, T. Parkin mitochondrial translocation is achieved through a novel catalytic activity coupled mechanism. Cell Res. 2013, 23, 886–897. [Google Scholar] [CrossRef]
- Seirafi, M.; Kozlov, G.; Gehring, K. Parkin structure and function. FEBS J. 2015, 282, 2076–2088. [Google Scholar] [CrossRef]
- Lim, K.L.; Tan, J.M. Role of the ubiquitin proteasome system in Parkinson’s disease. BMC Biochem. 2007, 8 (Suppl. S1), S13. [Google Scholar] [CrossRef]
- Bard, J.A.M.; Goodall, E.A.; Greene, E.R.; Jonsson, E.; Dong, K.C.; Martin, A. Structure and function of the 26S Proteasome. Annu. Rev. Biochem. 2018, 87, 697–724. [Google Scholar] [CrossRef] [PubMed]
- Geisler, S.; Holmström, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.-F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.; Tanaka, A.; Suen, D.-F.; Youle, R.J. Parkin-induced mitophagy in the pathogenesis of Parkinson disease. Autophagy 2009, 5, 706–708. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.-S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef]
- Harper, J.W.; Ordureau, A.; Heo, J.-M. Building and decoding ubiquitin chains for mitophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 93–108. [Google Scholar] [CrossRef]
- Becker, D.; Richter, J.; Tocilescu, M.A.; Przedborski, S.; Voos, W. Pink1 kinase and its membrane potential (Deltaψ)-dependent cleavage product both localize to outer mitochondrial membrane by unique targeting mode. J. Biol. Chem. 2012, 287, 22969–22987. [Google Scholar] [CrossRef]
- Yamano, K.; Youle, R.J. PINK1 is degraded through the N-end rule pathway. Autophagy 2013, 9, 1758–1769. [Google Scholar] [CrossRef]
- Greene, A.W.; Grenier, K.; Aguileta, M.A.; Muise, S.; Farazifard, R.; Haque, M.E.; McBride, H.M.; Park, D.S.; Fon, E.A. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 2012, 13, 378–385. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.-F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef]
- Kondapalli, C.; Kazlauskaite, A.; Zhang, N.; Woodroof, H.I.; Campbell, D.G.; Gourlay, R.; Burchell, L.; Walden, H.; Macartney, T.J.; Deak, M.; et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012, 2, 120080. [Google Scholar] [CrossRef] [PubMed]
- Shiba-Fukushima, K.; Imai, Y.; Yoshida, S.; Ishihama, Y.; Kanao, T.; Sato, S.; Hattori, N. PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci. Rep. 2012, 2, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Weil, R.; Laplantine, E.; Curic, S.; Génin, P. Role of optineurin in the mitochondrial dysfunction: Potential implications in neurodegenerative diseases and cancer. Front. Immunol. 2018, 9, 1243. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Davis, R.L.; Sue, C.M. Mitochondrial dysfunction in Parkinson’s disease: New mechanistic insights and therapeutic perspectives. Curr. Neurol. Neurosci. Rep. 2018, 18, 21. [Google Scholar] [CrossRef]
- Hang, L.; Thundyil, J.; Lim, K.-L. Mitochondrial dysfunction and Parkinson disease: A Parkin-AMPK alliance in neuroprotection. Ann. N. Y. Acad. Sci. 2015, 1350, 37–47. [Google Scholar] [CrossRef]
- Shin, J.-H.; Ko, H.S.; Kang, H.; Lee, Y.; Lee, Y.-I.; Pletinkova, O.; Troconso, J.C.; Dawson, V.L.; Dawson, T.M. PARIS (ZNF746) repression of PGC-1α contributes to neurodegeneration in Parkinson’s disease. Cell 2011, 144, 689–702. [Google Scholar] [CrossRef]
- Stevens, D.A.; Lee, Y.; Kang, H.C.; Lee, B.D.; Lee, Y.I.; Bower, A.; Jiang, H.; Kang, S.U.; Andrabi, S.A.; Dawson, V.L.; et al. Parkin loss leads to PARIS-dependent declines in mitochondrial mass and respiration. Proc. Natl. Acad. Sci. USA 2015, 112, 11696–11701. [Google Scholar] [CrossRef]
- Bogetofte, H.; Jensen, P.; Ryding, M.; Schmidt, S.I.; Okarmus, J.; Ritter, L.; Worm, C.S.; Hohnholt, M.C.; Azevedo, C.; Roybon, L.; et al. PARK2 mutation causes metabolic disturbances and impaired survival of human iPSC-derived neurons. Front. Cell. Neurosci. 2019, 13, 297. [Google Scholar] [CrossRef]
- Klein, C.; Lohmann-Hedrich, K.; Rogaeva, E.; Schlossmacher, M.G.; Lang, A.E. Deciphering the role of heterozygous mutations in genes associated with parkinsonism. Lancet Neurol. 2007, 6, 652–662. [Google Scholar] [CrossRef]
- Huttenlocher, J.; Stefansson, H.; Steinberg, S.; Helgadottir, H.T.; Sveinbjörnsdóttir, S.; Riess, O.; Bauer, P.; Stefansson, K. Heterozygote carriers for CNVs in PARK2 are at increased risk of Parkinson’s disease. Hum. Mol. Genet. 2015, 24, 5637–5643. [Google Scholar] [CrossRef]
- Farrer, M.; Chan, P.; Chen, R.; Tan, L.; Lincoln, S.; Hernandez, D.; Forno, L.; Gwinn-Hardy, K.; Petrucelli, L.; Hussey, J.; et al. Lewy bodies and parkinsonism in families with parkin mutations. Ann. Neurol. 2001, 50, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Pramstaller, P.P.; Schlossmacher, M.G.; Jacques, T.S.; Scaravilli, F.; Eskelson, C.; Pepivani, I.; Hedrich, K.; Adel, S.; Gonzales-McNeal, M.; Hilker, R.; et al. Lewy body Parkinson’s disease in a large pedigree with 77 Parkin mutation carriers. Ann. Neurol. 2005, 58, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Ruffmann, C.; Zini, M.; Goldwurm, S.; Bramerio, M.; Spinello, S.; Rusconi, D.; Gambacorta, M.; Tagliavini, F.; Pezzoli, G.; Giaccone, G. Lewy body pathology and typical Parkinson disease in a patient with a heterozygous (R275W) mutation in the Parkin gene (PARK2). Acta Neuropathol. 2012, 123, 901–903. [Google Scholar] [CrossRef] [PubMed]
- Miyakawa, S.; Ogino, M.; Funabe, S.; Uchino, A.; Shimo, Y.; Hattori, N.; Ichinoe, M.; Mikami, T.; Saegusa, M.; Nishiyama, K.; et al. Lewy body pathology in a patient with a homozygous parkin deletion. Mov. Disord. 2013, 28, 388–391. [Google Scholar] [CrossRef] [PubMed]
- Doherty, K.M.; Silveira-Moriyama, L.; Parkkinen, L.; Healy, D.G.; Farrell, M.; Mencacci, N.E.; Ahmed, Z.; Brett, F.M.; Hardy, J.; Quinn, N.; et al. Parkin disease: A clinicopathologic entity? JAMA Neurol. 2013, 70, 571–579. [Google Scholar] [CrossRef]
- Selikhova, M.; Kempster, P.A.; Revesz, T.; Holton, J.L.; Lees, A.J. Neuropathological findings in benign tremulous parkinsonism. Mov. Disord. 2013, 28, 145–152. [Google Scholar] [CrossRef]
- Sharp, M.E.; Marder, K.S.; Côté, L.; Clark, L.N.; Nichols, W.C.; Vonsattel, J.-P.; Alcalay, R.N. Parkinson’s disease with Lewy bodies associated with a heterozygous PARKIN dosage mutation. Mov. Disord. 2014, 29, 566–568. [Google Scholar] [CrossRef]
- Sasaki, S.; Shirata, A.; Yamane, K.; Iwata, M. Parkin-positive autosomal recessive juvenile Parkinsonism with alpha-synuclein-positive inclusions. Neurology 2004, 63, 678–682. [Google Scholar] [CrossRef]
- Takahashi, H.; Ohama, E.; Suzuki, S.; Horikawa, Y.; Ishikawa, A.; Morita, T.; Tsuji, S.; Ikuta, F. Familial juvenile parkinsonism: Clinical and pathologic study in a family. Neurology 1994, 44, 437–441. [Google Scholar] [CrossRef]
- Yamamura, Y.; Arihiro, K.; Kohriyama, T.; Nakamura, S. Early-onset parkinsonism with diurnal fluctuation—Clinical and pathological studies. Rinsho Shinkeigaku 1993, 33, 491–496. [Google Scholar]
- Yamamura, Y.; Kuzuhara, S.; Kondo, K.; Yanagi, T.; Uchida, M.; Matsumine, H.; Mizuno, Y. Clinical, pathologic and genetic studies on autosomal recessive early-onset parkinsonism with diurnal fluctuation. Parkinsonism Relat. Disord. 1998, 4, 65–72. [Google Scholar] [CrossRef]
- Hayashi, S.; Wakabayashi, K.; Ishikawa, A.; Nagai, H.; Saito, M.; Maruyama, M.; Takahashi, T.; Ozawa, T.; Tsuji, S.; Takahashi, H. An autopsy case of autosomal-recessive juvenile parkinsonism with a homozygous exon 4 deletion in the parkin gene. Mov. Disord. 2000, 15, 884–888. [Google Scholar] [CrossRef]
- Van de Warrenburg, B.P.; Lammens, M.; Lücking, C.B.; Denèfle, P.; Wesseling, P.; Booij, J.; Praamstra, P.; Quinn, N.; Brice, A.; Horstink, M.W. Clinical and pathologic abnormalities in a family with parkinsonism and parkin gene mutations. Neurology 2001, 56, 555–557. [Google Scholar] [CrossRef] [PubMed]
- Gouider-Khouja, N.; Larnaout, A.; Amouri, R.; Sfar, S.; Belal, S.; Ben Hamida, C.; Ben Hamida, M.; Hattori, N.; Mizuno, Y.; Hentati, F. Autosomal recessive parkinsonism linked to parkin gene in a Tunisian family. Clinical, genetic and pathological study. Parkinsonism Relat. Disord. 2003, 9, 247–251. [Google Scholar] [CrossRef]
- Orimo, S.; Amino, T.; Yokochi, M.; Kojo, T.; Uchihara, T.; Takahashi, A.; Wakabayashi, K.; Takahashi, H.; Hattori, N.; Mizuno, Y. Preserved cardiac sympathetic nerve accounts for normal cardiac uptake of MIBG in PARK2. Mov. Disord. 2005, 20, 1350–1353. [Google Scholar] [CrossRef]
- Cornejo-Olivas, M.R.; Torres, L.; Mata, I.F.; Mazzetti, P.; Rivas, D.; Cosentino, C.; Inca-Martinez, M.; Cuba, J.M.; Zabetian, C.P.; Leverenz, J.B. A Peruvian family with a novel PARK2 mutation: Clinical and pathological characteristics. Parkinsonism Relat. Disord. 2015, 21, 444–448. [Google Scholar] [CrossRef]
- Johansen, K.K.; Torp, S.H.; Farrer, M.J.; Gustavsson, E.K.; Aasly, J.O. A case of Parkinson’s disease with no Lewy body pathology due to a homozygous exon deletion in Parkin. Case Rep. Neurol. Med. 2018, 2018, 6838965. [Google Scholar] [CrossRef]
- Yamamura, Y. The long journey to the discovery of PARK2: The 50th anniversary of Japanese society of neuropathology. Neuropathology 2010, 30, 495–500. [Google Scholar] [CrossRef]
- Yamamura, Y.; Sobue, I.; Ando, K.; Iida, M.; Yanagi, T. Paralysis agitans of early onset with marked diurnal fluctuation of symptoms. Neurology 1973, 23, 239–244. [Google Scholar] [CrossRef]
- Matsumine, H.; Saito, M.; Shimoda-Matsubayashi, S.; Tanaka, H.; Ishikawa, A.; Nakagawa-Hattori, Y.; Yokochi, M.; Kobayashi, T.; Igarashi, S.; Takano, H.; et al. Localization of a gene for an autosomal recessive form of juvenile Parkinsonism to chromosome 6q25.2-27. Am. J. Hum. Genet. 1997, 60, 588–596. [Google Scholar]
- Hattori, N.; Mizuno, Y. Twenty years since the discovery of the parkin gene. J. Neural Transm. (Vienna Austria 1996) 2017, 124, 1037–1054. [Google Scholar] [CrossRef] [PubMed]
- Dickson, D.W.; Braak, H.; Duda, J.E.; Duyckaerts, C.; Gasser, T.; Halliday, G.M.; Hardy, J.; Leverenz, J.B.; Del Tredici, K.; Wszolek, Z.K.; et al. Neuropathological assessment of Parkinson’s disease: Refining the diagnostic criteria. Lancet Neurol. 2009, 8, 1150–1157. [Google Scholar] [CrossRef]
- Daniel, S.E.; Lees, A.J. Parkinson’s disease society brain bank, London: Overview and research. J. Neural Transm. Suppl. 1993, 39, 165–172. [Google Scholar] [PubMed]
- Gelb, D.J.; Oliver, E.; Gilman, S. Diagnostic criteria for Parkinson disease. Arch. Neurol. 1999, 56, 33–39. [Google Scholar] [CrossRef]
- Adler, C.H.; Beach, T.G.; Hentz, J.G.; Shill, H.A.; Caviness, J.N.; Driver-Dunckley, E.; Sabbagh, M.N.; Sue, L.I.; Jacobson, S.A.; Belden, C.M.; et al. Low clinical diagnostic accuracy of early vs. advanced Parkinson disease: Clinicopathologic study. Neurology 2014, 83, 406–412. [Google Scholar] [CrossRef]
- Schneider, S.A.; Alcalay, R.N. Neuropathology of genetic synucleinopathies with parkinsonism: Review of the literature. Mov. Disord. 2017, 32, 1504–1523. [Google Scholar] [CrossRef]
- Markesbery, W.R.; Jicha, G.A.; Liu, H.; Schmitt, F.A. Lewy body pathology in normal elderly subjects. J. Neuropathol. Exp. Neurol. 2009, 68, 816–822. [Google Scholar] [CrossRef]
- Parkkinen, L.; Soininen, H.; Laakso, M.; Alafuzoff, I. Alpha-synuclein pathology is highly dependent on the case selection. Neuropathol. Appl. Neurobiol. 2001, 27, 314–325. [Google Scholar] [CrossRef]
- Fearnley, J.M.; Lees, A.J. Ageing and Parkinson’s disease: Substantia nigra regional selectivity. Brain J. Neurol. 1991, 114 Pt 5, 2283–2301. [Google Scholar] [CrossRef]
- Doherty, K.M.; Hardy, J. Parkin disease and the Lewy body conundrum. Mov. Disord. 2013, 28, 702–704. [Google Scholar] [CrossRef]
- Beasley, S.A.; Hristova, V.A.; Shaw, G.S. Structure of the Parkin in-between-ring domain provides insights for E3-ligase dysfunction in autosomal recessive Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2007, 104, 3095–3100. [Google Scholar] [CrossRef] [PubMed]
- Sriram, S.R.; Li, X.; Ko, H.S.; Chung, K.K.; Wong, E.; Lim, K.L.; Dawson, V.L.; Dawson, T.M. Familial-associated mutations differentially disrupt the solubility, localization, binding and ubiquitination properties of parkin. Hum. Mol. Genet. 2005, 14, 2571–2586. [Google Scholar] [CrossRef]
- Cookson, M.R.; Lockhart, P.J.; McLendon, C.; O’Farrell, C.; Schlossmacher, M.; Farrer, M.J. RING finger 1 mutations in Parkin produce altered localization of the protein. Hum. Mol. Genet. 2003, 12, 2957–2965. [Google Scholar] [CrossRef]
- Calne, D.B.; Mizuno, Y. The neuromythology of Parkinson’s disease. Parkinsonism Relat. Disord. 2004, 10, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.L.; Chew, K.C.; Tan, J.M.; Wang, C.; Chung, K.K.; Zhang, Y.; Tanaka, Y.; Smith, W.; Engelender, S.; Ross, C.A.; et al. Parkin mediates nonclassical, proteasomal-independent ubiquitination of synphilin-1: Implications for Lewy body formation. J. Neurosci. 2005, 25, 2002–2009. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.L.; Dawson, V.L.; Dawson, T.M. Parkin-mediated lysine 63-linked polyubiquitination: A link to protein inclusions formation in Parkinson’s and other conformational diseases? Neurobiol. Aging 2006, 27, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.K.; Zhang, Y.; Lim, K.L.; Tanaka, Y.; Huang, H.; Gao, J.; Ross, C.A.; Dawson, V.L.; Dawson, T.M. Parkin ubiquitinates the alpha-synuclein-interacting protein, synphilin-1: Implications for Lewy-body formation in Parkinson disease. Nat. Med. 2001, 7, 1144–1150. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A. Linking ubiquitin, parkin and synphilin-1. Nat. Med. 2001, 7, 1108–1109. [Google Scholar] [CrossRef]
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The many faces of alpha-synuclein: From structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 2013, 14, 38–48. [Google Scholar] [CrossRef]
- Anderson, J.P.; Walker, D.E.; Goldstein, J.M.; de Laat, R.; Banducci, K.; Caccavello, R.J.; Barbour, R.; Huang, J.; Kling, K.; Lee, M.; et al. Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J. Biol. Chem. 2006, 281, 29739–29752. [Google Scholar] [CrossRef]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. alpha-synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, T.; Watanabe, S.T.; Iwatsubo, T.; Hasegawa, M. Seeded aggregation and toxicity of α-synuclein and tau: Cellular models of neurodegenerative diseases. J. Biol. Chem. 2010, 285, 34885–34898. [Google Scholar] [CrossRef] [PubMed]
- Desplats, P.; Lee, H.J.; Bae, E.J.; Patrick, C.; Rockenstein, E.; Crews, L.; Spencer, B.; Masliah, E.; Lee, S.J. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc. Natl. Acad. Sci. USA 2009, 106, 13010–13015. [Google Scholar] [CrossRef] [PubMed]
- Hansen, C.; Angot, E.; Bergström, A.L.; Steiner, J.A.; Pieri, L.; Paul, G.; Outeiro, T.F.; Melki, R.; Kallunki, P.; Fog, K.; et al. α-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J. Clin. Investig. 2011, 121, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012, 338, 949–953. [Google Scholar] [CrossRef]
- Espay, A.J.; Vizcarra, J.A.; Marsili, L.; Lang, A.E.; Simon, D.K.; Merola, A.; Josephs, K.A.; Fasano, A.; Morgante, F.; Savica, R.; et al. Revisiting protein aggregation as pathogenic in sporadic Parkinson and Alzheimer diseases. Neurology 2019, 92, 329–337. [Google Scholar] [CrossRef]
- Zhang, J.; Li, X.; Li, J.D. The roles of post-translational modifications on α-synuclein in the pathogenesis of Parkinson’s diseases. Front. Neurosci. 2019, 13, 381. [Google Scholar] [CrossRef]
- Lee, K.W.; Chen, W.; Junn, E.; Im, J.Y.; Grosso, H.; Sonsalla, P.K.; Feng, X.; Ray, N.; Fernandez, J.R.; Chao, Y.; et al. Enhanced phosphatase activity attenuates alpha-synucleinopathy in a mouse model. J. Neurosci. 2011, 31, 6963–6971. [Google Scholar] [CrossRef]
- Park, H.J.; Lee, K.W.; Park, E.S.; Oh, S.; Yan, R.; Zhang, J.; Beach, T.G.; Adler, C.H.; Voronkov, M.; Braithwaite, S.P.; et al. Dysregulation of protein phosphatase 2A in parkinson disease and dementia with lewy bodies. Ann. Clin. Transl. Neurol. 2016, 3, 769–780. [Google Scholar] [CrossRef]
- Ogris, E.; Du, X.; Nelson, K.C.; Mak, E.K.; Yu, X.X.; Lane, W.S.; Pallas, D.C. A protein phosphatase methylesterase (PME-1) is one of several novel proteins stably associating with two inactive mutants of protein phosphatase 2A. J. Biol. Chem. 1999, 274, 14382–14391. [Google Scholar] [CrossRef]
- Tolstykh, T.; Lee, J.; Vafai, S.; Stock, J.B. Carboxyl methylation regulates phosphoprotein phosphatase 2A by controlling the association of regulatory B subunits. EMBO J. 2000, 19, 5682–5691. [Google Scholar] [CrossRef] [PubMed]
- Khandelwal, P.J.; Dumanis, S.B.; Feng, L.R.; Maguire-Zeiss, K.; Rebeck, G.; Lashuel, H.A.; Moussa, C.E. Parkinson-related parkin reduces alpha-synuclein phosphorylation in a gene transfer model. Mol. Neurodegener. 2010, 5, 47. [Google Scholar] [CrossRef] [PubMed]
- Burai, R.; Ait-Bouziad, N.; Chiki, A.; Lashuel, H.A. Elucidating the role of site-specific nitration of α-synuclein in the pathogenesis of Parkinson’s disease via protein semisynthesis and mutagenesis. J. Am. Chem. Soc. 2015, 137, 5041–5052. [Google Scholar] [CrossRef] [PubMed]
- Stone, D.K.; Kiyota, T.; Mosley, R.L.; Gendelman, H.E. A model of nitric oxide induced α-synuclein misfolding in Parkinson’s disease. Neurosci. Lett. 2012, 523, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Giasson, B.I.; Duda, J.E.; Murray, I.V.; Chen, Q.; Souza, J.M.; Hurtig, H.I.; Ischiropoulos, H.; Trojanowski, J.Q.; Lee, V.M. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science 2000, 290, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Danielson, S.R.; Held, J.M.; Schilling, B.; Oo, M.; Gibson, B.W.; Andersen, J.K. Preferentially increased nitration of alpha-synuclein at tyrosine-39 in a cellular oxidative model of Parkinson’s disease. Anal. Chem. 2009, 81, 7823–7828. [Google Scholar] [CrossRef]
- Jiang, H.; Jiang, Q.; Liu, W.; Feng, J. Parkin suppresses the expression of monoamine oxidases. J. Biol. Chem. 2006, 281, 8591–8599. [Google Scholar] [CrossRef]
- Jiang, H.; Ren, Y.; Yuen, E.Y.; Zhong, P.; Ghaedi, M.; Hu, Z.; Azabdaftari, G.; Nakaso, K.; Yan, Z.; Feng, J. Parkin controls dopamine utilization in human midbrain dopaminergic neurons derived from induced pluripotent stem cells. Nat. Commun. 2012, 3, 668. [Google Scholar] [CrossRef]
- Lee, V.M.; Goedert, M.; Trojanowski, J.Q. Neurodegenerative tauopathies. Annu. Rev. Neurosci. 2001, 24, 1121–1159. [Google Scholar] [CrossRef]
- Moussaud, S.; Jones, D.R.; Moussaud-Lamodière, E.L.; Delenclos, M.; Ross, O.A.; McLean, P.J. Alpha-synuclein and tau: Teammates in neurodegeneration? Mol. Neurodegener. 2014, 9, 43. [Google Scholar] [CrossRef]
- Arima, K.; Mizutani, T.; Alim, M.A.; Tonozuka-Uehara, H.; Izumiyama, Y.; Hirai, S.; Uéda, K. NACP/alpha-synuclein and tau constitute two distinctive subsets of filaments in the same neuronal inclusions in brains from a family of parkinsonism and dementia with Lewy bodies: Double-immunolabeling fluorescence and electron microscopic studies. Acta Neuropathol. 2000, 100, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Ishizawa, T.; Mattila, P.; Davies, P.; Wang, D.; Dickson, D.W. Colocalization of tau and alpha-synuclein epitopes in Lewy bodies. J. Neuropathol. Exp. Neurol. 2003, 62, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Biernat, J.; Gustke, N.; Drewes, G.; Mandelkow, E.M.; Mandelkow, E. Phosphorylation of Ser262 strongly reduces binding of tau to microtubules: Distinction between PHF-like immunoreactivity and microtubule binding. Neuron 1993, 11, 153–163. [Google Scholar] [CrossRef]
- Qureshi, H.Y.; Paudel, H.K. Parkinsonian neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and alpha-synuclein mutations promote Tau protein phosphorylation at Ser262 and destabilize microtubule cytoskeleton in vitro. J. Biol. Chem. 2011, 286, 5055–5068. [Google Scholar] [CrossRef]
- Duka, T.; Rusnak, M.; Drolet, R.E.; Duka, V.; Wersinger, C.; Goudreau, J.L.; Sidhu, A. Alpha-synuclein induces hyperphosphorylation of Tau in the MPTP model of parkinsonism. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2006, 20, 2302–2312. [Google Scholar] [CrossRef]
- Meredith, G.E.; Rademacher, D.J. MPTP mouse models of Parkinson’s disease: An update. J. Parkinsons Dis. 2011, 1, 19–33. [Google Scholar] [CrossRef]
- Duka, T.; Duka, V.; Joyce, J.N.; Sidhu, A. Alpha-Synuclein contributes to GSK-3beta-catalyzed Tau phosphorylation in Parkinson’s disease models. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2009, 23, 2820–2830. [Google Scholar] [CrossRef]
- Moussa, C.E. Parkin attenuates wild-type tau modification in the presence of beta-amyloid and alpha-synuclein. J. Mol. Neurosci. 2009, 37, 25–36. [Google Scholar] [CrossRef]
- Tompkins, M.M.; Basgall, E.J.; Zamrini, E.; Hill, W.D. Apoptotic-like changes in Lewy-body-associated disorders and normal aging in substantia nigral neurons. Am. J. Pathol. 1997, 150, 119–131. [Google Scholar]
- Erekat, N.S. Apoptosis and its role in Parkinson’s disease. In Parkinson’s Disease: Pathogenesis and Clinical Aspects; Stoker, T.B., Greenland, J.C., Eds.; Codon Publications: Brisbane, Australia, 2018. [Google Scholar] [CrossRef]
- Viswanath, V.; Wu, Y.; Boonplueang, R.; Chen, S.; Stevenson, F.F.; Yantiri, F.; Yang, L.; Beal, M.F.; Andersen, J.K. Caspase-9 activation results in downstream caspase-8 activation and bid cleavage in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinson’s disease. J. Neurosci. 2001, 21, 9519–9528. [Google Scholar] [CrossRef]
- Mogi, M.; Togari, A.; Kondo, T.; Mizuno, Y.; Komure, O.; Kuno, S.; Ichinose, H.; Nagatsu, T. Caspase activities and tumor necrosis factor receptor R1 (p55) level are elevated in the substantia nigra from parkinsonian brain. J. Neural Transm. (Vienna Austria 1996) 2000, 107, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, A.; Hunot, S.; Michel, P.P.; Muriel, M.P.; Vyas, S.; Faucheux, B.A.; Mouatt-Prigent, A.; Turmel, H.; Srinivasan, A.; Ruberg, M.; et al. Caspase-3: A vulnerability factor and final effector in apoptotic death of dopaminergic neurons in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2000, 97, 2875–2880. [Google Scholar] [CrossRef] [PubMed]
- da Costa, C.A.; Sunyach, C.; Giaime, E.; West, A.; Corti, O.; Brice, A.; Safe, S.; Abou-Sleiman, P.M.; Wood, N.W.; Takahashi, H.; et al. Transcriptional repression of p53 by parkin and impairment by mutations associated with autosomal recessive juvenile Parkinson’s disease. Nat. Cell Biol. 2009, 11, 1370–1375. [Google Scholar] [CrossRef] [PubMed]
- Alves da Costa, C.; Duplan, E.; Checler, F. α-synuclein and p53 functional interplay in physiopathological contexts. Oncotarget 2017, 8, 9001–9002. [Google Scholar] [CrossRef]
- Yuan, Y.; Jin, J.; Yang, B.; Zhang, W.; Hu, J.; Zhang, Y.; Chen, N.H. Overexpressed alpha-synuclein regulated the nuclear factor-kappaB signal pathway. Cell. Mol. Neurobiol. 2008, 28, 21–33. [Google Scholar] [CrossRef]
- Li, D.W.; Liu, Z.Q.; Chen, W.; Yao, M.; Li, G.R. Association of glycogen synthase kinase-3β with Parkinson’s disease (review). Mol. Med. Rep. 2014, 9, 2043–2050. [Google Scholar] [CrossRef]
- Linseman, D.A.; Butts, B.D.; Precht, T.A.; Phelps, R.A.; Le, S.S.; Laessig, T.A.; Bouchard, R.J.; Florez-McClure, M.L.; Heidenreich, K.A. Glycogen synthase kinase-3beta phosphorylates Bax and promotes its mitochondrial localization during neuronal apoptosis. J. Neurosci. 2004, 24, 9993–10002. [Google Scholar] [CrossRef]
- Watcharasit, P.; Bijur, G.N.; Song, L.; Zhu, J.; Chen, X.; Jope, R.S. Glycogen synthase kinase-3beta (GSK3beta) binds to and promotes the actions of p53. J. Biol. Chem. 2003, 278, 48872–48879. [Google Scholar] [CrossRef]
- Maurer, U.; Charvet, C.; Wagman, A.S.; Dejardin, E.; Green, D.R. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol. Cell 2006, 21, 749–760. [Google Scholar] [CrossRef]
- Zou, H.; Li, Y.; Liu, X.; Wang, X. An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J. Biol. Chem. 1999, 274, 11549–11556. [Google Scholar] [CrossRef]
- Rosen, K.M.; Moussa, C.E.; Lee, H.K.; Kumar, P.; Kitada, T.; Qin, G.; Fu, Q.; Querfurth, H.W. Parkin reverses intracellular beta-amyloid accumulation and its negative effects on proteasome function. J. Neurosci. Res. 2010, 88, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Hyun, D.H.; Lee, M.; Hattori, N.; Kubo, S.; Mizuno, Y.; Halliwell, B.; Jenner, P. Effect of wild-type or mutant Parkin on oxidative damage, nitric oxide, antioxidant defenses, and the proteasome. J. Biol. Chem. 2002, 277, 28572–28577. [Google Scholar] [CrossRef] [PubMed]
- Ardley, H.C.; Scott, G.B.; Rose, S.A.; Tan, N.G.; Markham, A.F.; Robinson, P.A. Inhibition of proteasomal activity causes inclusion formation in neuronal and non-neuronal cells overexpressing Parkin. Mol. Biol. Cell 2003, 14, 4541–4556. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Fujiwara, H.; Nonaka, T.; Wakabayashi, K.; Takahashi, H.; Lee, V.M.; Trojanowski, J.Q.; Mann, D.; Iwatsubo, T. Phosphorylated alpha-synuclein is ubiquitinated in alpha-synucleinopathy lesions. J. Biol. Chem. 2002, 277, 49071–49076. [Google Scholar] [CrossRef] [PubMed]
- Videira, P.A.Q.; Castro-Caldas, M. Linking glycation and glycosylation with inflammation and mitochondrial dysfunction in Parkinson’s disease. Front. Neurosci. 2018, 12, 381. [Google Scholar] [CrossRef] [PubMed]
- Petrucelli, L.; O’Farrell, C.; Lockhart, P.J.; Baptista, M.; Kehoe, K.; Vink, L.; Choi, P.; Wolozin, B.; Farrer, M.; Hardy, J.; et al. Parkin protects against the toxicity associated with mutant alpha-synuclein: Proteasome dysfunction selectively affects catecholaminergic neurons. Neuron 2002, 36, 1007–1019. [Google Scholar] [CrossRef]
- Yang, Y.; Nishimura, I.; Imai, Y.; Takahashi, R.; Lu, B. Parkin suppresses dopaminergic neuron-selective neurotoxicity induced by Pael-R in Drosophila. Neuron 2003, 37, 911–924. [Google Scholar] [CrossRef]
- Haywood, A.F.; Staveley, B.E. Parkin counteracts symptoms in a Drosophila model of Parkinson’s disease. BMC Neurosci. 2004, 5, 14. [Google Scholar] [CrossRef]
- Lo Bianco, C.; Schneider, B.L.; Bauer, M.; Sajadi, A.; Brice, A.; Iwatsubo, T.; Aebischer, P. Lentiviral vector delivery of parkin prevents dopaminergic degeneration in an alpha-synuclein rat model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 17510–17515. [Google Scholar] [CrossRef]
- Yamada, M.; Mizuno, Y.; Mochizuki, H. Parkin gene therapy for alpha-synucleinopathy: A rat model of Parkinson’s disease. Hum. Gene Ther. 2005, 16, 262–270. [Google Scholar] [CrossRef]
- Haywood, A.F.; Staveley, B.E. Mutant alpha-synuclein-induced degeneration is reduced by parkin in a fly model of Parkinson’s disease. Genome 2006, 49, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, T.; Miyachi, S.; Kitagawa, R.; Wada, K.; Nihira, T.; Ren, Y.R.; Hirai, Y.; Ageyama, N.; Terao, K.; Shimada, T.; et al. Neuronal specificity of alpha-synuclein toxicity and effect of Parkin co-expression in primates. Neuroscience 2007, 144, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Fournier, M.; Vitte, J.; Garrigue, J.; Langui, D.; Dullin, J.P.; Saurini, F.; Hanoun, N.; Perez-Diaz, F.; Cornilleau, F.; Joubert, C.; et al. Parkin deficiency delays motor decline and disease manifestation in a mouse model of synucleinopathy. PLoS ONE 2009, 4, e6629. [Google Scholar] [CrossRef] [PubMed]
- Van Rompuy, A.S.; Oliveras-Salva, M.; Van der Perren, A.; Corti, O.; Van den Haute, C.; Baekelandt, V. Nigral overexpression of alpha-synuclein in the absence of parkin enhances alpha-synuclein phosphorylation but does not modulate dopaminergic neurodegeneration. Mol. Neurodegener. 2015, 10, 23. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wilkaniec, A.; Lenkiewicz, A.M.; Czapski, G.A.; Jęśko, H.M.; Hilgier, W.; Brodzik, R.; Gąssowska-Dobrowolska, M.; Culmsee, C.; Adamczyk, A. Extracellular alpha-synuclein oligomers induce parkin s-nitrosylation: Relevance to sporadic Parkinson’s disease etiopathology. Mol. Neurobiol. 2019, 56, 125–140. [Google Scholar] [CrossRef]
- Goldberg, M.S.; Fleming, S.M.; Palacino, J.J.; Cepeda, C.; Lam, H.A.; Bhatnagar, A.; Meloni, E.G.; Wu, N.; Ackerson, L.C.; Klapstein, G.J.; et al. Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J. Biol. Chem. 2003, 278, 43628–43635. [Google Scholar] [CrossRef]
- Lorenzetti, D.; Antalffy, B.; Vogel, H.; Noveroske, J.; Armstrong, D.; Justice, M. The neurological mutant quaking(viable) is Parkin deficient. Mamm. Genome 2004, 15, 210–217. [Google Scholar] [CrossRef]
- Ko, H.S.; von Coelln, R.; Sriram, S.R.; Kim, S.W.; Chung, K.K.; Pletnikova, O.; Troncoso, J.; Johnson, B.; Saffary, R.; Goh, E.L.; et al. Accumulation of the authentic parkin substrate aminoacyl-tRNA synthetase cofactor, p38/JTV-1, leads to catecholaminergic cell death. J. Neurosci. 2005, 25, 7968–7978. [Google Scholar] [CrossRef]
- von Coelln, R.; Thomas, B.; Andrabi, S.A.; Lim, K.L.; Savitt, J.M.; Saffary, R.; Stirling, W.; Bruno, K.; Hess, E.J.; Lee, M.K.; et al. Inclusion body formation and neurodegeneration are parkin independent in a mouse model of alpha-synucleinopathy. J. Neurosci. 2006, 26, 3685–3696. [Google Scholar] [CrossRef]
- Tanaka, K.; Suzuki, T.; Hattori, N.; Mizuno, Y. Ubiquitin, proteasome and parkin. Biochim. Biophys. Acta 2004, 1695, 235–247. [Google Scholar] [CrossRef]
- Narendra, D.; Walker, J.E.; Youle, R. Mitochondrial quality control mediated by PINK1 and Parkin: Links to parkinsonism. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.L.; Ng, X.H.; Grace, L.G.; Yao, T.P. Mitochondrial dynamics and Parkinson’s disease: Focus on parkin. Antioxid. Redox Signal. 2012, 16, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Palacino, J.J.; Sagi, D.; Goldberg, M.S.; Krauss, S.; Motz, C.; Wacker, M.; Klose, J.; Shen, J. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J. Biol. Chem. 2004, 279, 18614–18622. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, K.; Tanji, K.; Mori, F.; Takahashi, H. The Lewy body in Parkinson’s disease: Molecules implicated in the formation and degradation of alpha-synuclein aggregates. Neuropathology 2007, 27, 494–506. [Google Scholar] [CrossRef] [PubMed]
- Terry, R.D. Do neuronal inclusions kill the cell? J. Neural Transm. Suppl. 2000, 59, 91–93. [Google Scholar] [CrossRef]
- Farrer, M.J. Genetics of Parkinson disease: Paradigm shifts and future prospects. Nat. Rev. Genet. 2006, 7, 306–318. [Google Scholar] [CrossRef]
- Khandelwal, P.J.; Moussa, C.E. The Relationship between Parkin and Protein Aggregation in Neurodegenerative Diseases. Front. Psychiatry 2010, 1, 15. [Google Scholar] [CrossRef]
- Rodríguez-Navarro, J.A.; Casarejos, M.J.; Menéndez, J.; Solano, R.M.; Rodal, I.; Gómez, A.; Yébenes, J.G.; Mena, M.A. Mortality, oxidative stress and tau accumulation during ageing in parkin null mice. J. Neurochem. 2007, 103, 98–114. [Google Scholar] [CrossRef]
- Hong, X.; Liu, J.; Zhu, G.; Zhuang, Y.; Suo, H.; Wang, P.; Huang, D.; Xu, J.; Huang, Y.; Yu, M.; et al. Parkin overexpression ameliorates hippocampal long-term potentiation and β-amyloid load in an Alzheimer’s disease mouse model. Hum. Mol. Genet. 2014, 23, 1056–1072. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Clinical Phenotype | |
|---|---|
| Motor features | Bradykinesia |
| Resting tremor | |
| Non-motor features | Anxiety |
| Psychosis | |
| Panic attack | |
| Depression | |
| Other clinical features | Normal cognitive function |
| Hyperreflexia | |
| Frequent focal dystonia | |
| Sleep benefit | |
| Benign disease course | |
| Excellent response to low dose Levodopa | |
| Prone to develop Levodopa-induced dyskinesias | |
| References | PARK2 Genotype | Age of Onset | Age of Death | Neuropathology | Lewy Body Pathology |
|---|---|---|---|---|---|
| [139] | No details reported | NA | 67 | Loss of neurons and gliosis in SNpc and locus coeruleus. | No |
| [140,141] | Homozygous deletion between exon 3 and 7. | 20 | 52 | Loss of neurons in SNpc and gliosis. | No |
| [101] | Homozygous exon 4 deletion. | 24 | 62 | Moderate loss of neurons in SNpc and locus coeruleus. Tau pathology in hippocampus and neurofibrillary tangles in the frontal, temporal, and parietal cortices. | No |
| [142] | Homozygous exon 4 deletion | 32 | 70 | Loss of neurons and gliosis in SNpc and locus coeruleus. Few Neurofibrillary tangles observed. | No |
| [131] | Exon 7 R275W missense mutation and exon 3 deletion | 41 | 52 | Loss of neurons in SNpc and locus coeruleus. | Yes |
| [143] | Exon 6 K211N missense mutation and exon 3 deletion. | 18 | 75 | Loss of neurons in SNpc and the spinocerebellar system. Tau pathology in caudate nucleus, putamen, subthalamic nucleus, and substantia nigra was reported, but neurofibrillary tangles were absent. | No |
| [144] | Homozygous two-base AG deletion in exon 2 | 34 | 47 | Loss of neurons in SNpc and SNpr with astrocytic gliosis. | No |
| [138] | Homozygous exon 3 deletion | 33 | 70 | Moderate to severe depletion of neurons in SNpc and gliosis. Only a mild decrease of neurons in locus coeruleus. | LB in the pedunculopontine nucleus. |
| [132] | Exon 7 deletion + T1072 deletion | 49 | 73 | Reactive gliosis and loss of neurons in SNpc and locus coeruleus. | Yes |
| [145] | Homozygous exon 4 deletion | 24 | 73 | Loss of neurons and gliosis in SNpc and locus coeruleus. | No |
| [133] | Heterozygous R275W mutation in exon 7. | 62 | 80 | Severe loss of neurons in SNpc and locus coeruleus. Pre-tangles observed in subiculum, transentorhinal, and entorhinal cortex. | Yes |
| [134] | Homozygous deletion of exon 2 to 4. | 61 | 72 | A marked decrease in neurons of SNpc and locus coeruleus. Tau pathology was observed in the entorhinal cortex. | Yes |
| [135] | Case 1: R275W + exon 6 deletion | 36 | 86 | Moderate to severe loss of neurons in SNpc in all cases and mild to moderate loss of neurons in locus coeruleus. Hyperphosphorylated tau deposition observed in case 3. | Only in case 3 and 5. |
| Case 2: R275W + pro113fs | 25 | 62 | |||
| Case 3: R275W + G430W | 33 | 60 | |||
| Case 4: G430D + pro113fs | 32 | 68 | |||
| Case 5: R275W + exon 6 deletion | 46 | 82 | |||
| [136] | Exon 7 R275W missense mutation and exon 6 deletion | 48 | 82 | Moderate loss of neurons in SNpc. | Yes |
| [137] | Heterozygous exon 3–4 deletion. | 44 | 76 | Severe loss of neurons in SNpc and locus coeruleus. Presence of neurofibrillary tangles. | Yes |
| [146] | Splice site mutation in intron 5 (IVS5-1G>A) and exon 7 deletion. | 16 | 60 | Severe loss of neurons in SNpc. Neurofibrillary tangle pathology was observed. | No |
| [147] | Homozygous deletion of exon 3–4. | 20 | 79 | Severe neuronal loss and gliosis in SNpc. Scanty tau pathology was observed. | No |
| References | Method | Organism | Finding | |
|---|---|---|---|---|
| [217] | Overexpression of parkin with A53T and A30P mutated -synuclein | Primary cell culture | Increased parkin expression can mitigate mutated -synuclein induced toxicity. | Yes |
| [227] | Quantification of wild-type -synuclein pathology in parkin−/− mice | Mouse model | No loss of nigral dopaminergic neurons and no accumulation of -synuclein. | No |
| [218] | Co-expression of parkin with A53T, A30P, or wild-type -synuclein | Drosophila melanogaster | Increased parkin expression can mitigate mutated -synuclein induced toxicity. | Yes |
| [219] | Co-expression of parkin with wild-type -synuclein. | Drosophila melanogaster | Increased parkin expression can suppress -synuclein-induced loss of climbing ability. | Yes |
| [228] | Quantification of wild-type -synuclein pathology in parkin−/− mice | Mouse mutant quaking (viable) model | No loss of dopaminergic neurons and no accumulation of -synuclein. | No |
| [220] | Overexpression of wild-type rat parkin with human A30P -synuclein using lentiviral vector delivery. | Rat model | Increased parkin expression can significantly reduce -synuclein-induced neuropathology. | Yes |
| [229] | Quantification of wild-type -synuclein pathology in parkin−/− mice | Mouse model | No accumulation of -synuclein. | No |
| [221] | Overexpression of human wild-type -synuclein and human parkin in the substantia nigra in rats using rAAV vector | Rat model | Increased parkin expression can mitigate -synuclein induced toxicity. | Yes |
| [222] | Co-expression of parkin with A30P -synuclein. | Drosophila melanogaster | Increased expression of parkin could counteract the effect of A30P mutated -synuclein. | Yes |
| [230] | Overexpression of human A53T -synuclein and deletion of parkin. | Mouse model | No effect of loss of parkin on neuropathology. | No |
| [223] | Co-expression of parkin with wild-type -synuclein. | Macaque monkeys. | Overexpression of parkin was associated with less accumulation of -synuclein and phosphorylation at S129. | Yes |
| [224] | A transgenic mouse model expressing human A30P -synuclein and no parkin. | Mouse model | A lower amount of pSer129 -synuclein in parkin−/− mice compared with parkin+/+ mice. | Yes |
| [182] | Co-expression of parkin with wild-type -synuclein. | Sprague Dawley rats | Increased expression of parkin resulted in a decreased expression of pSer87 and pSer129 -synuclein. | Yes |
| [225] | Overexpression of human wild-type -synuclein by rAAV vectors in parkin−/− and wild type mice. | Mouse model | Increased pSer129 -synuclein but no loss of dopaminergic neurons in parkin knockout mice. | Yes |
| [226] | Addition of exogenous -synuclein oligomers in a primary cell culture with a parkin knock-down or overexpression. | Rat pheochromocytoma (PC12) cells | Parkin overexpression protected against the extracellular -synuclein oligomer-mediated toxicity. | Yes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Madsen, D.A.; Schmidt, S.I.; Blaabjerg, M.; Meyer, M. Interaction between Parkin and α-Synuclein in PARK2-Mediated Parkinson’s Disease. Cells 2021, 10, 283. https://doi.org/10.3390/cells10020283
Madsen DA, Schmidt SI, Blaabjerg M, Meyer M. Interaction between Parkin and α-Synuclein in PARK2-Mediated Parkinson’s Disease. Cells. 2021; 10(2):283. https://doi.org/10.3390/cells10020283
Chicago/Turabian StyleMadsen, Daniel Aghaie, Sissel Ida Schmidt, Morten Blaabjerg, and Morten Meyer. 2021. "Interaction between Parkin and α-Synuclein in PARK2-Mediated Parkinson’s Disease" Cells 10, no. 2: 283. https://doi.org/10.3390/cells10020283
APA StyleMadsen, D. A., Schmidt, S. I., Blaabjerg, M., & Meyer, M. (2021). Interaction between Parkin and α-Synuclein in PARK2-Mediated Parkinson’s Disease. Cells, 10(2), 283. https://doi.org/10.3390/cells10020283