Overproduction of Human Zip (SLC39) Zinc Transporters in Saccharomyces cerevisiae for Biophysical Characterization


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
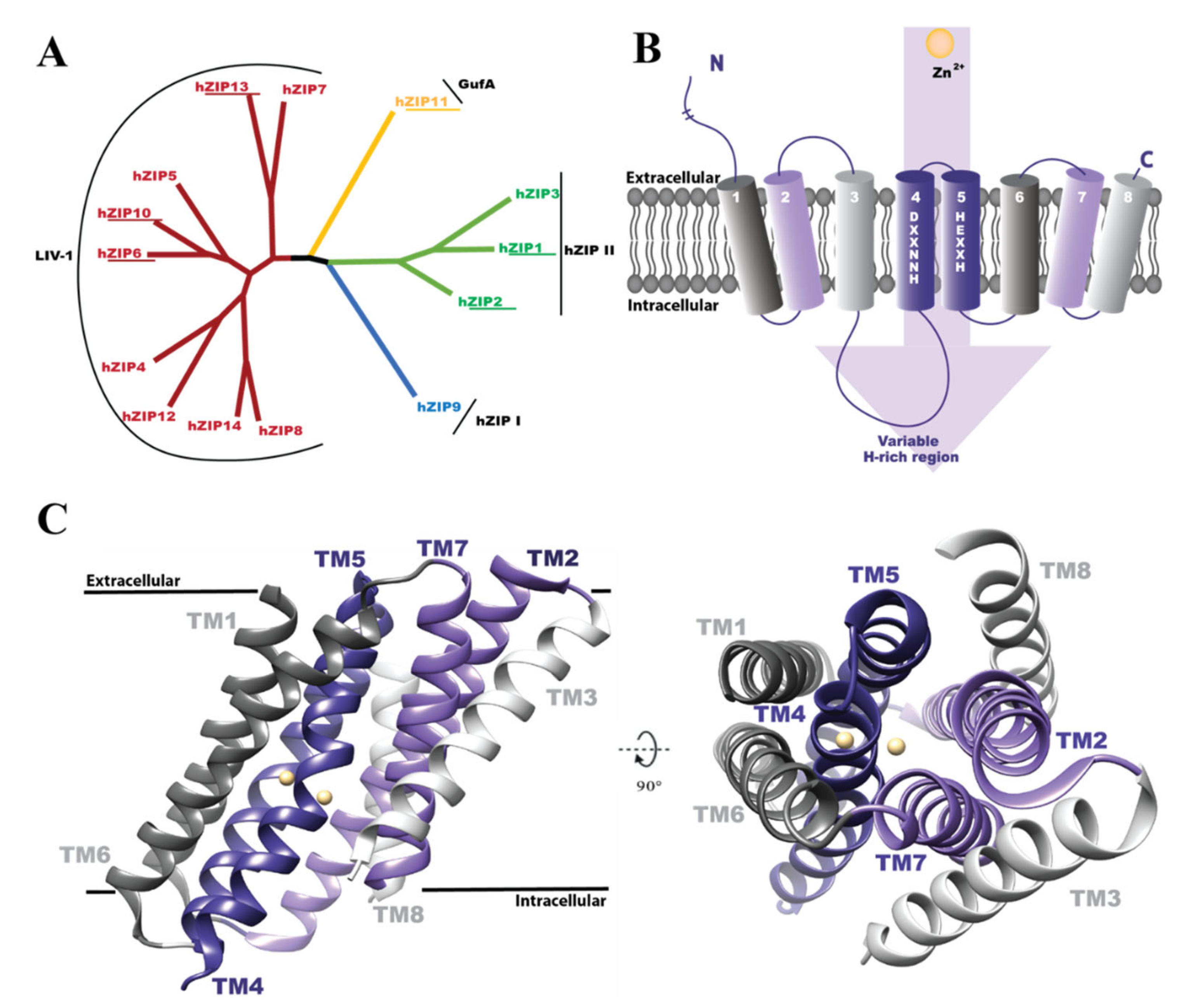
1. Introduction
2. Materials and Methods
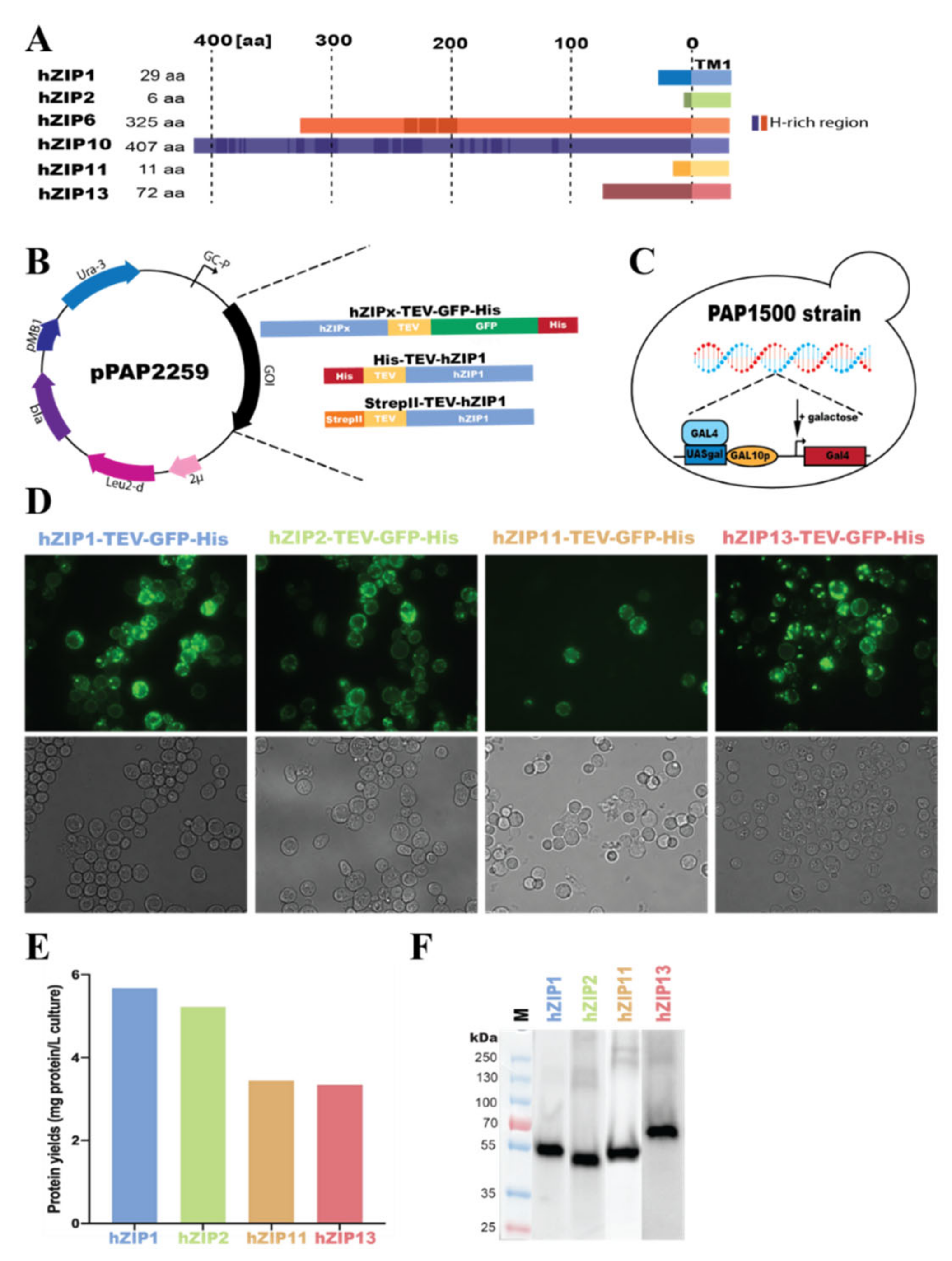
2.1. Engineering of Expression Plasmids
2.2. Overproduction of hZIPs, Live-Cell Bioimaging and In-Gel GFP Fluorescence
2.3. Preparation of Crude Membranes
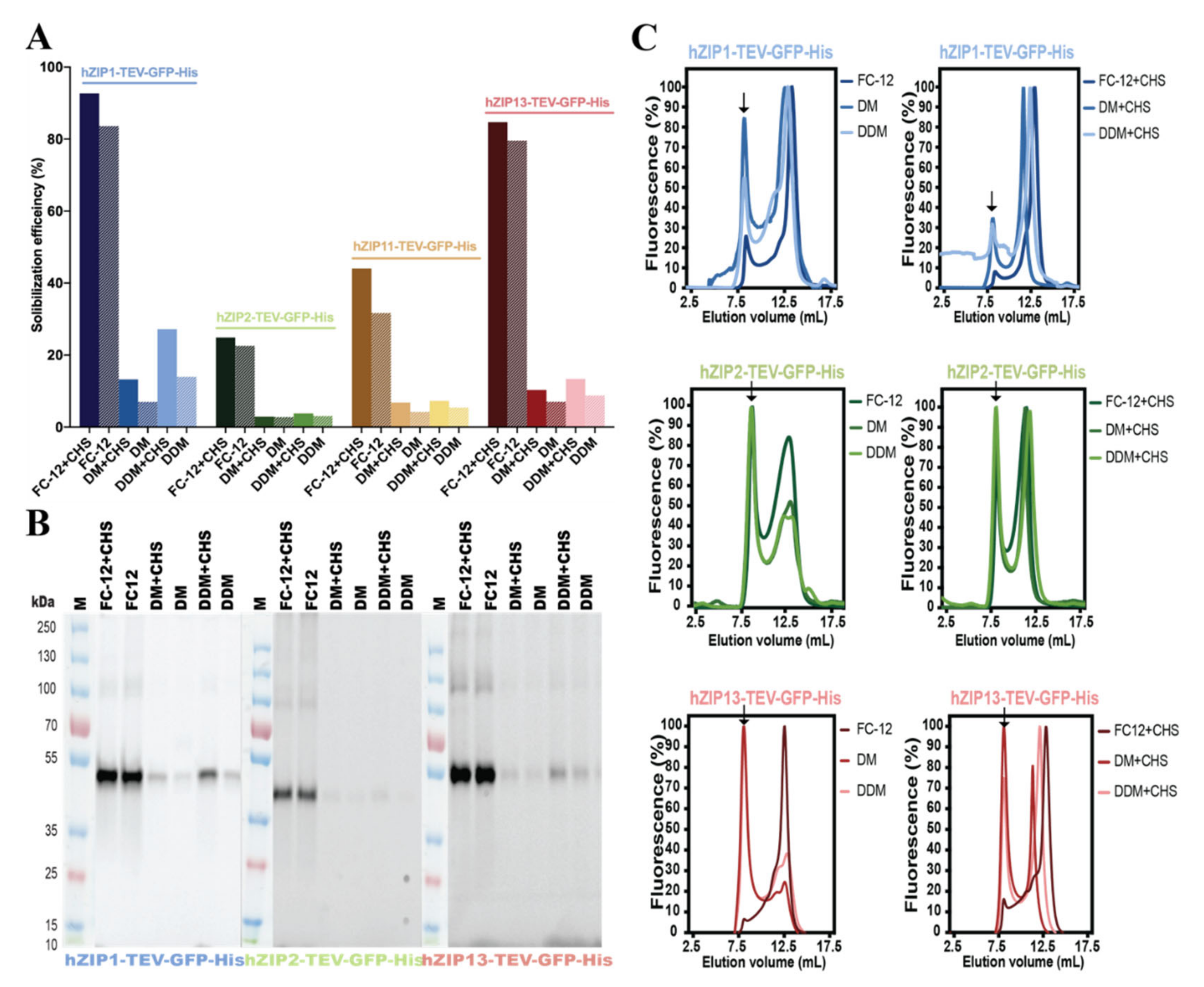
2.4. Detergent Screening, Immunoblotting, and F-SEC Analysis
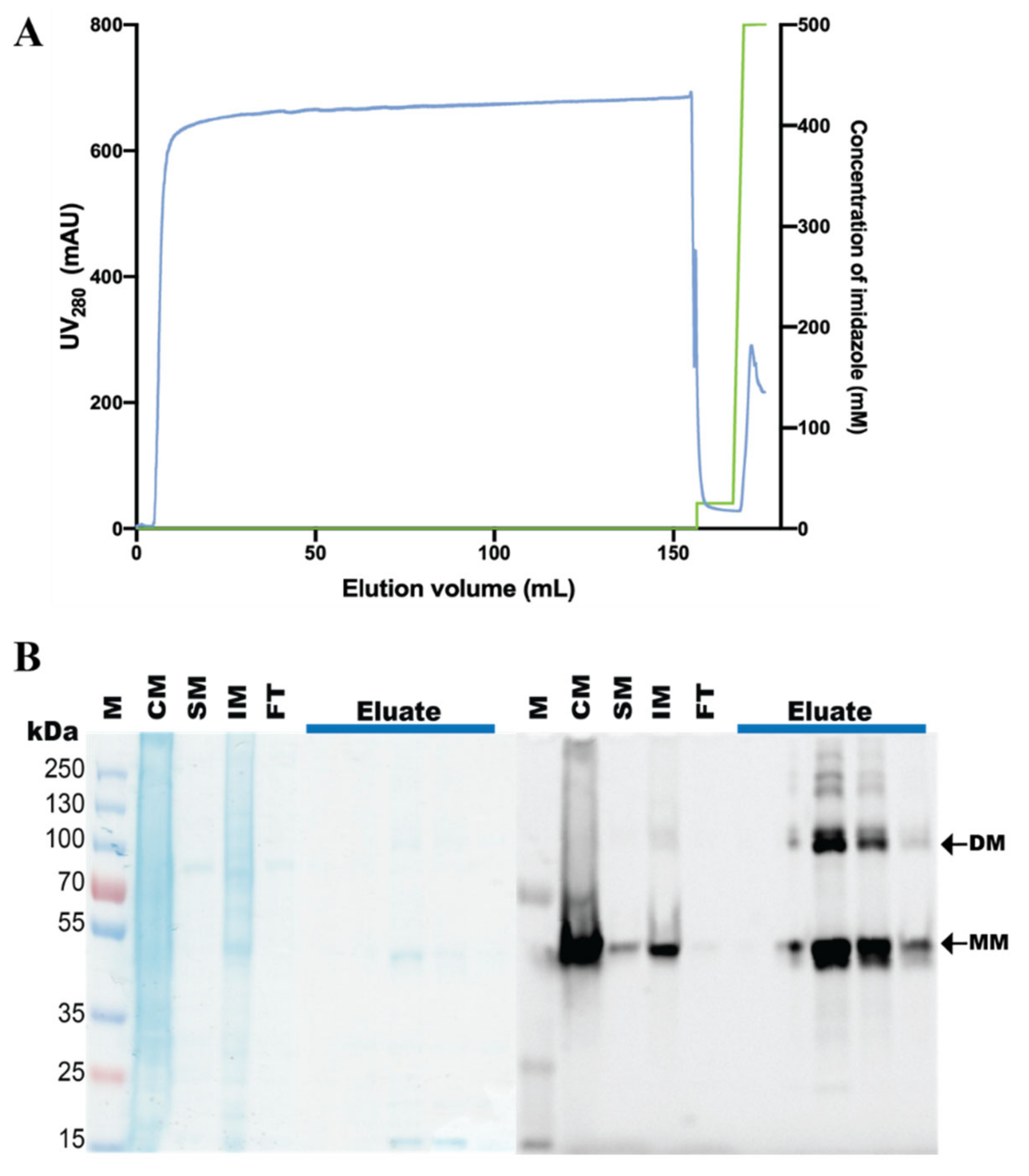
2.5. Purification of hZIP1, TEV Protease Cleavage, and SEC Analysis
3. Results
3.1. Overproduction of hZIP-TEV-GFP-His Fusions in S. cerevisiae-Based System
3.2. Detergent Screening and F-SEC Analysis of Solubilized hZIP-TEV-GFP-His Fusions
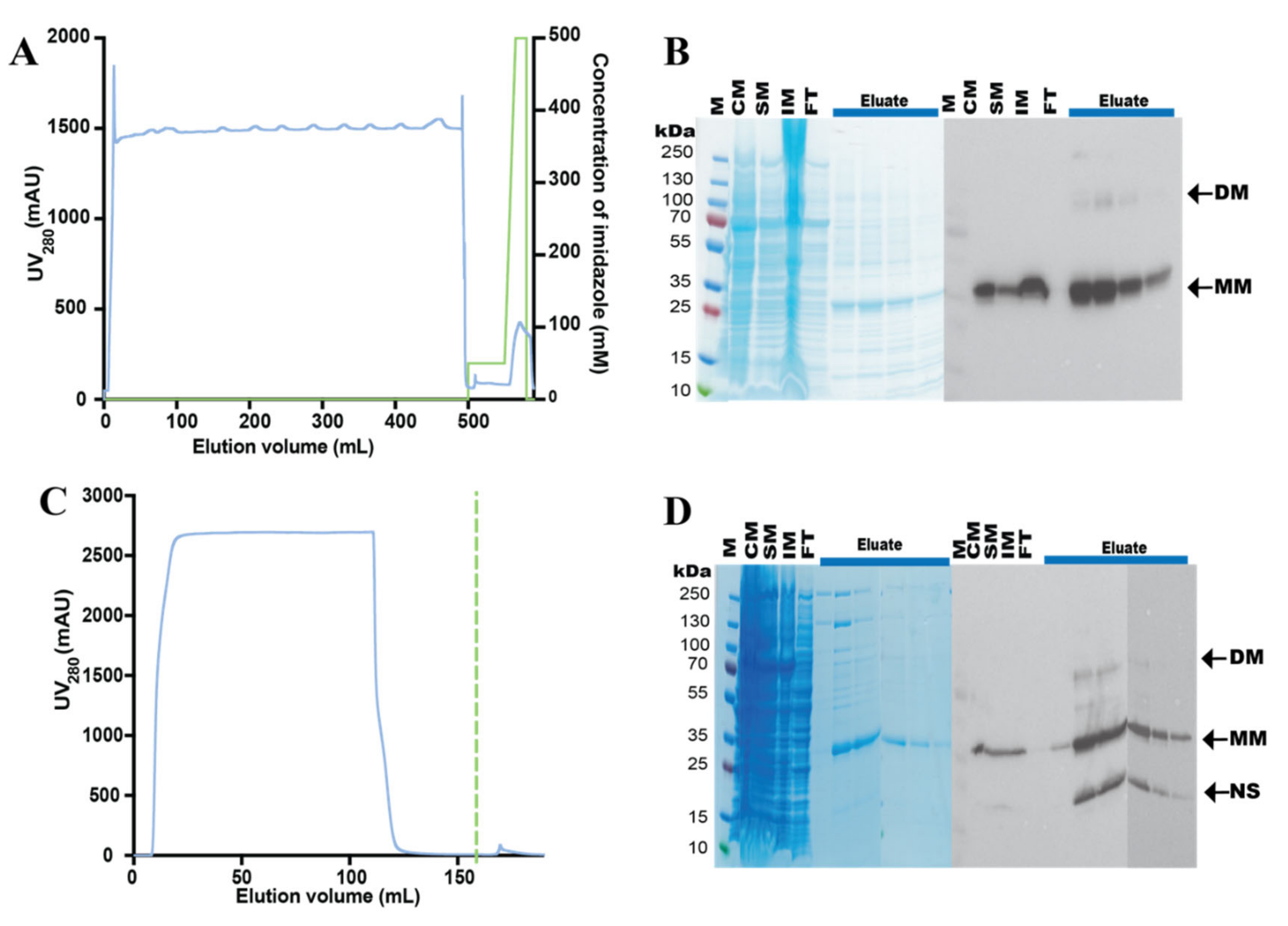
3.3. Purification of the hZIP1-TEV-GFP-His Fusion
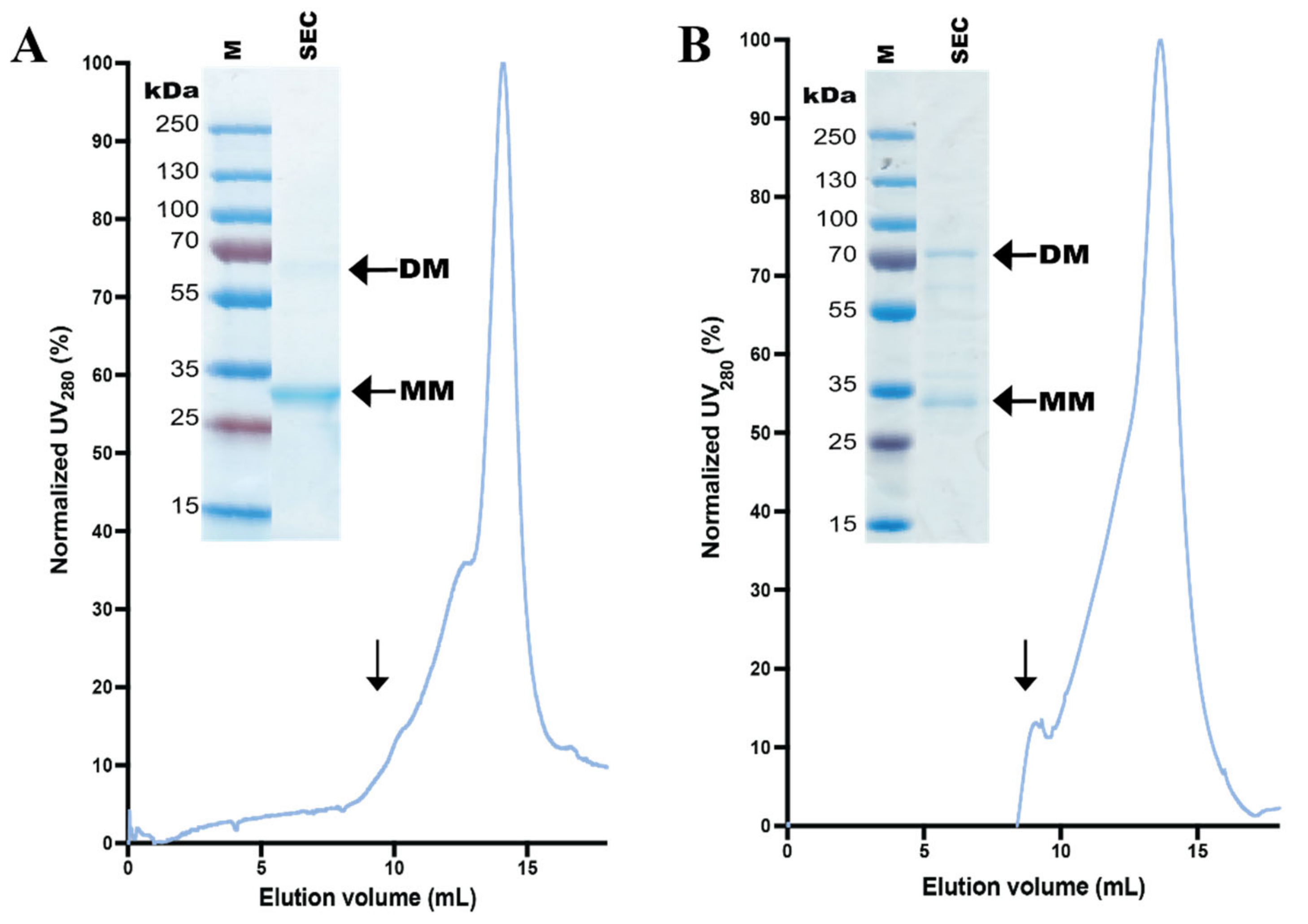
3.4. Design and Purification of N-Terminal hZIP1 Fusions
3.5. Stability of Affinity-Pure N-Terminal hZIP1 Fusions
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Andreini, C.; Banci, L.; Bertini, I.; Rosato, A. Counting the Zinc-Proteins Encoded in the Human Genome. J. Proteome Res. 2006, 5, 196–201. [Google Scholar] [CrossRef]
- Chasapis, C.T.; Loutsidou, A.C.; Spiliopoulou, C.A.; Stefanidou, M. Zinc and human health: An update. Arch. Toxicol. 2012, 86, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Thingholm, T.E.; Rönnstrand, L.; Rosenberg, P.A. Why and how to investigate the role of protein phosphorylation in ZIP and ZnT zinc transporter activity and regulation. Cell. Mol. Life Sci. 2020, 77, 3085–3102. [Google Scholar] [CrossRef] [PubMed]
- Guerinot, M.L. The ZIP Family of Metal Transporters. Biochim. Biophys. Acta Biomembr. 2000, 1465, 190–198. [Google Scholar] [CrossRef]
- Bafaro, E.; Liu, Y.; Xu, Y.; Dempski, R.E. The emerging role of zinc transporters in cellular homeostasis and cancer. Signal Transduct. Target. Ther. 2017, 2, 17029. [Google Scholar] [CrossRef] [PubMed]
- Kambe, T.; Hashimoto, A.; Fujimoto, S. Current understanding of ZIP and ZnT zinc transporters in human health and diseases. Cell. Mol. Life Sci. 2014, 71, 3281–3295. [Google Scholar] [CrossRef] [PubMed]
- Kambe, T.; Tsuji, T.; Hashimoto, A.; Itsumura, N. The Physiological, Biochemical, and Molecular Roles of Zinc Transporters in Zinc Homeostasis and Metabolism. Physiol. Rev. 2015, 95, 749–784. [Google Scholar] [CrossRef]
- Dufner-Beattie, J.; Langmade, S.J.; Wang, F.; Eide, D.; Andrews, G.K. Structure, Function, and Regulation of a Subfamily of Mouse Zinc Transporter Genes. J. Biol. Chem. 2003, 278, 50142–50150. [Google Scholar] [CrossRef]
- Gaither, L.A.; Eide, D.J. Eukaryotic zinc transporters and their regulation. BioMetals 2001, 14, 251–270. [Google Scholar] [CrossRef]
- Bowers, K.; Srai, S.K.S. The trafficking of metal ion transporters of the Zrt- and Irt-like protein family. Traffic 2018, 19, 813–822. [Google Scholar] [CrossRef]
- Kimura, T.; Kambe, T. The Functions of Metallothionein and ZIP and ZnT Transporters: An Overview and Perspective. Int. J. Mol. Sci. 2016, 17, 336. [Google Scholar] [CrossRef] [PubMed]
- Hennigar, S.R.; McClung, J.P. Zinc Transport in the Mammalian Intestine. Compr. Physiol. 2018, 9, 59–74. [Google Scholar] [CrossRef]
- Bin, B.-H.; Seo, J.; Kim, S.T. Function, Structure, and Transport Aspects of ZIP and ZnT Zinc Transporters in Immune Cells. J. Immunol. Res. 2018, 2018, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Higashi, Y.; Segawa, S.; Matsuo, T.; Nakamura, S.; Kikkawa, Y.; Nishida, K.; Nagasawa, K. Microglial zinc uptake via zinc transporters induces ATP release and the activation of microglia. Glia 2011, 59, 1933–1945. [Google Scholar] [CrossRef]
- Inoue, Y.; Hasegawa, S.; Ban, S.; Yamada, T.; Date, Y.; Mizutani, H.; Nakata, S.; Tanaka, M.; Hirashima, N. ZIP2 Protein, a Zinc Transporter, Is Associated with Keratinocyte Differentiation. J. Biol. Chem. 2014, 289, 21451–21462. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhou, B. Dietary zinc absorption: A play of Zips and ZnTs in the gut. IUBMB Life 2010, 62, 176–182. [Google Scholar] [CrossRef]
- Miyai, T.; Hojyo, S.; Ikawa, T.; Kawamura, M.; Irié, T.; Ogura, H.; Hijikata, A.; Bin, B.-H.; Yasuda, T.; Kitamura, H.; et al. Zinc transporter SLC39A10/ZIP10 facilitates antiapoptotic signaling during early B-cell development. Proc. Natl. Acad. Sci. USA 2014, 111, 11780–11785. [Google Scholar] [CrossRef]
- Franz, M.C.; Pujol-Giménez, J.; Montalbetti, N.; Fernandez-Tenorio, M.; DeGrado, T.R.; Niggli, E.; Romero, M.F.; Hediger, M.A. Reassessment of the Transport Mechanism of the Human Zinc Transporter SLC39A2. Biochemistry 2018, 57, 3976–3986. [Google Scholar] [CrossRef]
- Franklin, L.C.C.R.B. Evidence that Human Prostate Cancer is a ZIP1-Deficient Malignancy that could be Effectively Treated with a Zinc Ionophore (Clioquinol) Approach. Chemother. Open Access 2014, 4, 1–10. [Google Scholar] [CrossRef]
- Li, M.; Zhang, Y.; Liu, Z.; Bharadwaj, U.; Wang, H.; Wang, X.; Zhang, S.; Liuzzi, J.P.; Chang, S.-M.; Cousins, R.J.; et al. Aberrant expression of zinc transporter ZIP4 (SLC39A4) significantly contributes to human pancreatic cancer pathogenesis and progression. Proc. Natl. Acad. Sci. USA 2007, 104, 18636–18641. [Google Scholar] [CrossRef]
- Taylor, K.M.; Morgan, H.E.; Smart, K.; Zahari, N.M.; Pumford, S.; Ellis, I.O.; Robertson, J.F.R.; Nicholson, R.I. The Emerging Role of the LIV-1 Subfamily of Zinc Transporters in Breast Cancer. Mol. Med. 2007, 13, 396–406. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Chaffee, K.G.; Parker, A.S.; Sicotte, H.; Petersen, G.M. Zinc transporter genes and urological cancers: Integrated analysis suggests a role for ZIP11 in bladder cancer. Tumor Biol. 2015, 36, 7431–7437. [Google Scholar] [CrossRef] [PubMed]
- Hoch, E.; Levy, M.; Hershfinkel, M.; Sekler, I. Elucidating the H+ Coupled Zn2+ Transport Mechanism of ZIP4; Implications in Acrodermatitis Enteropathica. Int. J. Mol. Sci. 2020, 21, 734. [Google Scholar] [CrossRef] [PubMed]
- Bin, B.-H.; Fukada, T.; Hosaka, T.; Yamasaki, S.; Ohashi, W.; Hojyo, S.; Miyai, T.; Nishida, K.; Yokoyama, S.; Hirano, T. Biochemical characterization of human ZIP13 protein a homo-dimerized zinc transporter involved in the spondylocheiro dysplastic ehlers-danlos syndrome. J. Biol. Chem. 2011, 286, 40255–40265. [Google Scholar] [CrossRef]
- Dusanic, M.; Dekomien, G.; Lucke, T.; Vorgerd, M.; Weiß, J.; Epplen, J.T.; Köhler, C.; Hoffjan, S. Novel Nonsense Mutation in SLC39A13 Initially Presenting as Myopathy: Case Report and Review of the Literature. Mol. Syndr. 2018, 9, 100–109. [Google Scholar] [CrossRef]
- Zhang, T.; Sui, D.; Zhang, C.; Cole, L.; Hu, J. Asymmetric functions of a binuclear metal center within the transport pathway of a human zinc transporter ZIP4. FASEB J. 2020, 34, 237–247. [Google Scholar] [CrossRef]
- Zhang, T.; Liu, J.; Fellner, M.; Zhang, C.; Sui, D.; Hu, J. Crystal structures of a ZIP zinc transporter reveal a binuclear metal center in the transport pathway. Sci. Adv. 2017, 3, e1700344. [Google Scholar] [CrossRef]
- Zhang, T.; Sui, D.; Hu, J. Structural insights of ZIP4 extracellular domain critical for optimal zinc transport. Nat. Commun. 2016, 7, 11979. [Google Scholar] [CrossRef]
- Dempski, R.E. The Cation Selectivity of the ZIP Transporters. In Na Channels from Phyla to Function; Elsevier: Amsterdam, The Netherlands, 2012; Volume 69, pp. 221–245. [Google Scholar]
- Blindauer, C.A. Advances in the molecular understanding of biological zinc transport. Chem. Commun. 2015, 51, 4544–4563. [Google Scholar] [CrossRef]
- Lin, W.; Chai, J.; Love, J.; Fu, D. Selective Electrodiffusion of Zinc Ions in a Zrt-, Irt-like Protein, ZIPB. J. Biol. Chem. 2010, 285, 39013–39020. [Google Scholar] [CrossRef]
- Ma, C.; Hao, Z.; Huysmans, G.; Lesiuk, A.; Bullough, P.; Wang, Y.; Bartlam, M.; Phillips, S.E.; Young, J.D.; Goldman, A.; et al. A Versatile Strategy for Production of Membrane Proteins with Diverse Topologies: Application to Investigation of Bacterial Homologues of Human Divalent Metal Ion and Nucleoside Transporters. PLoS ONE 2015, 10, e0143010. [Google Scholar] [CrossRef] [PubMed]
- Jia, B.; Jeon, C.O. High-Throughput Recombinant Protein Expression in Escherichia Coli: Current Status and Future Perspec-tives. Open Biol. 2016, 6, 2046–2441. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.; Shin, K.; Patterson, R.E.; Liu, X.Q.; Rainey, J.K. Current Strategies for Protein Production and Purification Ena-bling Membrane Protein Structural Biology1. Biochem. Cell Biol. 2016, 94, 507–527. [Google Scholar] [CrossRef] [PubMed]
- Terpe, K. Overview of bacterial expression systems for heterologous protein production: From molecular and biochemical fundamentals to commercial systems. Appl. Microbiol. Biotechnol. 2006, 72, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.R.; Byregowda, S.M.; Veeregowda, B.M.; Balamurugan, V. An Overview of Heterologous Expression Host Systems for the Production of Recombinant Proteins. Adv. Anim. Veter- Sci. 2016, 4, 346–356. [Google Scholar] [CrossRef]
- Liu, Z.; Tyo, K.E.; Martínez, J.L.; Petranovic, D.; Nielsen, J. Different expression systems for production of recombinant proteins in Saccharomyces cerevisiae. Biotechnol. Bioeng. 2012, 109, 1259–1268. [Google Scholar] [CrossRef]
- Hou, J.; Tyo, K.E.J.; Liu, Z.; Petranovic, D.; Nielsen, J. Metabolic Engineering of Recombinant Protein Secretion by Saccharo-myces Cerevisiae. FEMS Yeast Res. 2012, 12, 491–510. [Google Scholar] [CrossRef]
- Gotfryd, K.; Mósca, A.F.; Missel, J.W.; Truelsen, S.F.; Wang, K.; Spulber, M.; Krabbe, S.L.; Nielsen, C.; Laforenza, U.; Soveral, G.; et al. Human adipose glycerol flux is regulated by a pH gate in AQP10. Nat. Commun. 2018, 9, 4749. [Google Scholar] [CrossRef]
- Wang, K.; Preisler, S.S.; Zhang, L.; Cui, Y.; Missel, J.W.; Grønberg, C.; Gotfryd, K.; Lindahl, E.; Andersson, M.; Calloe, K.; et al. Structure of the human ClC-1 chloride channel. PLoS Biol. 2019, 17, e3000218. [Google Scholar] [CrossRef]
- Molbaek, K.; Scharff-Poulsen, P.; Nielsen, C.; Klaerke, D.A.; Pedersen, P.A. High yield purification of full-length functional hERG K+ channels produced in Saccharomyces cerevisiae. Microb. Cell Factories 2015, 14, 15. [Google Scholar] [CrossRef]
- Bjørkskov, F.B.; Krabbe, S.L.; Nurup, C.N.; Missel, J.W.; Spulber, M.; Bomholt, J.; Molbaek, K.; Helix-Nielsen, C.; Gotfryd, K.; Gourdon, P.E.; et al. Purification and functional comparison of nine human Aquaporins produced in Saccharomyces cerevisiae for the purpose of biophysical characterization. Sci. Rep. 2017, 7, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, K.; Klaerke, D.A.; Calloe, K.; Lowrey, L.; Pedersen, P.A.; Gourdon, P.; Gotfryd, K. Purification of Functional Human TRP Channels Recombinantly Produced in Yeast. Cells 2019, 8, 148. [Google Scholar] [CrossRef] [PubMed]
- Cesareni, G.; Murray, J.A.H. Plasmid Vectors Carrying the Replication Origin of Filamentous Single-Stranded Phages. In Genetic Engineering; Springer: Berlin, Germany, 1987; pp. 135–154. [Google Scholar]
- Erhart, E.; Hollenberg, C.P. The presence of a defective LEU2 gene on 2 mu DNA recombinant plasmids of Saccharomyces cerevisiae is responsible for curing and high copy number. J. Bacteriol. 1983, 156, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Stecher, G.; Tamura, K.; Kumar, S. Molecular Evolutionary Genetics Analysis (MEGA) for macOS. Mol. Biol. Evol. 2020, 37, 1237–1239. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, P.A.; Rasmussen, J.H.; Jørgensen, P.L. Expression in High Yield of Pig α1β1 Na,K-ATPase and Inactive Mutants D369N and D807N in Saccharomyces cerevisiae. J. Biol. Chem. 1996, 271, 2514–2522. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-Dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Drew, D.; Newstead, S.; Sonoda, Y.; Kim, H.; Von Heijne, G.; Iwata, S. GFP-based optimization scheme for the overexpression and purification of eukaryotic membrane proteins in Saccharomyces cerevisiae. Nat. Protoc. 2008, 3, 784–798. [Google Scholar] [CrossRef]
- Tsirigos, K.D.; Peters, C.; Shu, N.; Käll, L.; Elofsson, A. The TOPCONS web server for consensus prediction of membrane protein topology and signal peptides. Nucleic Acids Res. 2015, 43, W401–W407. [Google Scholar] [CrossRef]
- The UniProt Consortium UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [CrossRef]
- Arachea, B.T.; Sun, Z.; Potente, N.; Malik, R.; Isailovic, D.; Viola, R.E. Detergent selection for enhanced extraction of membrane proteins. Protein Expr. Purif. 2012, 86, 12–20. [Google Scholar] [CrossRef]
- Stetsenko, A.; Guskov, A. An Overview of the Top Ten Detergents Used for Membrane Protein Crystallization. Crystals 2017, 7, 197. [Google Scholar] [CrossRef]
- Kawate, T.; Gouaux, E. Fluorescence-Detection Size-Exclusion Chromatography for Precrystallization Screening of Integral Membrane Proteins. Structure 2006, 14, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Geertsma, E.R.; Groeneveld, M.; Slotboom, D.-J.; Poolman, B. Quality control of overexpressed membrane proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 5722–5727. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.G.M.; Skerra, A. The Strep-tag system for one-step purification and high-affinity detection or capturing of proteins. Nat. Protoc. 2007, 2, 1528–1535. [Google Scholar] [CrossRef] [PubMed]
- Arinaminpathy, Y.; Khurana, E.; Engelman, D.M.; Gerstein, M.B. Computational analysis of membrane proteins: The largest class of drug targets. Drug Discov. Today 2009, 14, 1130–1135. [Google Scholar] [CrossRef]
- Edwards, A.M.; Berman, J.; Sundström, M. Structural Genomics and Drug Discovery. Annu. Rep. Med. Chem. 2005, 40, 349–369. [Google Scholar] [CrossRef]
- Kotov, V.; Bartels, K.; Veith, K.; Josts, I.; Subhramanyam, U.K.T.; Günther, C.; Labahn, J.; Marlovits, T.C.; Moraes, I.; Tidow, H.; et al. High-throughput stability screening for detergent-solubilized membrane proteins. Sci. Rep. 2019, 9, 1–19. [Google Scholar] [CrossRef]
- Almeida, J.G.; Preto, A.J.; Koukos, P.I.; Bonvin, A.M.J.J.; Moreira, I.S. Membrane Proteins Structures: A Review on Computa-tional Modeling Tools. Biochim. Biophys. Acta Biomembr. 2017, 1859, 2021–2039. [CrossRef]
- Andréll, J.; Tate, C.G. Overexpression of membrane proteins in mammalian cells for structural studies. Mol. Membr. Biol. 2012, 30, 52–63. [Google Scholar] [CrossRef]
- Lin, C.-Y.; Huang, Z.; Wen, W.; Wu, A.; Wang, C.; Niu, L. Enhancing Protein Expression in HEK-293 Cells by Lowering Culture Temperature. PLoS ONE 2015, 10, e0123562. [Google Scholar] [CrossRef]
- Rana, M.S.; Wang, X.; Banerjee, A. An Improved Strategy for Fluorescent Tagging of Membrane Proteins for Overexpression and Purification in Mammalian Cells. Biochemistry 2018, 57, 6741–6751. [Google Scholar] [CrossRef] [PubMed]
- Molbaek, K.; Tejada, M.; Ricke, C.H.; Scharff-Poulsen, P.; Ellekvist, P.; Helix-Nielsen, C.; Kumar, N.; Klaerke, D.A.; Pedersen, P.A. Purification and initial characterization of Plasmodium falciparum K+ channels, PfKch1 and PfKch2 produced in Saccharomyces cerevisiae. Microb. Cell Factories 2020, 19, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Drew, D.; Lerch, M.; Kunji, E.R.S.; Slotboom, D.-J.; De Gier, J.-W. Optimization of membrane protein overexpression and purification using GFP fusions. Nat. Chem. Biol. 2006, 3, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Fantini, J.; Barrantes, F.J. How cholesterol interacts with membrane proteins: An exploration of cholesterol-binding sites including CRAC, CARC, and tilted domains. Front. Physiol. 2013, 4, 31. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, A.K.; Wiener, M.C. Membrane protein expression and production: Effects of polyhistidine tag length and position. Protein Expr. Purif. 2004, 33, 311–325. [Google Scholar] [CrossRef] [PubMed]
- Booth, W.T.; Schlachter, C.R.; Pote, S.; Ussin, N.; Mank, N.J.; Klapper, V.; Offermann, L.R.; Tang, C.; Hurlburt, B.K.; Chruszcz, M. Impact of an N-terminal Polyhistidine Tag on Protein Thermal Stability. ACS Omega 2018, 3, 760–768. [Google Scholar] [CrossRef]
- Chun, E.; Thompson, A.A.; Liu, W.; Roth, C.B.; Griffith, M.T.; Katritch, V.; Kunken, J.; Xu, F.; Cherezov, V.; Hanson, M.A.; et al. Fusion Partner Toolchest for the Stabilization and Crystallization of G Protein-Coupled Receptors. Structure 2012, 20, 967–976. [Google Scholar] [CrossRef]
- Wallin, E.; Von Heijne, G. Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Sci. 1998, 7, 1029–1038. [Google Scholar] [CrossRef]
- Locatelli-Hoops, S.C.; Yeliseev, A.A. Use of tandem affinity chromatography for purification of cannabinoid receptor CB2. Breast Cancer 2014, 1177, 107–120. [Google Scholar] [CrossRef]
- Newby, Z.E.R.; O’Connell, J.D.; Gruswitz, F.; Hays, F.A.; Harries, W.E.C.; Harwood, I.M.; Ho, J.D.; Lee, J.K.; Savage, D.F.; Miercke, L.J.W.; et al. A general protocol for the crystallization of membrane proteins for X-ray structural investigation. Nat. Protoc. 2009, 4, 619–637. [Google Scholar] [CrossRef]
- Tonggu, L.; Wang, L. Cryo-EM sample preparation method for extremely low concentration liposomes. Ultramicroscopy 2020, 208, 112849. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Becares, E.R.; Pedersen, P.A.; Gourdon, P.; Gotfryd, K. Overproduction of Human Zip (SLC39) Zinc Transporters in Saccharomyces cerevisiae for Biophysical Characterization. Cells 2021, 10, 213. https://doi.org/10.3390/cells10020213
Becares ER, Pedersen PA, Gourdon P, Gotfryd K. Overproduction of Human Zip (SLC39) Zinc Transporters in Saccharomyces cerevisiae for Biophysical Characterization. Cells. 2021; 10(2):213. https://doi.org/10.3390/cells10020213
Chicago/Turabian StyleBecares, Eva Ramos, Per Amstrup Pedersen, Pontus Gourdon, and Kamil Gotfryd. 2021. "Overproduction of Human Zip (SLC39) Zinc Transporters in Saccharomyces cerevisiae for Biophysical Characterization" Cells 10, no. 2: 213. https://doi.org/10.3390/cells10020213
APA StyleBecares, E. R., Pedersen, P. A., Gourdon, P., & Gotfryd, K. (2021). Overproduction of Human Zip (SLC39) Zinc Transporters in Saccharomyces cerevisiae for Biophysical Characterization. Cells, 10(2), 213. https://doi.org/10.3390/cells10020213