
Early Life Inflammation and the Developing Hematopoietic and Immune Systems: The Cochlea as a Sensitive Indicator of Disruption

 ,
, 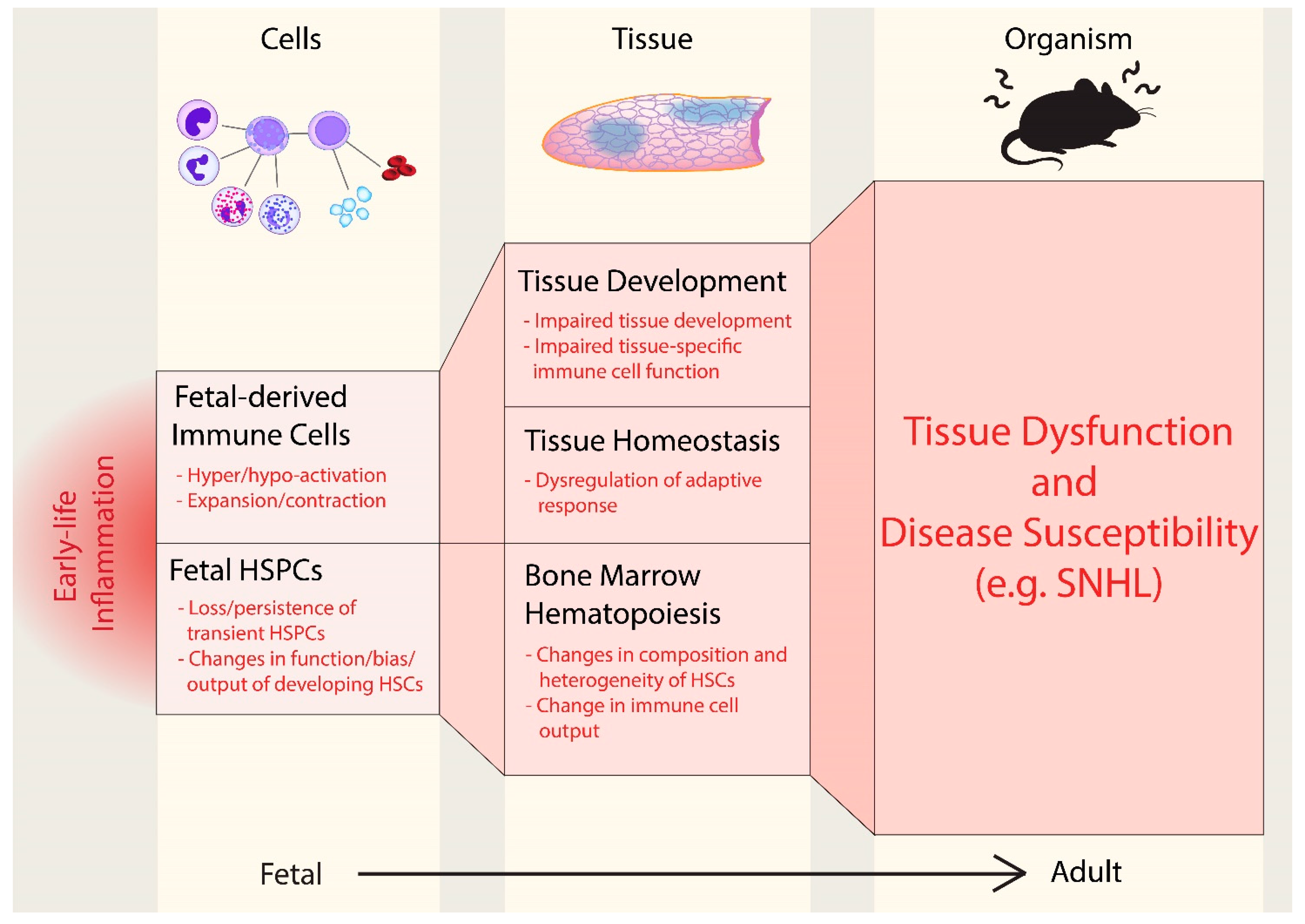{kind=link}
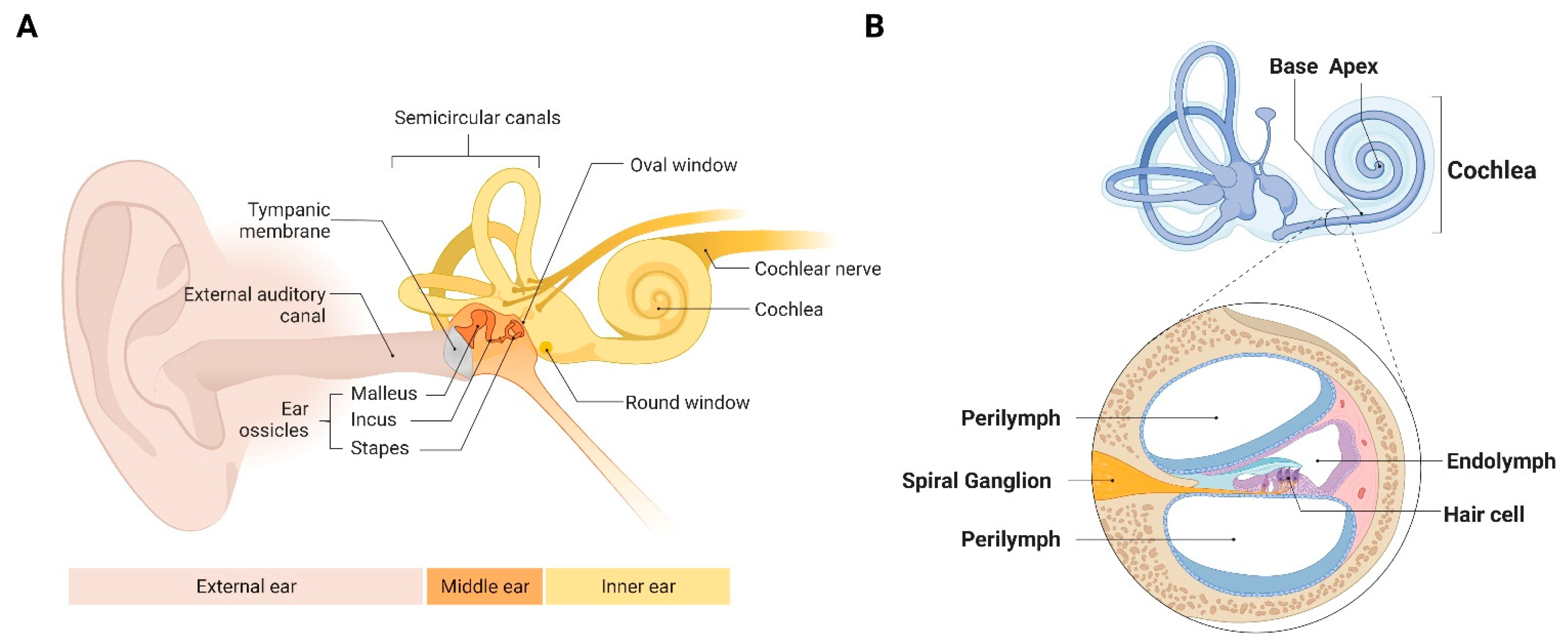{kind=link}
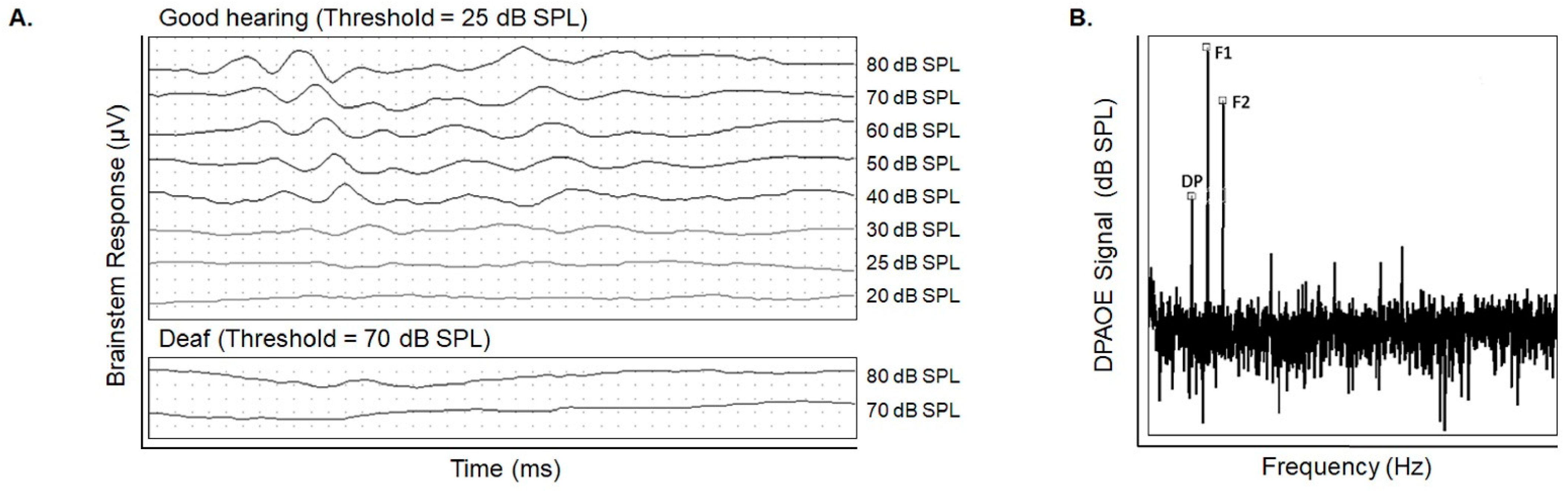{kind=link}
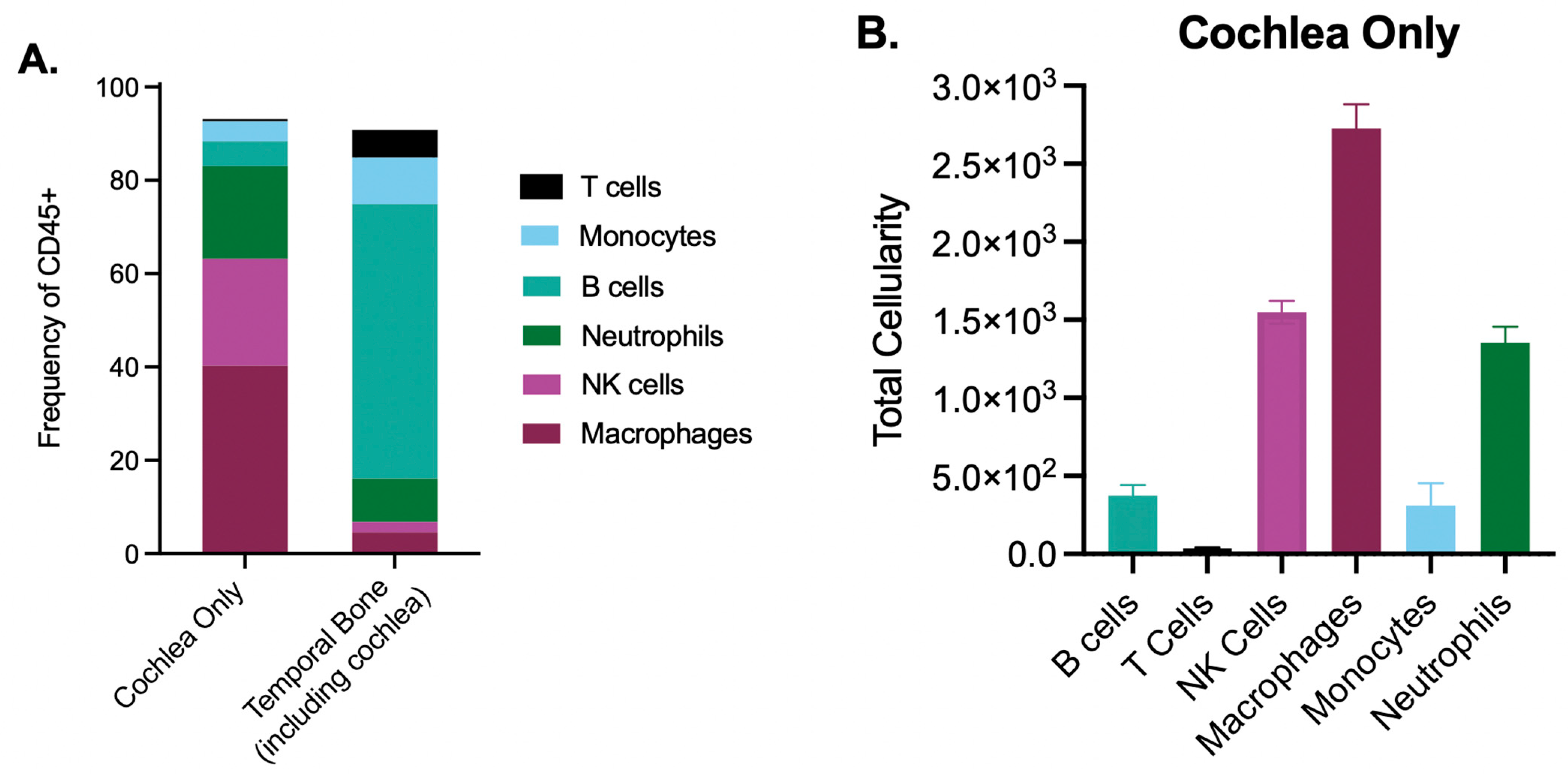{kind=link}
{kind=link}
Abstract
:1. Perinatal Inflammation Shapes Offspring Immunity
1.1. Contribution of Fetal-Derived Immune Cells to Tissue Immunity
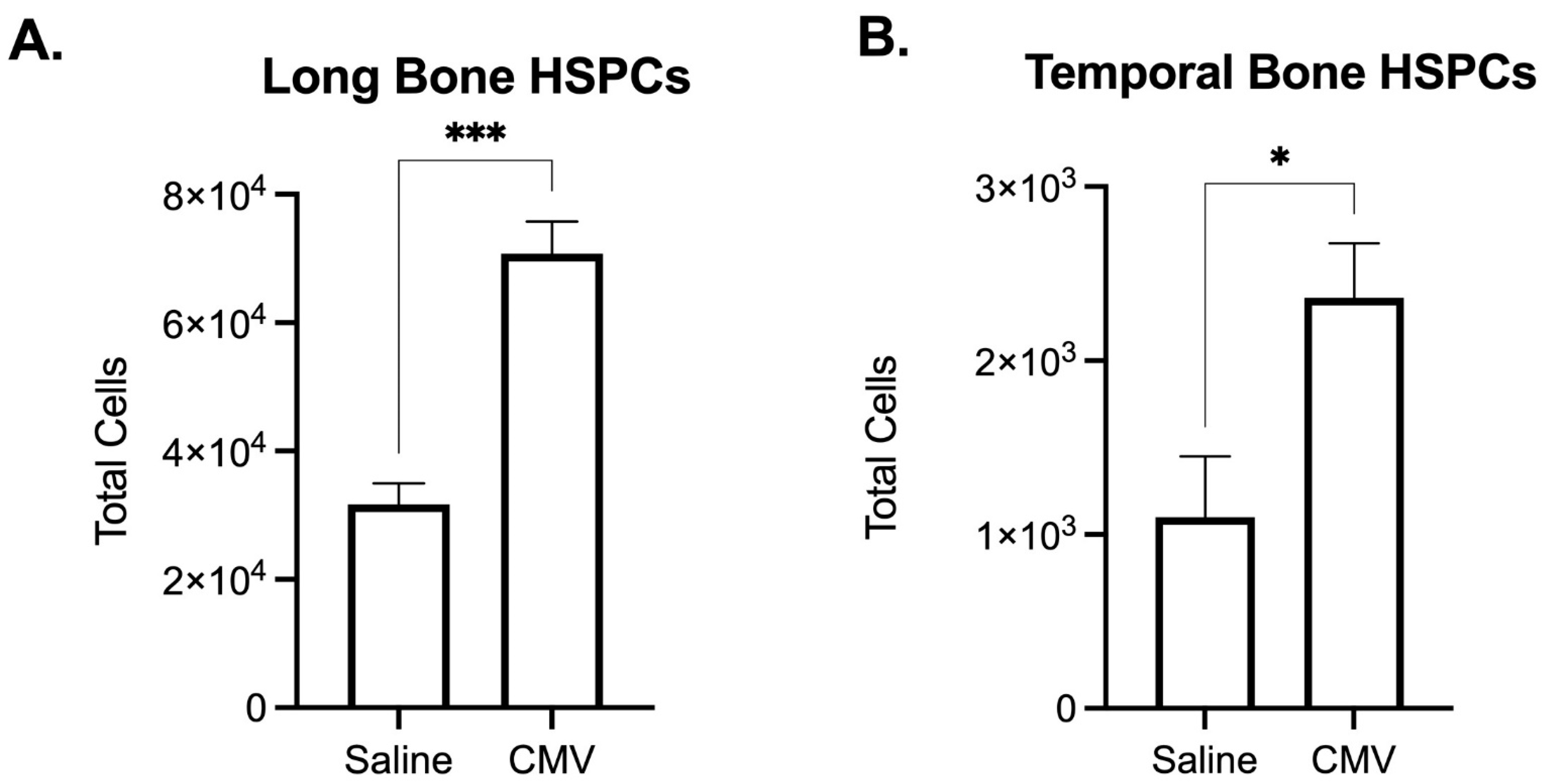
1.2. Susceptibility of Hematopoietic Stem Cell Development to Perinatal Inflammation
2. The Cochlea as a Model Organ to Study the Effect of Early Life Inflammation on the Developing Hematopoietic and Immune Systems
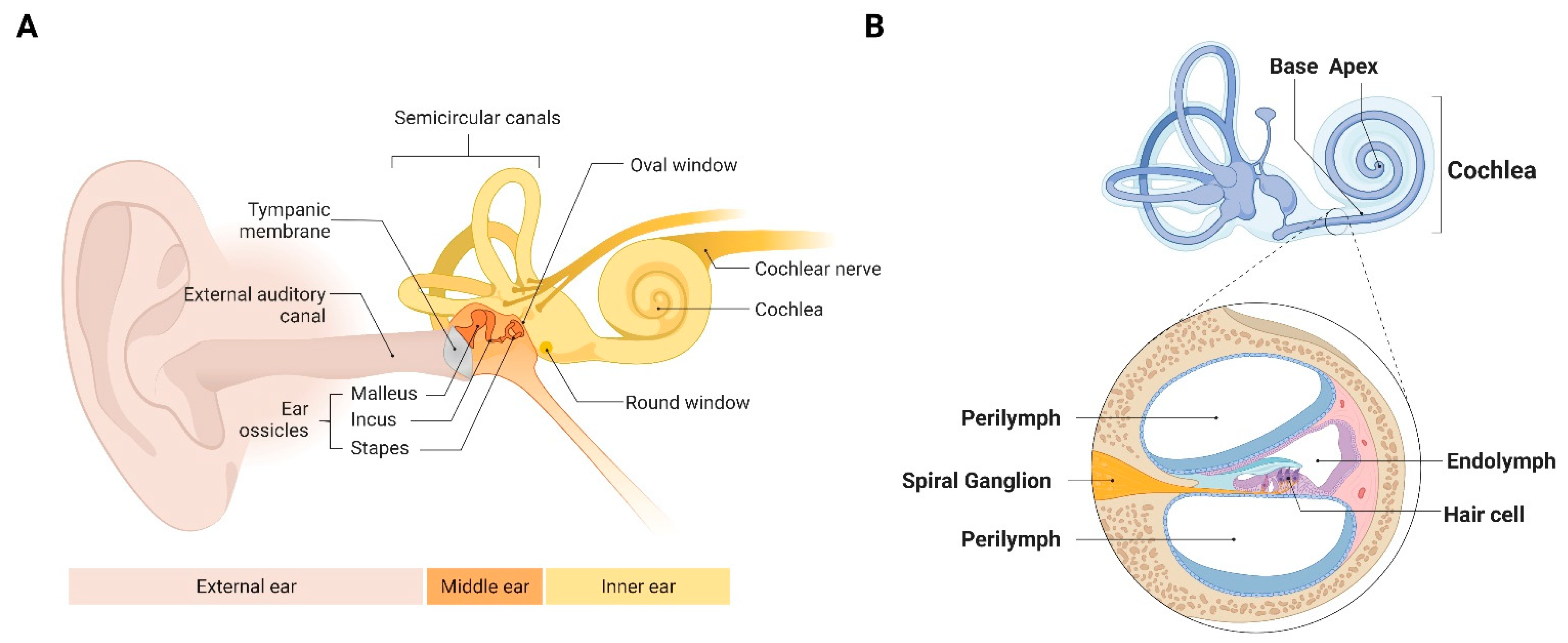
2.1. The Cochlea Is Sensitive to a Broad Range of Inputs
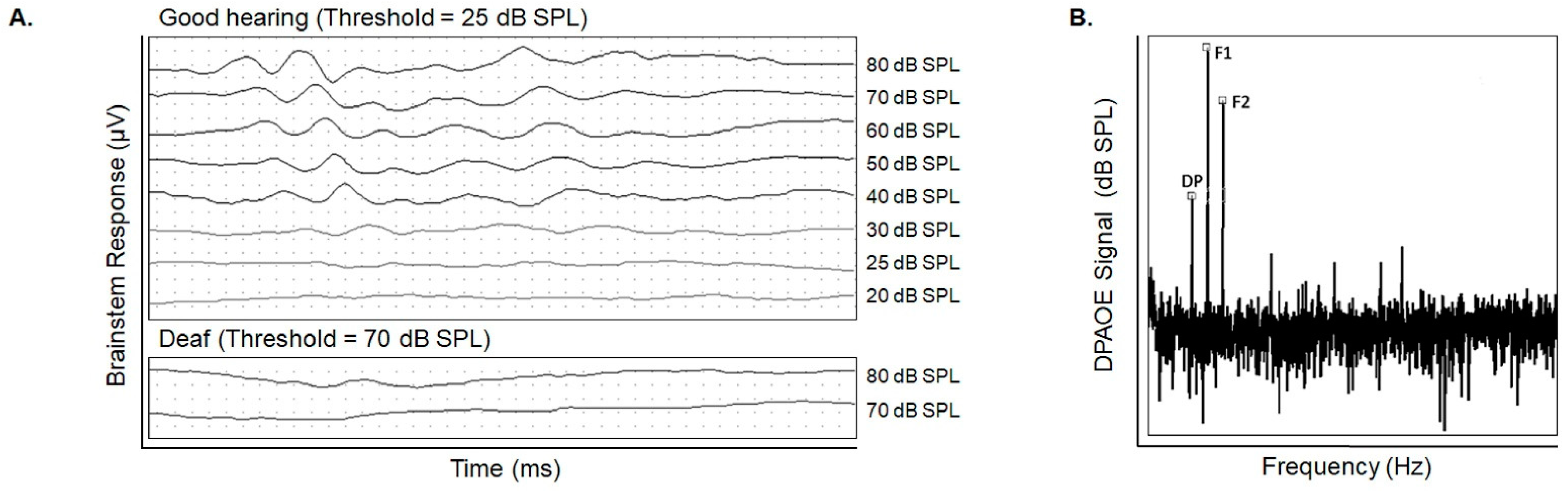
2.2. Hearing Testing Accurately Assesses Cochlear Damage
2.3. The Cochlea Contains a Rich, Unexplored Immune Milieu
2.4. The Developing Cochlea Contains Both Immune and Hematopoietic Stem and Progenitor Cells
2.5. Persistent Inflammation May Contribute to SNHL
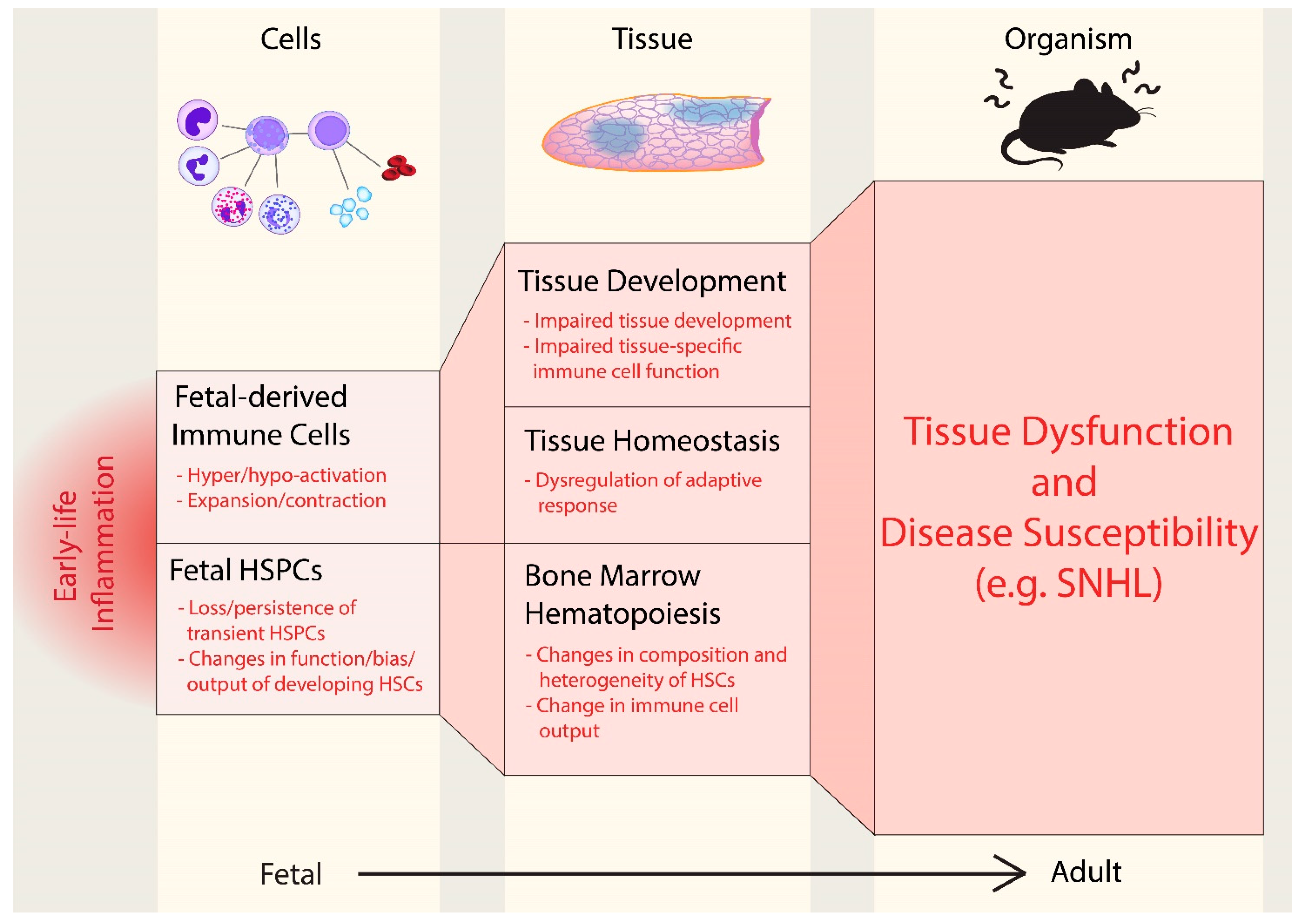
3. Conclusions: Perturbation of Fetal Immunity as a Driver of SNHL
4. Materials and Methods
4.1. Mice
4.2. Viruses
4.3. Viral Inoculation
4.4. Cochlea Tissue Processing
4.5. Temporal Bone Processing
4.6. Long Bone Processing
4.7. Flow Cytometry
4.8. Gating Strategies for Flow Cytometry
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti, A.; Little, P. Nature, nurture and human disease. Nature 2003, 421, 412–414. [Google Scholar] [CrossRef] [PubMed]
- Hemminki, K.; Lorenzo Bermejo, J.; Forsti, A. The balance between heritable and environmental aetiology of human disease. Nat. Rev. Genet. 2006, 7, 958–965. [Google Scholar] [CrossRef]
- Barker, D.J.; Osmond, C. Infant mortality, childhood nutrition, and ischaemic heart disease in England and Wales. Lancet 1986, 1, 1077–1081. [Google Scholar] [CrossRef]
- Barker, D.J.; Eriksson, J.G.; Forsen, T.; Osmond, C. Fetal origins of adult disease: Strength of effects and biological basis. Int. J. Epidemiol. 2002, 31, 1235–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barker, D.J.; Winter, P.D.; Osmond, C.; Margetts, B.; Simmonds, S.J. Weight in infancy and death from ischaemic heart disease. Lancet 1989, 2, 577–580. [Google Scholar] [CrossRef]
- Wadhwa, P.D.; Buss, C.; Entringer, S.; Swanson, J.M. Developmental origins of health and disease: Brief history of the approach and current focus on epigenetic mechanisms. Semin. Reprod. Med. 2009, 27, 358–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdiguero, E.G.; Geissmann, F. The development and maintenance of resident macrophages. Nat. Immunol. 2016, 17, 2–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kristiansen, T.A.; Jaensson Gyllenback, E.; Zriwil, A.; Bjorklund, T.; Daniel, J.A.; Sitnicka, E.; Soneji, S.; Bryder, D.; Yuan, J. Cellular Barcoding Links B-1a B Cell Potential to a Fetal Hematopoietic Stem Cell State at the Single-Cell Level. Immunity 2016, 45, 346–357. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.; Lee, J.; Koga, S.; Ricardo-Gonzalez, R.R.; Nussbaum, J.C.; Smith, L.K.; Villeda, S.A.; Liang, H.E.; Locksley, R.M. Tissue-Resident Group 2 Innate Lymphoid Cells Differentiate by Layered Ontogeny and In Situ Perinatal Priming. Immunity 2019, 50, 1425–1438. [Google Scholar] [CrossRef] [PubMed]
- Squarzoni, P.; Oller, G.; Hoeffel, G.; Pont-Lezica, L.; Rostaing, P.; Low, D.; Bessis, A.; Ginhoux, F.; Garel, S. Microglia modulate wiring of the embryonic forebrain. Cell Rep. 2014, 8, 1271–1279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munro, D.A.D.; Wineberg, Y.; Tarnick, J.; Vink, C.S.; Li, Z.; Pridans, C.; Dzierzak, E.; Kalisky, T.; Hohenstein, P.; Davies, J.A. Macrophages restrict the nephrogenic field and promote endothelial connections during kidney development. eLife 2019, 8, e43271. [Google Scholar] [CrossRef]
- Shigeta, A.; Huang, V.; Zuo, J.; Besada, R.; Nakashima, Y.; Lu, Y.; Ding, Y.; Pellegrini, M.; Kulkarni, R.P.; Hsiai, T.; et al. Endocardially Derived Macrophages Are Essential for Valvular Remodeling. Dev. Cell 2019, 48, 617–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeFalco, T.; Bhattacharya, I.; Williams, A.V.; Sams, D.M.; Capel, B. Yolk-sac-derived macrophages regulate fetal testis vascularization and morphogenesis. Proc. Natl. Acad. Sci. USA 2014, 111, E2384–E2393. [Google Scholar] [CrossRef] [Green Version]
- Nobs, S.P.; Kopf, M. Tissue-resident macrophages: Guardians of organ homeostasis. Trends Immunol. 2021, 42, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.M.; Hamidzadeh, K.; Goncalves, R. Macrophages and the maintenance of homeostasis. Cell Mol. Immunol. 2021, 18, 579–587. [Google Scholar] [CrossRef]
- Gordon, S.; Pluddemann, A. Tissue macrophages: Heterogeneity and functions. BMC Biol. 2017, 15, 53. [Google Scholar] [CrossRef] [PubMed]
- Strachan, D.P. Hay fever, hygiene, and household size. BMJ 1989, 299, 1259–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apostol, A.C.; Jensen, K.D.C.; Beaudin, A.E. Training the Fetal Immune System Through Maternal Inflammation-A Layered Hygiene Hypothesis. Front. Immunol. 2020, 11, 123. [Google Scholar] [CrossRef] [PubMed]
- Saso, A.; Kampmann, B. Vaccine responses in newborns. Semin. Immunopathol. 2017, 39, 627–642. [Google Scholar] [CrossRef] [Green Version]
- Ege, M.J.; Mayer, M.; Normand, A.C.; Genuneit, J.; Cookson, W.O.; Braun-Fahrlander, C.; Heederik, D.; Piarroux, R.; von Mutius, E. Exposure to environmental microorganisms and childhood asthma. N. Engl. J. Med. 2011, 364, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J.L.; Shi, H.N.; Walker, W.A. The role of microbes in developmental immunologic programming. Pediatr. Res. 2011, 69, 465–472. [Google Scholar] [CrossRef] [Green Version]
- Dauby, N.; Goetghebuer, T.; Kollmann, T.R.; Levy, J.; Marchant, A. Uninfected but not unaffected: Chronic maternal infections during pregnancy, fetal immunity, and susceptibility to postnatal infections. Lancet Infect. Dis. 2012, 12, 330–340. [Google Scholar] [CrossRef]
- Hadland, B.; Yoshimoto, M. Many layers of embryonic hematopoiesis: New insights into B-cell ontogeny and the origin of hematopoietic stem cells. Exp. Hematol. 2018, 60, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Gentek, R.; Ghigo, C.; Hoeffel, G.; Jorquera, A.; Msallam, R.; Wienert, S.; Klauschen, F.; Ginhoux, F.; Bajénoff, M. Epidermal γδ T cells originate from yolk sac hematopoiesis and clonally self-renew in the adult. J. Exp. Med. 2018, 215, 2994–3005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentek, R.; Ghigo, C.; Hoeffel, G.; Bulle, M.J.; Msallam, R.; Gautier, G.; Launay, P.; Chen, J.; Ginhoux, F.; Bajénoff, M. Hemogenic Endothelial Fate Mapping Reveals Dual Developmental Origin of Mast Cells. Immunity 2018, 48, 1160–1171.e5. [Google Scholar] [CrossRef] [Green Version]
- Ginhoux, F.; Guilliams, M. Tissue-Resident Macrophage Ontogeny and Homeostasis. Immunity 2016, 44, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Bian, Z.; Gong, Y.; Huang, T.; Lee, C.Z.W.; Bian, L.; Bai, Z.; Shi, H.; Zeng, Y.; Liu, C.; He, J.; et al. Deciphering human macrophage development at single-cell resolution. Nature 2020, 582, 571–576. [Google Scholar] [CrossRef]
- Lavin, Y.; Mortha, A.; Rahman, A.; Merad, M. Regulation of macrophage development and function in peripheral tissues. Nat. Rev. Immunol. 2015, 15, 731–744. [Google Scholar] [CrossRef] [Green Version]
- Lavin, Y.; Winter, D.; Blecher-Gonen, R.; David, E.; Keren-Shaul, H.; Merad, M.; Jung, S.; Amit, I. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 2014, 159, 1312–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amit, I.; Winter, D.R.; Jung, S. The role of the local environment and epigenetics in shaping macrophage identity and their effect on tissue homeostasis. Nat. Immunol. 2016, 17, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, D.; Chow, A.; Noizat, C.; Teo, P.; Beasley, M.B.; Leboeuf, M.; Becker, C.D.; See, P.; Price, J.; Lucas, D.; et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 2013, 38, 792–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalmau, I.; Finsen, B.; Tønder, N.; Zimmer, J.; González, B.; Castellano, B. Development of microglia in the prenatal rat hippocampus. J. Comp. Neurol. 1997, 377, 70–84. [Google Scholar] [CrossRef]
- Alain, B.; Echade, C.; Delphin, B.; Anne, R. Microglial Control of Neuronal Death and Synaptic Properties. Glia 2007, 55, 233–238. [Google Scholar]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef] [Green Version]
- De Schepper, S.; Verheijden, S.; Aguilera-Lizarraga, J.; Viola, M.F.; Boesmans, W.; Stakenborg, N.; Voytyuk, I.; Schmidt, I.; Boeckx, B.; Dierckx de Casterle, I.; et al. Self-Maintaining Gut Macrophages Are Essential for Intestinal Homeostasis. Cell 2018, 175, 400–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, N.R.; Mohan, J.; Kopecky, B.J.; Guo, S.; Du, L.; Leid, J.; Feng, G.; Lokshina, I.; Dmytrenko, O.; Luehmann, H.; et al. Resident cardiac macrophages mediate adaptive myocardial remodeling. Immunity 2021, 54, 2072–2088. [Google Scholar] [CrossRef] [PubMed]
- Hulsmans, M.; Clauss, S.; Xiao, L.; Aguirre, A.D.; King, K.R.; Hanley, A.; Hucker, W.J.; Wülfers, E.M.; Seemann, G.; Courties, G.; et al. Macrophages Facilitate Electrical Conduction in the Heart. Cell 2017, 169, 510–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guilliams, M.; De Kleer, I.; Henri, S.; Post, S.; Vanhoutte, L.; De Prijck, S.; Deswarte, K.; Malissen, B.; Hammad, H.; Lambrecht, B.N. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J. Exp. Med. 2013, 210, 1977–1992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dick, S.A.; Macklin, J.A.; Nejat, S.; Momen, A.; Clemente-Casares, X.; Althagafi, M.G.; Chen, J.; Kantores, C.; Hosseinzadeh, S.; Aronoff, L.; et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat. Immunol. 2019, 20, 29–39. [Google Scholar] [CrossRef] [PubMed]
- van de Laar, L.; Saelens, W.; De Prijck, S.; Martens, L.; Scott, C.L.; Van Isterdael, G.; Hoffmann, E.; Beyaert, R.; Saeys, Y.; Lambrecht, B.N.; et al. Yolk Sac Macrophages, Fetal Liver, and Adult Monocytes Can Colonize an Empty Niche and Develop into Functional Tissue-Resident Macrophages. Immunity 2016, 44, 755–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cronk, J.C.; Filiano, A.J.; Louveau, A.; Marin, I.; Marsh, R.; Ji, E.; Goldman, D.H.; Smirnov, I.; Geraci, N.; Acton, S.; et al. Peripherally derived macrophages can engraft the brain independent of irradiation and maintain an identity distinct from microglia. J. Exp. Med. 2018, 215, 1627–1647. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Herndon, J.M.; Sojka, D.K.; Kim, K.W.; Knolhoff, B.L.; Zuo, C.; Cullinan, D.R.; Luo, J.; Bearden, A.R.; Lavine, K.J.; et al. Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate from Embryonic Hematopoiesis and Promote Tumor Progression. Immunity 2017, 47, 597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, M.A.; Metcalf, D. Ontogeny of the haemopoietic system: Yolk sac origin of in vivo and in vitro colony forming cells in the developing mouse embryo. Br. J. Haematol. 1970, 18, 279–296. [Google Scholar] [CrossRef] [PubMed]
- Palis, J. Hematopoietic stem cell-independent hematopoiesis: Emergence of erythroid, megakaryocyte, and myeloid potential in the mammalian embryo. FEBS Lett. 2016, 590, 3965–3974. [Google Scholar] [CrossRef] [PubMed]
- Muller, A.M.; Medvinsky, A.; Strouboulis, J.; Grosveld, F.; Dzierzak, E. Development of hematopoietic stem cell activity in the mouse embryo. Immunity 1994, 1, 291–301. [Google Scholar] [CrossRef] [Green Version]
- Medvinsky, A.; Dzierzak, E. Definitive hematopoiesis is autonomously initiated by the AGM region. Cell 1996, 86, 897–906. [Google Scholar] [CrossRef] [Green Version]
- McGrath, K.; Palis, J. Ontogeny of erythropoiesis in the mammalian embryo. Curr. Top. Dev. Biol. 2008, 82, 1–22. [Google Scholar] [CrossRef]
- Herzenberg, L.A. Toward a layered immune system. Cell 1989, 59, 953–954. [Google Scholar] [CrossRef]
- Smith, N.L.; Patel, R.K.; Reynaldi, A.; Grenier, J.K.; Wang, J.; Watson, N.B.; Nzingha, K.; Yee Mon, K.J.; Peng, S.A.; Grimson, A.; et al. Developmental Origin Governs CD8. Cell 2018, 174, 117–130.e114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Ramos, A.; Chapman, B.; Johnnidis, J.B.; Le, L.; Ho, Y.J.; Klein, A.; Hofmann, O.; Camargo, F.D. Clonal dynamics of native haematopoiesis. Nature 2014, 514, 322–327. [Google Scholar] [CrossRef]
- Busch, K.; Klapproth, K.; Barile, M.; Flossdorf, M.; Holland-Letz, T.; Schlenner, S.M.; Reth, M.; Höfer, T.; Rodewald, H.R. Fundamental properties of unperturbed haematopoiesis from stem cells in vivo. Nature 2015, 518, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Beaudin, A.E.; Boyer, S.W.; Perez-Cunningham, J.; Hernandez, G.E.; Derderian, S.C.; Jujjavarapu, C.; Aaserude, E.; MacKenzie, T.; Forsberg, E.C. A Transient Developmental Hematopoietic Stem Cell Gives Rise to Innate-like B and T Cells. Cell Stem Cell 2016, 19, 768–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietras, E.M. Inflammation: A key regulator of hematopoietic stem cell fate in health and disease. Blood 2017, 130, 1693–1698. [Google Scholar] [CrossRef] [Green Version]
- Haas, S.; Hansson, J.; Klimmeck, D.; Loeffler, D.; Velten, L.; Uckelmann, H.; Wurzer, S.; Prendergast, Á.; Schnell, A.; Hexel, K.; et al. Inflammation-Induced Emergency Megakaryopoiesis Driven by Hematopoietic Stem Cell-like Megakaryocyte Progenitors. Cell Stem Cell 2015, 17, 422–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matatall, K.A.; Shen, C.C.; Challen, G.A.; King, K.Y. Type II interferon promotes differentiation of myeloid-biased hematopoietic stem cells. Stem Cells 2014, 32, 3023–3030. [Google Scholar] [CrossRef] [Green Version]
- Pietras, E.M.; Mirantes-Barbeito, C.; Fong, S.; Loeffler, D.; Kovtonyuk, L.V.; Zhang, S.; Lakshminarasimhan, R.; Chin, C.P.; Techner, J.M.; Will, B.; et al. Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat. Cell Biol. 2016, 18, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Benz, C.; Copley, M.R.; Kent, D.G.; Wohrer, S.; Cortes, A.; Aghaeepour, N.; Ma, E.; Mader, H.; Rowe, K.; Day, C.; et al. Hematopoietic stem cell subtypes expand differentially during development and display distinct lymphopoietic programs. Cell Stem Cell 2012, 10, 273–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowie, M.B.; McKnight, K.D.; Kent, D.G.; McCaffrey, L.; Hoodless, P.A.; Eaves, C.J. Hematopoietic stem cells proliferate until after birth and show a reversible phase-specific engraftment defect. J. Clin. Investig. 2006, 116, 2808–2816. [Google Scholar] [CrossRef] [PubMed]
- Bowie, M.B.; Kent, D.G.; Dykstra, B.; McKnight, K.D.; McCaffrey, L.; Hoodless, P.A.; Eaves, C.J. Identification of a new intrinsically timed developmental checkpoint that reprograms key hematopoietic stem cell properties. Proc. Natl. Acad. Sci. USA 2007, 104, 5878–5882. [Google Scholar] [CrossRef] [Green Version]
- Matsuoka, S.; Ebihara, Y.; Xu, M.; Ishii, T.; Sugiyama, D.; Yoshino, H.; Ueda, T.; Manabe, A.; Tanaka, R.; Ikeda, Y.; et al. CD34 expression on long-term repopulating hematopoietic stem cells changes during developmental stages. Blood 2001, 97, 419–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christensen, J.L.; Weissman, I.L. Flk-2 is a marker in hematopoietic stem cell differentiation: A simple method to isolate long-term stem cells. Proc. Natl. Acad. Sci. USA 2001, 98, 14541–14546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopinath, B.; Rochtchina, E.; Wang, J.J.; Schneider, J.; Leeder, S.R.; Mitchell, P. Prevalence of age-related hearing loss in older adults: Blue Mountains Study. Arch. Intern. Med. 2009, 169, 415–416. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, D.L.; Lucas, J.W.; Clarke, T.C. Summary health statistics for U.S. adults: National health interview survey, 2012. Vital Health Stat. 2014, 10, 1–161. [Google Scholar]
- Centers for Disease Control and Prevention. Identifying infants with hearing loss—United States, 1999–2007. MMWR Morb. Mortal. Wkly. Rep. 2010, 59, 220–223. [Google Scholar]
- Smith, R.J.; Bale, J.F., Jr.; White, K.R. Sensorineural hearing loss in children. Lancet 2005, 365, 879–890. [Google Scholar] [CrossRef]
- Leal, M.C.; Muniz, L.F.; Ferreira, T.S.; Santos, C.M.; Almeida, L.C.; Van Der Linden, V.; Ramos, R.C.; Rodrigues, L.C.; Neto, S.S. Hearing Loss in Infants with Microcephaly and Evidence of Congenital Zika Virus Infection - Brazil, November 2015–May 2016. MMWR Morb. Mortal. Wkly. Rep. 2016, 65, 917–919. [Google Scholar] [CrossRef] [Green Version]
- Engman, M.L.; Malm, G.; Engstrom, L.; Petersson, K.; Karltorp, E.; Tear Fahnehjelm, K.; Uhlen, I.; Guthenberg, C.; Lewensohn-Fuchs, I. Congenital CMV infection: Prevalence in newborns and the impact on hearing deficit. Scand. J. Infect. Dis. 2008, 40, 935–942. [Google Scholar] [CrossRef]
- Kenneson, A.; Cannon, M.J. Review and meta-analysis of the epidemiology of congenital cytomegalovirus (CMV) infection. Rev. Med. Virol. 2007, 17, 253–276. [Google Scholar] [CrossRef]
- Stehel, E.K.; Shoup, A.G.; Owen, K.E.; Jackson, G.L.; Sendelbach, D.M.; Boney, L.F.; Sanchez, P.J. Newborn hearing screening and detection of congenital cytomegalovirus infection. Pediatrics 2008, 121, 970–975. [Google Scholar] [CrossRef]
- Cannon, M.J. Congenital cytomegalovirus (CMV) epidemiology and awareness. J. Clin. Virol. 2009, 46 (Suppl. 4), S6–S10. [Google Scholar] [CrossRef] [PubMed]
- Stratton, K.R.; Durch, J.S.; Lawrence, R.S. (Eds.) Vaccines for the 21st Century: A Tool for Decisionmaking; National Academies Press: Washington, DC, USA, 2001. [Google Scholar]
- Torre, P., 3rd; Zeldow, B.; Hoffman, H.J.; Buchanan, A.; Siberry, G.K.; Rice, M.; Sirois, P.A.; Williams, P.L. Hearing loss in perinatally HIV-infected and HIV-exposed but uninfected children and adolescents. Pediatr. Infect Dis. J. 2012, 31, 835–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, E.; Cradock-Watson, J.E.; Pollock, T.M. Consequences of confirmed maternal rubella at successive stages of pregnancy. Lancet 1982, 2, 781–784. [Google Scholar] [CrossRef]
- Foulon, I.; De Brucker, Y.; Buyl, R.; Lichtert, E.; Verbruggen, K.; Piérard, D.; Camfferman, F.A.; Gucciardo, L.; Gordts, F. Hearing Loss With Congenital Cytomegalovirus Infection. Pediatrics 2019, 144, e20183095. [Google Scholar] [CrossRef]
- Lanzieri, T.M.; Chung, W.; Leung, J.; Caviness, A.C.; Baumgardner, J.L.; Blum, P.; Bialek, S.R.; Demmler-Harrison, G. Hearing Trajectory in Children with Congenital Cytomegalovirus Infection. Otolaryngol..–Head Neck Surg. 2018, 158, 736–744. [Google Scholar] [CrossRef]
- Lanzieri, T.M.; Chung, W.; Flores, M.; Blum, P.; Caviness, A.C.; Bialek, S.R.; Grosse, S.D.; Miller, J.A.; Demmler-Harrison, G. Hearing Loss in Children With Asymptomatic Congenital Cytomegalovirus Infection. Pediatrics 2017, 139, e20162610. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Gomez, E.; Perez-Carpena, P.; Flook, M.; Lopez-Escamez, J.A. A Systematic Review on the Association of Acquired Human Cytomegalovirus Infection with Hearing Loss. J. Clin. Med. 2020, 9, 4011. [Google Scholar] [CrossRef]
- Nomoto, H.; Ishikane, M.; Nakamoto, T.; Ohta, M.; Morioka, S.; Yamamoto, K.; Kutsuna, S.; Tezuka, S.; Kunimatsu, J.; Ohmagari, N. Conjunctivitis, the key clinical characteristic of adult rubella in Japan during two large outbreaks, 2012–2013 and 2018–2019. PLoS ONE 2020, 15, e0231966. [Google Scholar] [CrossRef]
- Wild, N.J.; Sheppard, S.; Smithells, R.W.; Holzel, H.; Jones, G. Onset and severity of hearing loss due to congenital rubella infection. Arch. Dis. Child. 1989, 64, 1280–1283. [Google Scholar] [CrossRef] [Green Version]
- Williamson, W.D.; Demmler, G.J.; Percy, A.K.; Catlin, F.I. Progressive hearing loss in infants with asymptomatic congenital cytomegalovirus infection. Pediatrics 1992, 90, 862–866. [Google Scholar] [CrossRef]
- Fowler, K.B.; McCollister, F.P.; Dahle, A.J.; Boppana, S.; Britt, W.J.; Pass, R.F. Progressive and fluctuating sensorineural hearing loss in children with asymptomatic congenital cytomegalovirus infection. J. Pediatr. 1997, 130, 624–630. [Google Scholar] [CrossRef]
- Korver, A.M.; Smith, R.J.; Van Camp, G.; Schleiss, M.R.; Bitner-Glindzicz, M.A.; Lustig, L.R.; Usami, S.I.; Boudewyns, A.N. Congenital hearing loss. Nat. Rev. Dis. Primers. 2017, 3, 16094. [Google Scholar] [CrossRef] [Green Version]
- Brown, E.D.; Chau, J.K.; Atashband, S.; Westerberg, B.D.; Kozak, F.K. A systematic review of neonatal toxoplasmosis exposure and sensorineural hearing loss. Int. J. Pediatr. Otorhinolaryngol. 2009, 73, 707–711. [Google Scholar] [CrossRef] [PubMed]
- McLeod, R.; Boyer, K.; Karrison, T.; Kasza, K.; Swisher, C.; Roizen, N.; Jalbrzikowski, J.; Remington, J.; Heydemann, P.; Noble, A.G.; et al. Outcome of Treatment for Congenital Toxoplasmosis, 1981–2004: The National Collaborative Chicago-Based, Congenital Toxoplasmosis Study. Clin. Infect. Dis. 2006, 42, 1383–1394. [Google Scholar] [CrossRef] [Green Version]
- Zoller, M.; Wilson, W.R.; Nadol, J.B. Treatment of Syphilitic Hearing Loss:Combined Penicillin and Steroid Therapy in 29 Patients. Ann. Otol. Rhinol. Laryngol. 1979, 88, 160–165. [Google Scholar] [CrossRef] [PubMed]
- van den Boogaard, J.; de Gier, B.; de Oliveira Bressane Lima, P.; Desai, S.; de Melker, H.E.; Hahné, S.J.M.; Veldhuijzen, I.K. Immunogenicity, duration of protection, effectiveness and safety of rubella containing vaccines: A systematic literature review and meta-analysis. Vaccine 2021, 39, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Rawlinson, W.D.; Boppana, S.B.; Fowler, K.B.; Kimberlin, D.W.; Lazzarotto, T.; Alain, S.; Daly, K.; Doutré, S.; Gibson, L.; Giles, M.L.; et al. Congenital cytomegalovirus infection in pregnancy and the neonate: Consensus recommendations for prevention, diagnosis, and therapy. Lancet Infect. Dis. 2017, 17, e177–e188. [Google Scholar] [CrossRef]
- Bradford, R.D.; Yoo, Y.-G.; Golemac, M.; Pugel, E.P.; Jonjic, S.; Britt, W.J. Murine CMV-induced hearing loss is associated with inner ear inflammation and loss of spiral ganglia neurons. PLoS Pathog. 2015, 11, e1004774. [Google Scholar] [CrossRef] [Green Version]
- Schachtele, S.J.; Mutnal, M.B.; Schleiss, M.R.; Lokensgard, J.R. Cytomegalovirus-induced sensorineural hearing loss with persistent cochlear inflammation in neonatal mice. J. Neurovirol. 2011, 17, 201–211. [Google Scholar] [CrossRef] [Green Version]
- Schraff, S.A.; Schleiss, M.R.; Brown, D.K.; Meinzen-Derr, J.; Choi, K.Y.; Greinwald, J.H.; Choo, D.I. Macrophage inflammatory proteins in cytomegalovirus-related inner ear injury. Otolaryngol.–Head Neck Surg. 2007, 137, 612–618. [Google Scholar] [CrossRef]
- Huygens, A.; Dauby, N.; Vermijlen, D.; Marchant, A. Immunity to cytomegalovirus in early life. Front. Immunol. 2014, 5, 552. [Google Scholar] [CrossRef] [PubMed]
- Slavuljica, I.; Kvestak, D.; Huszthy, P.C.; Kosmac, K.; Britt, W.J.; Jonjic, S. Immunobiology of congenital cytomegalovirus infection of the central nervous system-the murine cytomegalovirus model. Cell Mol. Immunol. 2015, 12, 180–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baiduc, R.R.; Poling, G.L.; Hong, O.; Dhar, S. Clinical measures of auditory function: The cochlea and beyond. Dis. Mon. 2013, 59, 147–156. [Google Scholar] [CrossRef] [Green Version]
- Juhn, S.K.; Hunter, B.A.; Odland, R.M. Blood-labyrinth barrier and fluid dynamics of the inner ear. Int. Tinnitus. J. 2001, 7, 72–83. [Google Scholar] [PubMed]
- Kemp, D.T. Evidence of mechanical nonlinearity and frequency selective wave amplification in the cochlea. Arch. Otorhinolaryngol. 1979, 224, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Gravel, J.S.; Traquina, D.N. Experience with the audiologic assessment of infants and toddlers. Int. J. Pediatr. Otorhinolaryngol. 1992, 23, 59–71. [Google Scholar] [CrossRef]
- Liden, G.; Kankkunen, A. Visual reinforcement audiometry. Acta Otolaryngol. 1969, 67, 281–292. [Google Scholar] [CrossRef]
- Hickox, A.E.; Larsen, E.; Heinz, M.G.; Shinobu, L.; Whitton, J.P. Translational issues in cochlear synaptopathy. Hear. Res. 2017, 349, 164–171. [Google Scholar] [CrossRef]
- Kujawa, S.G.; Liberman, M.C. Adding insult to injury: Cochlear nerve degeneration after “temporary” noise-induced hearing loss. J. Neurosci. 2009, 29, 14077–14085. [Google Scholar] [CrossRef] [Green Version]
- Patel, R.J.; Wang, Y.; Park, A.H. Development of a Murine Model for CMV-Induced Hearing Loss. Otolaryngol.–Head Neck Surg. 2012, 147, P230. [Google Scholar] [CrossRef]
- Almishaal, A.; Mathur, P.D.; Franklin, L.; Shi, K.; Haller, T.; Martinovic, A.; Hirschmugl, K.; Earl, B.R.; Zhang, C.; Yang, J.; et al. Role of cochlear synaptopathy in cytomegalovirus infected mice and in children. Int. J. Pediatr. Otorhinolaryngol. 2020, 138, 110275. [Google Scholar] [CrossRef] [PubMed]
- Almishaal, A.A.; Mathur, P.D.; Hillas, E.; Chen, L.; Zhang, A.; Yang, J.; Wang, Y.; Yokoyama, W.M.; Firpo, M.A.; Park, A.H. Natural killer cells attenuate cytomegalovirus-induced hearing loss in mice. PLoS Pathog. 2017, 13, e1006599. [Google Scholar] [CrossRef] [PubMed]
- Carraro, M.; Almishaal, A.; Hillas, E.; Firpo, M.; Park, A.; Harrison, R.V. Cytomegalovirus (CMV) Infection Causes Degeneration of Cochlear Vasculature and Hearing Loss in a Mouse Model. J. Assoc. Res. Otolaryngol. 2017, 18, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.P. Immunology of the inner ear: Evidence of local antibody production. Ann. Otol. Rhinol. Laryngol. 1984, 93, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Nyberg, S.; Abbott, N.J.; Shi, X.; Steyger, P.S.; Dabdoub, A. Delivery of therapeutics to the inner ear: The challenge of the blood-labyrinth barrier. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Bielefeld, E.C.; Kobel, M.J. Advances and challenges in pharmaceutical therapies to prevent and repair cochlear injuries from noise. Front. Cell. Neurosci. 2019, 13, 285. [Google Scholar] [CrossRef]
- Hirose, K.; Li, S.-Z. The role of monocytes and macrophages in the dynamic permeability of the blood-perilymph barrier. Physiol. Behav. 2019, 374, 49–57. [Google Scholar] [CrossRef]
- Hirose, K.; Discolo, C.M.; Keasler, J.R.; Ransohoff, R. Mononuclear phagocytes migrate into the murine cochlea after acoustic trauma. J. Comp. Neurol. 2005, 489, 180–194. [Google Scholar] [CrossRef]
- Rai, V.; Wood, M.B.; Feng, H.; Schabla, N.M.; Tu, S.; Zuo, J. The immune response after noise damage in the cochlea is characterized by a heterogeneous mix of adaptive and innate immune cells. Sci. Rep. 2020, 10, 15167. [Google Scholar] [CrossRef]
- Shi, X. Resident macrophages in the cochlear blood-labyrinth barrier and their renewal via migration of bone-marrow-derived cells. Cell Tissue Res. 2010, 342, 21–30. [Google Scholar] [CrossRef]
- Okano, T.; Nakagawa, T.; Kita, T.; Kada, S.; Yoshimoto, M.; Nakahata, T.; Ito, J. Bone marrow-derived cells expressing Iba1 are constitutively present as resident tissue macrophages in the mouse cochlea. J. Neurosci. Res. 2008, 86, 1758–1767. [Google Scholar] [CrossRef] [PubMed]
- Matern, M.; Vijayakumar, S.; Margulies, Z.; Milon, B.; Song, Y.; Elkon, R.; Zhang, X.; Jones, S.M.; Hertzano, R. Gfi1Cre mice have early onset progressive hearing loss and induce recombination in numerous inner ear non-hair cells. Sci. Rep. 2017, 7, 42079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishimoto, I.; Okano, T.; Nishimura, K.; Motohashi, T.; Omori, K. Early Development of Resident Macrophages in the Mouse Cochlea Depends on Yolk Sac Hematopoiesis. Front. Neurol. 2019, 10, 1115. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Kaga, K. Development of blood-labyrinth barrier in the semicircular canal ampulla of the rat. Hear. Res. 1999, 129, 27–34. [Google Scholar] [CrossRef]
- Dai, C.F.; Mangiardi, D.; Cotanche, D.A.; Steyger, P.S. Uptake of fluorescent gentamicin by vertebrate sensory cells in vivo. Hear. Res. 2006, 213, 64–78. [Google Scholar] [CrossRef] [Green Version]
- Okano, T.; Kishimoto, I. Csf1 Signaling Regulates Maintenance of Resident Macrophages and Bone Formation in the Mouse Cochlea. Front. Neurol. 2019, 10, 1244. [Google Scholar] [CrossRef] [Green Version]
- Booth, J.S.; Toapanta, F.R. B and T Cell Immunity in Tissues and Across the Ages. Vaccines 2021, 9, 24. [Google Scholar] [CrossRef]
- Hashemi, E.; Malarkannan, S. Tissue-Resident NK Cells: Development, Maturation, and Clinical Relevance. Cancers 2020, 12, 1553. [Google Scholar] [CrossRef]
- Masopust, D.; Soerens, A.G. Tissue-Resident T Cells and Other Resident Leukocytes. Annu. Rev. Immunol. 2019, 37, 521–546. [Google Scholar] [CrossRef]
- Ng, L.G.; Qin, J.S.; Roediger, B.; Wang, Y.; Jain, R.; Cavanagh, L.L.; Smith, A.L.; Jones, C.A.; de Veer, M.; Grimbaldeston, M.A.; et al. Visualizing the neutrophil response to sterile tissue injury in mouse dermis reveals a three-phase cascade of events. J. Investig. Dermatol. 2011, 131, 2058–2068. [Google Scholar] [CrossRef] [Green Version]
- Sojka, D.K.; Plougastel-Douglas, B.; Yang, L.; Pak-Wittel, M.A.; Artyomov, M.N.; Ivanova, Y.; Zhong, C.; Chase, J.M.; Rothman, P.B.; Yu, J.; et al. Tissue-resident natural killer (NK) cells are cell lineages distinct from thymic and conventional splenic NK cells. eLife 2014, 3, e01659. [Google Scholar] [CrossRef] [PubMed]
- Dogra, P.; Rancan, C.; Ma, W.; Toth, M.; Senda, T.; Carpenter, D.J.; Kubota, M.; Matsumoto, R.; Thapa, P.; Szabo, P.A.; et al. Tissue Determinants of Human NK Cell Development, Function, and Residence. Cell 2020, 180, 749–763. [Google Scholar] [CrossRef] [Green Version]
- Luangrath, M.A.; Schmidt, M.E.; Hartwig, S.M.; Varga, S.M. Tissue-Resident Memory T Cells in the Lungs Protect against Acute Respiratory Syncytial Virus Infection. Immunohorizons 2021, 5, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Lebrun, A.; Kean, R.B.; Hooper, D.C. Brain tissue-resident immune memory cells are required for long-term protection against CNS infection with rabies virus. Future Virol. 2020, 15, 755–761. [Google Scholar] [CrossRef]
- Johnson, R.M.; Brunham, R.C. Tissue-Resident T Cells as the Central Paradigm of Chlamydia Immunity. Infect. Immun. 2016, 84, 868–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casanova-Acebes, M.; Nicolas-Avila, J.A.; Li, J.L.; Garcia-Silva, S.; Balachander, A.; Rubio-Ponce, A.; Weiss, L.A.; Adrover, J.M.; Burrows, K.; N, A.G.; et al. Neutrophils instruct homeostatic and pathological states in naive tissues. J. Exp. Med. 2018, 215, 2778–2795. [Google Scholar] [CrossRef]
- Jan, T.A.; Chai, R.; Sayyid, Z.N.; Cheng, A.G. Isolating LacZ-expressing Cells from Mouse Inner Ear Tissues using Flow Cytometry. JoVE 2011, e3432. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Patel, R.; Ren, C.; Taggart, M.G.; Firpo, M.A.; Schleiss, M.R.; Park, A.H. A comparison of different murine models for cytomegalovirus-induced sensorineural hearing loss. Laryngoscope 2013, 123, 2801–2806. [Google Scholar] [CrossRef]
- Sung, C.Y.W.; Seleme, M.C.; Payne, S.; Jonjic, S.; Hirose, K.; Britt, W. Virus-induced cochlear inflammation in newborn mice alters auditory function. JCI Insight 2019, 4, e128878. [Google Scholar] [CrossRef]
- Pecha, P.P.; Almishaal, A.A.; Mathur, P.D.; Hillas, E.; Johnson, T.; Price, M.S.; Haller, T.; Yang, J.; Rajasekaran, N.S.; Firpo, M.A.; et al. Role of Free Radical Formation in Murine Cytomegalovirus-Induced Hearing Loss. Otolaryngol.–Head Neck Surg. 2020, 162, 709–717. [Google Scholar] [CrossRef]
- Tsuprun, V.; Keskin, N.; Schleiss, M.R.; Schachern, P.; Cureoglu, S. Cytomegalovirus-induced pathology in human temporal bones with congenital and acquired infection. Am. J. Otolaryngol. 2019, 40, 102270. [Google Scholar] [CrossRef] [PubMed]
- Teissier, N.; Delezoide, A.L.; Mas, A.E.; Khung-Savatovsky, S.; Bessières, B.; Nardelli, J.; Vauloup-Fellous, C.; Picone, O.; Houhou, N.; Oury, J.F.; et al. Inner ear lesions in congenital cytomegalovirus infection of human fetuses. Acta Neuropathol. 2011, 122, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Haller, T.J.; Price, M.S.; Lindsay, S.R.; Hillas, E.; Seipp, M.; Firpo, M.A.; Park, A.H. Effects of ganciclovir treatment in a murine model of cytomegalovirus-induced hearing loss. Laryngoscope 2020, 130, 1064–1069. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Otsuka, K.S.; Nielson, C.; Firpo, M.A.; Park, A.H.; Beaudin, A.E. Early Life Inflammation and the Developing Hematopoietic and Immune Systems: The Cochlea as a Sensitive Indicator of Disruption. Cells 2021, 10, 3596. https://doi.org/10.3390/cells10123596
Otsuka KS, Nielson C, Firpo MA, Park AH, Beaudin AE. Early Life Inflammation and the Developing Hematopoietic and Immune Systems: The Cochlea as a Sensitive Indicator of Disruption. Cells. 2021; 10(12):3596. https://doi.org/10.3390/cells10123596
Chicago/Turabian StyleOtsuka, Kelly S., Christopher Nielson, Matthew A. Firpo, Albert H. Park, and Anna E. Beaudin. 2021. "Early Life Inflammation and the Developing Hematopoietic and Immune Systems: The Cochlea as a Sensitive Indicator of Disruption" Cells 10, no. 12: 3596. https://doi.org/10.3390/cells10123596
APA StyleOtsuka, K. S., Nielson, C., Firpo, M. A., Park, A. H., & Beaudin, A. E. (2021). Early Life Inflammation and the Developing Hematopoietic and Immune Systems: The Cochlea as a Sensitive Indicator of Disruption. Cells, 10(12), 3596. https://doi.org/10.3390/cells10123596